Mark Cuban built his fortune by spotting inefficient markets and charging in with a simpler model. The Dallas Mavericks. HDNet. Broadcast.com. Each time, he identified a bloated incumbent and undercut it on cost and transparency. When he turned his attention to pharmaceutical pricing in 2022, the same instinct drove him: the supply chain is absurd, the middlemen are extractive, and the solution is to cut them out.
He was right—and he was also, in a specific and consequential way, tackling the easier half of the problem.
Cost Plus Drugs—formally the Mark Cuban Cost Plus Drug Company (MCCPDC)—has delivered genuine results for patients. Imatinib, a leukemia drug that retails at more than $2,500 per month at a traditional pharmacy, runs $14.40 at Cost Plus Drugs. Albendazole, a parasitic infection treatment that Shkreli-era pharma repriced to $500 per course, sells for $35. The numbers are real. The savings are real. The model—purchase at net acquisition cost, add a 15% markup, $5 pharmacy labor fee, $5 shipping—is transparent in a way the rest of the industry decidedly is not.
But here is the structural reality underneath those headlines: every drug in that catalog is a generic. Off-patent. Post-exclusivity. Subject to commodity-style competition from dozens of manufacturers. And that matters enormously when you try to understand both what Cuban’s model accomplishes and what it cannot touch.
Generic drugs account for roughly 90% of all prescriptions dispensed in the United States. They also account for roughly 20% of total drug expenditure. Brand-name drugs fill the other 10% of prescriptions—and consume 80% of the dollars. That inversion is the central economic fact of American pharmaceutical pricing, and it is the reason why even the most efficient possible generic distribution model leaves most of the price problem intact.
This article is not a critique of Cuban’s intentions or execution. It is an analysis of the structural divide between the generic drug market—where cost-plus transparency can meaningfully lower prices—and the branded drug market, where a patent-protected manufacturer, a rebate-dependent PBM ecosystem, and an increasingly aggressive regulatory environment interact in ways that no single distributor can resolve. Understanding that divide is not optional for anyone who invests in, negotiates with, or depends on the pharmaceutical industry.
Part I: The Generic Drug Market and Why Transparency Can Win There
How Generic Pricing Actually Works
A generic drug is a copy of a brand-name drug whose patents have expired. Under the Hatch-Waxman Act of 1984, a generic manufacturer can file an Abbreviated New Drug Application (ANDA) with the FDA, demonstrating that its product is bioequivalent to the original without replicating the full clinical trial process. Once approved, the generic can enter the market, and competition typically drives prices down sharply—the FDA estimates generics end up roughly 80% to 85% cheaper than their brand-name counterparts.
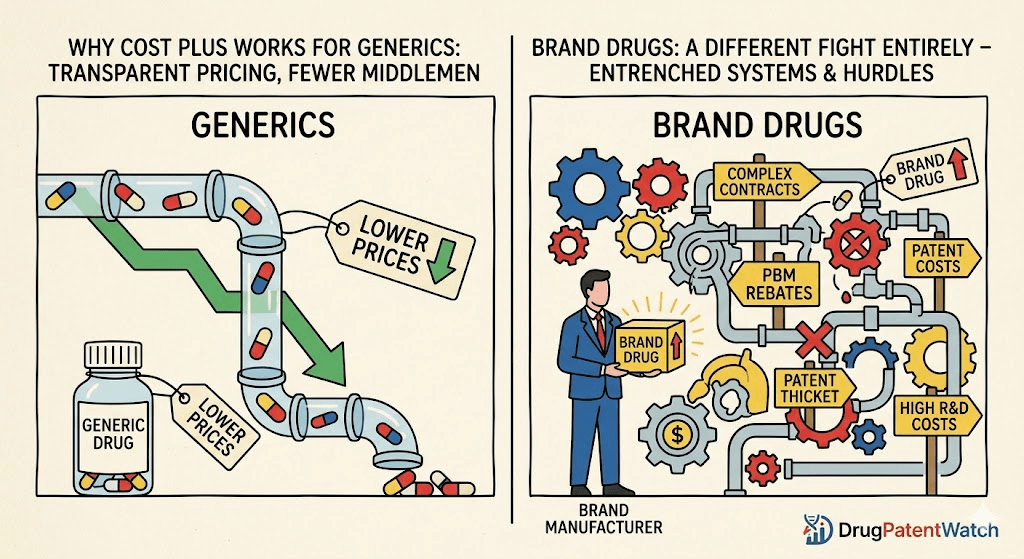
The key economic characteristic of the generic market is that exclusivity is gone. Without a patent preventing competition, any number of manufacturers can produce and sell the same molecule. This is a commodity market in the classical sense: multiple suppliers, standardized product, price as the primary competitive variable.
In that environment, the traditional pharmaceutical supply chain—manufacturer to wholesaler to pharmacy benefit manager (PBM) to insurer to retail pharmacy to patient—adds cost at every step without adding clinical value. PBMs are especially consequential here. Their business model on generic drugs differs from branded drugs: rather than negotiating rebates from manufacturers (who have little margin to give), PBMs profit from “spread pricing”—charging an insurance plan more for a drug than they pay the pharmacy to dispense it, pocketing the difference. That spread is invisible to patients and plan sponsors alike.
Cost Plus Drugs attacks this specific inefficiency directly. By negotiating directly with manufacturers, bypassing the PBM layer entirely, and publishing its full pricing stack—acquisition cost, markup, pharmacy fee, shipping—the company eliminates the information asymmetry that makes spread pricing possible. For a 30-day supply of a generic that costs $4 to manufacture and distribute, this is transformative.
The Real Limits of Cuban’s Generic Model
There are genuine constraints on Cost Plus Drugs’ impact even within generics, and they are worth understanding precisely because they are often glossed over in coverage of the company.
First, the company’s prices are not always the lowest available. A 2023 analysis of 211 Cost Plus Drugs generic offerings found that at least 141—67%—were priced higher than one or more competitors on GoodRx. Amazon Pharmacy, Costco Pharmacy, and various state Medicaid programs all operate in the generic space with considerable buying power. A June 2024 analysis found that switching to Cost Plus Drugs from existing insurance coverage would save money in fewer than 12% of generic drug cases.
Second, generics are already cheap. American generic drug prices average 15% lower than comparable products in other OECD countries. The crisis in U.S. pharmaceutical spending is not primarily a generic drug crisis. As Ezekiel Emanuel and John Connolly wrote in STAT News in 2024: “The more fundamental reason Cuban’s company will not really lower drug prices much is that it deals almost entirely in off-patent generic drugs. But generics are already low-priced.”
Third, Cost Plus Drugs’ reach is structurally limited. It operates as a direct-to-consumer mail-order pharmacy and accepts a limited number of health insurance plans. Most patients who use it pay out of pocket, and that spending does not count toward insurance deductibles or out-of-pocket maximums. For a patient with high-deductible coverage who is working toward their annual limit, paying $14 for imatinib at Cost Plus Drugs instead of $2,500 at a retail pharmacy is clearly the right financial choice—but a patient close to hitting their deductible gets more value from running the full retail price through their plan.
Where Cuban’s model does work, and works well, is in three specific segments: uninsured or underinsured patients, patients on high-deductible plans who haven’t met their deductible, and employers and health plans willing to build Cost Plus Drugs into their benefit design as a supplemental channel. The Humana partnership announced in late 2025 to target employer groups is a direct attempt to expand into that third category.
Drug Shortages: Where Cuban’s Manufacturing Bet May Pay Off Most
The most underappreciated dimension of Cost Plus Drugs is not its pharmacy business but its manufacturing investment. In March 2024, the company opened a 22,000-square-foot fill-and-finish facility in Dallas—a $11 million plant equipped with robotic filling lines capable of pivoting between drug types within four hours. Its initial products were epinephrine and norepinephrine, ICU drugs that had experienced repeated national shortages.
This matters because generic drug shortages are a market failure that retail pricing transparency cannot fix. The problem is not PBM spread pricing; it is that the economics of manufacturing cheap generic injectables are so thin that manufacturers chronically under-invest in capacity. When one supplier has a quality problem, the entire market can go short. The FDA currently lists more than 100 drugs in shortage, including antibiotics, chemotherapies, and anesthetics.
Cuban’s manufacturing investment addresses a real structural gap that his pharmacy cannot. “We don’t have unlimited capacity,” he told the HLTH conference in early 2024, “but if we do this right, over the next five years, there will no longer be any more shortages in sterile injectables.” That claim may be ambitious, but the investment rationale is sound. Domestic manufacturing with rapid changeover capability is the right structural response to a fragile generic supply chain dependent on overseas facilities with inconsistent quality standards.
The caveat is that manufacturing generics at scale requires capital, regulatory expertise, and lead time that stretch well beyond what a single facility can provide. Cuban himself acknowledged the company was “not making money yet” as of early 2024. Growing into a manufacturer capable of meaningfully affecting national generic supply is a multi-year, capital-intensive project—not a disruptive hack.
Part II: The Brand Drug Market—A Completely Different System
Why the Same Model Cannot Transfer
Take everything that makes Cost Plus Drugs work on generics—commodity competition, transparent acquisition costs, a willingness to sell without insurance involvement—and apply it to a branded drug still under patent protection. None of it works.
A brand-name drug with active patent coverage has exactly one legal supplier: the manufacturer. There is no competing source to buy from, no market-clearing price established by competition, no commodity pricing to be transparent about. The manufacturer sets the Wholesale Acquisition Cost (WAC)—the list price—and every other price in the system flows from that number. Cost Plus Drugs cannot negotiate a lower acquisition cost for Eliquis or Keytruda or Dupixent because AbbVie, Bristol-Myers Squibb/Pfizer, Merck, and Sanofi/Regeneron have the only supply and no legal obligation to sell at a lower price.
Dr. José Pagán, chair of the Department of Public Health Policy and Management at NYU, put it directly in 2022: “Cuban’s pharmacy currently focuses on lowering the costs of those generic drugs by side-stepping PBMs. Drugmakers usually only accept less money for their brand-name drugs when they offer rebates to those pharmacy benefit managers.”
That sentence contains the entire structural explanation. Brand manufacturers use the PBM rebate system not as a cost to be minimized but as a competitive tool. They offer large rebates to PBMs in exchange for preferred formulary placement—the tiered list of drugs that insurance plans cover and at what patient cost-sharing level. A manufacturer who pays a large rebate gets Tier 2 status (low patient copay, high utilization) while a competitor who pays less gets pushed to Tier 3 or excluded entirely. The rebate is, in effect, a placement fee paid backward through the supply chain.
This system is economically irrational in the way that only a highly regulated, third-party-payer market can sustain. The manufacturer sets a high list price. The PBM demands a large rebate on that list price. The manufacturer offers the rebate, which incentivizes PBMs to use that drug. The net price to the payer may actually be competitive—but the patient, who pays cost-sharing based on the list price, not the net price, sees a high number at the pharmacy counter regardless.
“Brand-name drugs make up a much smaller share of prescriptions than generic drugs, yet they accounted for nearly 80% of prescription drug expenditures in 2018. Generic drugs end up around 80% to 85% less expensive than their brand-name equivalents.”— U.S. Food and Drug Administration, as cited in CNBC coverage of Cost Plus Drugs [1]
The Scale of the Gross-to-Net Bubble
For context: that $356 billion is larger than the entire pharmaceutical revenue of any single country outside the United States. It is money that flows between manufacturers and PBMs in a system so opaque that patients and policymakers alike struggle to understand what drugs actually cost. The gross-to-net bubble has expanded by roughly 10% per year in recent years, even as list price growth slowed. Manufacturers raised list prices modestly; PBMs demanded larger rebates; the spread widened.
But this deflation is happening within the existing system, driven by the IRA’s Medicare negotiation program and market forces on specific high-profile products. It does not represent a structural change to how branded drug prices are set for the majority of the market. A cost-plus distributor cannot accelerate that process because the distributor has no leverage over a manufacturer who is the sole legal supplier of a patent-protected drug.
Patent Protection: The Foundation of Brand Drug Pricing Power
Understanding branded drug pricing requires understanding pharmaceutical patent law, because patents are the mechanism that creates the entire pricing dynamic. The government grants a 20-year exclusivity period from the filing date in exchange for public disclosure of the invention. During that window, the manufacturer is the only legal source of the molecule. Competition is prohibited by law.
In practice, “20 years” understates how long exclusivity can last. Brand manufacturers have developed a suite of IP management strategies that extend effective market protection well beyond the primary patent:
Patent thickets: Filing dozens or hundreds of secondary patents on formulations, delivery mechanisms, manufacturing processes, and minor molecular modifications. Each secondary patent expires later than the primary and must be litigated separately by any generic challenger. AbbVie built a 132-patent fortress around Humira (adalimumab), delaying U.S. biosimilar competition until January 2023—nearly 20 years after FDA approval in 2002 and seven years after biosimilar competition began in Europe. Over two decades, Humira accumulated more than $200 billion in lifetime sales. The primary patent expired in 2016. The patent thicket held competitors off for seven more years.
Product hopping: Reformulating the drug—often with a minor clinical improvement like reduced injection-site pain or a once-daily formulation—and switching the market to the new version before the old patent expires. When AbbVie faced biosimilar entry based on the original Humira formulation, it had already migrated much of the market to a citrate-free formulation covered by newer patents. Biosimilar developers who had spent years matching the original formulation now faced a product the market had moved away from.
Evergreening: The broader strategy of accumulating new IP protections on an aging drug to prevent or slow competitive entry. Revlimid (lenalidomide), Celgene’s multiple myeloma blockbuster, carried a portfolio of 30 patents with 18 terminal disclaimers, insulating it from generic competition well past the primary patent’s expiration date.
Orange Book gaming: The FDA’s Orange Book lists patents associated with approved drugs, and any generic challenger must file a Paragraph IV certification asserting those patents are invalid or not infringed. Brand manufacturers have incentive to list as many patents as possible in the Orange Book—including patents of questionable validity—because each listing potentially triggers a 30-month litigation stay upon Paragraph IV challenge. The FTC launched an enforcement campaign in 2023 targeting device patents listed in connection with combination products, which the agency viewed as an improper extension of Hatch-Waxman protections to patents the statute was not designed to cover.
Data from DrugPatentWatch, which tracks pharmaceutical patent portfolios and litigation activity across the industry, consistently shows that the most commercially significant branded drugs carry the densest and most strategically layered patent estates. Tracking those estates—understanding which patents are approaching expiration, which are subject to active Paragraph IV challenges, and which have been listed in settlement agreements with generic entry dates—is fundamental to anticipating when and how brand drug pricing power will actually erode.
Part III: Hatch-Waxman and the Patent Challenge Pipeline
How Generics Actually Get Into the Market
The Hatch-Waxman Act of 1984 created the modern U.S. generic drug market by establishing a pathway for approval without full clinical trials and by codifying a process for challenging brand-name patents before they expire. Before Hatch-Waxman, a generic manufacturer could only enter the market after all patents expired and had to conduct its own clinical trials. The economics made generic development prohibitive for most drugs.
The ANDA (Abbreviated New Drug Application) process changed that by allowing generic manufacturers to demonstrate bioequivalence—that their product delivers the same active ingredient at the same rate to the same sites in the body—without repeating the original clinical work. The critical innovation for patent purposes is the Paragraph IV certification: a generic manufacturer can file an ANDA before the brand’s patents expire, certifying that those patents are either invalid or not infringed by the generic product.
Filing a Paragraph IV certification is, by legal construction, an act of patent infringement. This matters because it gives the brand manufacturer standing to sue immediately, before the generic has sold a single pill. If the brand manufacturer sues within 45 days of receiving notice, the FDA cannot approve the ANDA for 30 months while litigation proceeds—the “30-month stay.” This mechanism gives brand companies up to 2.5 years of additional market protection on top of their patent term, even if the litigation ultimately fails.
The 180-day exclusivity period for the first generic filer is the carrot that makes Paragraph IV challenges economically viable. The first manufacturer to successfully challenge a brand patent gets a six-month window of generic market exclusivity before other generic manufacturers can enter. On a drug with $1 billion in annual brand sales, even a fraction of market share during a 180-day window is worth hundreds of millions of dollars. This creates substantial economic incentive for generic manufacturers to invest in patent challenges even when the odds of winning are uncertain and the legal costs are high.
The Litigation Battlefield
Paragraph IV litigation is where generic and brand manufacturers conduct their most consequential fights, and the dynamics of those fights directly determine when patients get access to lower-cost alternatives.
Brand manufacturers have developed increasingly sophisticated tactics to extend effective exclusivity beyond what their primary patents allow. Serial patent litigation is one: after an initial Hatch-Waxman case settles or concludes, the brand files new patent applications and initiates new rounds of litigation on those new patents against the same or different generic challengers. In the case of Astellas’s overactive bladder drug mirabegron (Myrbetriq), after an initial Hatch-Waxman case settled in 2020 with generic entry expected in 2024, Astellas pursued four additional lawsuits built on new but substantively similar patents—leaving only two manufacturers able to launch in 2024 under the threat of massive damages claims.
The Humira case is the clearest illustration of how effective a large enough patent estate is as a competitive weapon. Faced with more than 100 patents to litigate, every biosimilar challenger eventually settled. AbbVie offered European market licenses in 2018 but secured U.S. delay commitments until 2023. As part of those settlements, biosimilar manufacturers agreed to pay royalties to AbbVie on their future U.S. sales—meaning AbbVie continues to profit from Humira even after losing formal exclusivity. The strategy was not to win in court; it was to make the cost of fighting in court too high for any competitor to bear alone.
What does all of this mean for investors and plan sponsors tracking branded drug economics? It means that the nominal patent expiration date on any drug is not the date that competition arrives. The date that actually matters is the anticipated generic entry date—which depends on the outcome of Paragraph IV litigation, settlement agreements, and the strategic decisions of both the brand manufacturer and generic challengers. Resources like DrugPatentWatch specialize in exactly this kind of patent landscape intelligence, tracking the full IP estate around major drugs, monitoring active ANDA filings, and estimating likely entry timelines based on litigation progress. For a health system, insurer, or employer building a five-year drug spend forecast, generic entry timing is one of the highest-leverage variables in the model.
The Biosimilar Pathway and Its Additional Complications
Large-molecule biologic drugs—antibodies, proteins, and other complex molecules produced by living cells—are not governed by Hatch-Waxman. They operate under the Biologics Price Competition and Innovation Act (BPCIA) of 2009, which created a separate regulatory and IP framework with some important structural differences.
The BPCIA’s “patent dance” is notoriously complex: a multi-step process by which the biosimilar applicant and the reference product sponsor exchange information about patents and manufacturing processes before litigation begins. Critically, the BPCIA places no statutory limit on the number of patents a brand biologic manufacturer can assert in litigation, unlike the Orange Book system’s structural constraints under Hatch-Waxman. This is the legal loophole that enabled AbbVie’s 132-patent Humira strategy. The scale of litigation that would be procedurally difficult under Hatch-Waxman is entirely permissible under the BPCIA.
Biosimilar competition, when it arrives, tends to have a more gradual price impact than small-molecule generic competition. Under Hatch-Waxman, a successful Paragraph IV filer with 180-day exclusivity typically captures most of the generic market rapidly, driving the brand’s volume down and prices down with it. Biosimilars launch at smaller discounts—typically 15% to 35% below the brand list price initially—and market penetration is slower, partly because prescribers are more cautious about switching patients on complex biologics and partly because PBM formulary structures can still favor the brand when the manufacturer offers substantial rebates.
The Humira experience post-2023 is instructive. Despite the entry of multiple biosimilar competitors, AbbVie has maintained substantial market share through aggressive rebate offers to PBMs—essentially using the gross-to-net system it spent 20 years building to retain formulary preference even after exclusivity ended. The biosimilar manufacturers entered the market only to find that “winning” on patent litigation does not automatically translate into commercial success when the incumbent has locked up formulary positions through rebate contracts.
Part IV: The IRA and the New Branded Drug Battleground
Medicare Drug Price Negotiation: What It Actually Does
The Inflation Reduction Act of 2022 changed the structure of U.S. drug pricing more fundamentally than any legislation since Hatch-Waxman, and it did so entirely in the branded drug segment. The generic market was not the target, because the generic market was not the problem. Brand-name drugs, with their patent-protected pricing power and PBM rebate relationships, were.
The IRA’s Medicare Drug Price Negotiation Program (MDPNP) authorizes the Secretary of Health and Human Services to negotiate Maximum Fair Prices (MFPs) for certain high-expenditure, single-source brand drugs and biologics covered under Medicare Parts D and B. The qualifying criteria: no marketed generic or biosimilar substitute, at least seven years of FDA approval (for small-molecule drugs) or 11 years (for biologics), and placement among the top-spending Medicare drugs.
The first round of 10 drugs negotiated in 2023 will see negotiated prices take effect on January 1, 2026. These drugs—including Eliquis, Jardiance, Xarelto, Januvia, Entresto, Enbrel, Imbruvica, Stelara, Fiasp/NovoLog, and Farxiga—collectively accounted for roughly 20% of all Medicare Part D gross spending in 2022. The Congressional Budget Office estimates the drug negotiation provisions will save Medicare $98.5 billion over 10 years. The first round of prices is estimated to save $1.5 billion annually in patient out-of-pocket costs and $6 billion annually for the Medicare program.
The program expands each year: 15 additional drugs for 2027 and 2028, and 20 more for 2029 onward. The scope is deliberately calibrated to the brand-name, patent-protected segment. It does not touch generics because it does not need to—generics are already priced by competition. It targets exactly the drugs that the cost-plus transparency model cannot reach.
The IRA’s “Pill Penalty” Problem
One structural anomaly created by the IRA has generated significant pharmaceutical industry pushback and genuine policy debate: the differential treatment of small-molecule drugs versus biologics in determining negotiation eligibility.
Under the IRA, a small-molecule drug (traditional “pill”) becomes eligible for negotiation seven years after FDA approval. A biologic becomes eligible after 11 years. The pharmaceutical industry calls this the “pill penalty” because it creates an incentive structure that favors developing biologics over small molecules, even when small molecules might be the better scientific approach for a given condition. A small-molecule drug loses exclusivity value to CMS negotiation faster than a biologic with an equivalent commercial profile.
The Trump administration’s Executive Order 14273 of April 2025 directed HHS to work with Congress to align the treatment of small-molecule drugs with biologics—a policy change the industry has lobbied for heavily. As of mid-2026, no legislative fix has passed, though the regulatory conversation continues. For pharmaceutical R&D planning and investment strategy, the pill penalty is a real factor shaping early-stage portfolio decisions.
How Manufacturers Are Responding
The IRA has produced observable changes in brand drug pricing behavior that were not anticipated by early analyses. Research tracking price changes in the post-IRA period found that many manufacturers, anticipating negotiation, began raising prices more aggressively in their remaining pre-negotiation years to maximize the revenue base from which negotiated discounts would be calculated—essentially front-loading price increases before the CMS ceiling arrived.
In Medicare Part B, the median annual price growth rate doubled—from 6.5% to 13.3%—in the post-IRA period. This is not a coincidence. Manufacturers in Part B, where physician-administered drugs are reimbursed at a percentage of Average Sales Price, have more flexibility to raise prices ahead of their IRA eligibility window.
The gross-to-net bubble is deflating on high-profile insulin and a handful of other products where manufacturers have chosen to cut list prices, partly to avoid the rebate arithmetic that generates larger gross-to-net reductions as list prices rise. But the deflation is selective and strategic, not systemic. For the majority of branded drugs not yet subject to IRA negotiation, the pricing architecture remains intact.
Patent Expiry Cliffs and the $300 Billion Revenue Risk
Independent of the IRA, the branded pharmaceutical industry faces a structural challenge that will define the next decade: an unprecedented concentration of patent expirations. Over 190 drugs, including 69 blockbusters, are projected to lose exclusivity between 2024 and 2030, with approximately $300 billion in cumulative annual revenues at risk, according to DrugPatentWatch analysis.
Merck’s Keytruda (pembrolizumab), the PD-1 checkpoint inhibitor that generated $25 billion in 2023 sales, has core composition-of-matter patents expiring in 2028. Without ongoing lifecycle management, this represents the largest single-drug revenue cliff in pharmaceutical history. Merck’s response has been to invest heavily in subcutaneous formulations, companion diagnostics, and combination therapy approvals—each generating new IP and potentially resetting the exclusivity clock on portions of the Keytruda franchise.
AstraZeneca’s Farxiga, J&J’s Stelara, and Bristol-Myers Squibb/Pfizer’s Eliquis all face major patent challenges in the current or immediately upcoming period. For each of these, the timeline to generic or biosimilar competition is not determined by the nominal patent expiration date but by the outcome of Paragraph IV litigation, biosimilar development programs, and IRA negotiation timelines—a complex interplay that requires real-time patent intelligence to navigate.
This is precisely the domain where specialized patent monitoring services provide concrete commercial value. Tracking which drugs are approaching their primary patent expiry, which have filed Paragraph IV certifications, which patent thickets have been weakened by successful IPR (inter partes review) challenges at the USPTO, and which settlement agreements have established known generic entry dates gives commercial teams—whether on the brand side managing their franchise or on the generic/biosimilar side planning their launch—the foundation for accurate forecasting. DrugPatentWatch’s patent expiration databases and litigation tracking are built specifically for this kind of commercial intelligence work, offering granular data on the patent landscape for drugs across all therapeutic categories.
Part V: Why the Two Markets Require Two Different Strategies
The Generic Market Playbook
For stakeholders operating in the generic drug space—whether as manufacturers, payers, or employers—the Cost Plus Drugs model highlights principles that apply well beyond Cuban’s specific company.
The first principle is that PBM disintermediation works where the manufacturer has no pricing power. When multiple manufacturers compete to sell the same molecule, the acquisition cost is a true market price, and any intermediary who charges more than that acquisition cost plus legitimate distribution costs is extracting economic rent. Transparent cost-plus distribution models—whether from Cost Plus Drugs, Amazon Pharmacy’s flat-fee model, or direct employer purchasing programs—are structurally sound responses to this problem.
The second principle is that generics pricing requires surveillance. The generic drug market is not static: manufacturers enter and exit, prices spike when supply contracts, and PBM spread pricing exploits the opacity that persists even after patent exclusivity ends. Real-time generic pricing intelligence—knowing the actual acquisition cost of a molecule across multiple suppliers, understanding which drugs face shortages and why, monitoring ANDA filing activity that predicts future competitive entry—is valuable for any large health plan or employer pharmacy program.
The third principle is that the savings from generic optimization, while real, are capped. A 50% to 90% improvement on a category that represents 20% of drug spend is meaningful but not transformative. Cost Plus Drugs has demonstrated savings of 50% to 90% on plan generic spend for its employer group clients—impressive numbers that, applied to a large self-insured employer’s drug benefit, might save millions of dollars annually. But the bigger opportunity, by an order of magnitude, is in the branded space.
The Brand Drug Playbook
Brand drug economics require a fundamentally different set of tools and strategies. The levers available to payers and plan sponsors in the branded space fall into four categories: formulary management, utilization management, rebate negotiation, and market timing.
Formulary management: The most powerful tool a PBM or plan sponsor has in the branded space is formulary placement. By restricting coverage of a high-cost brand or requiring step therapy—mandating that patients try a less expensive alternative first—plans can shift utilization toward lower-net-cost options. This is why PBMs negotiate rebates in the first place: the leverage is real, and the manufacturer who wants preferred formulary placement must pay for it. The FTC’s 2024 report documented cases where PBMs steered patients toward drugs with higher list prices simply because those drugs offered larger rebates, creating perverse incentives in which clinical value was secondary to rebate economics.
Utilization management: Prior authorization requirements, quantity limits, and step therapy protocols all affect how branded drugs are used. These tools can be clinically appropriate or can reflect primarily financial considerations. Their financial impact on drug spend is significant—a specialty drug with prior authorization requirements dispenses at lower rates and in smaller quantities than one with open formulary access.
Rebate negotiation: For large payers, negotiating directly with manufacturers—or using a PBM with strong rebate negotiating capacity—is the primary mechanism for reducing effective branded drug costs. The gross-to-net bubble exists precisely because these negotiations produce large absolute dollar concessions. Understanding the net price of a branded drug—not just its list price—is essential for any honest comparison of formulary alternatives.
Market timing: Knowing when a brand drug will face generic or biosimilar competition, and planning formulary transitions accordingly, is the most underappreciated strategy in plan sponsor drug management. A plan that moves 80% of its Eliquis patients to generic apoxaban within 90 days of first generic entry can realize savings of $5,000 or more per patient per year. The challenge is that generic entry timelines are uncertain, often negotiated in patent settlements, and sometimes delayed by manufacturing problems or additional litigation. Monitoring the patent landscape on each high-spend branded drug in a plan’s formulary is not optional—it is a core function of responsible pharmacy benefit management.
The Competitive Intelligence Imperative
In a market where the difference between a drug’s list price and its effective net price can be 70% or more, where patent expiration dates printed in package inserts bear little relationship to actual generic entry dates, and where the IRA is gradually but structurally changing the pricing architecture for the highest-spend branded drugs, competitive intelligence is not a luxury function. It is operational infrastructure.
For pharmaceutical companies, that means understanding the patent landscapes of competitors and anticipating Paragraph IV filings before they arrive. For generic manufacturers and biosimilar developers, it means identifying the highest-value targets for patent challenges and tracking the settlement agreements that determine when their own launches become commercially viable. For health plans, employers, and PBMs, it means monitoring the pipeline of expected generic entries on high-spend drugs and building formulary transition planning around realistic timelines rather than nominal patent dates.
This is the analytical layer that resources like DrugPatentWatch are built to support. The platform tracks patent portfolios, ANDA filings, biosimilar approval status, litigation outcomes, and settlement agreement dates across thousands of drugs—providing the kind of real-time patent intelligence that determines not just when a generic will enter the market but how confident you can be in that timeline. For a health system modeling drug spend five years out, or for a specialty pharmacy building a contract based on expected biosimilar availability, that confidence interval is worth a great deal.
Part VI: The Structural Battlefield Ahead
GLP-1s and the Next Brand Drug Price War
No discussion of branded drug economics in 2025 and 2026 is complete without addressing the GLP-1 receptor agonist class—semaglutide (Ozempic, Wegovy), tirzepatide (Mounjaro, Zepbound), and the pipeline compounds following behind them. These drugs have become the fastest-growing category in pharmaceutical history, with combined annual sales across the class approaching $50 billion globally and projected to exceed $100 billion by 2030.
The pricing dynamics of GLP-1s illustrate every structural feature of the branded drug market in concentrated form. Novo Nordisk and Eli Lilly each hold broad patent estates covering their respective molecules, formulations, delivery devices, and manufacturing processes. Compounding pharmacies entered during periods when the branded products were on the FDA shortage list—producing semaglutide and tirzepatide compounds at prices far below the $800 to $1,400 per month list prices of the branded products. The FDA and the manufacturers have fought to close that compounding channel as shortage designations are removed.
Mounjaro was not selected for IRA negotiation in the first round effective 2026 and was not included in the 2027 round negotiations announced in early 2026. The Trump administration announced plans to lower GLP-1 costs through mechanisms separate from IRA negotiation, though the details remained unclear as of mid-2026. For employers and health plans managing skyrocketing GLP-1 spend—drugs that are clinically effective but list-priced well above any cost-plus model’s reach—the practical options are prior authorization, quantity limits, step therapy, and waiting for either IRA negotiation eligibility or generic entry, whichever comes first.
Generic semaglutide is at least several years away from market entry under current patent timelines. Tirzepatide is further still. Understanding the exact structure of Novo Nordisk’s and Eli Lilly’s patent estates on these compounds—which patents cover the primary molecule versus formulation and device patents, which are approaching Paragraph IV challenge windows—is fundamental intelligence for any payer or employer building a GLP-1 management strategy for the rest of the decade.
The Biosimilar Market: Progress and Persistence
The U.S. biosimilar market has matured considerably since the first biosimilar approval in 2015, but it remains structurally challenged relative to the small-molecule generic market. As of 2026, more than 50 biosimilars have received FDA approval and many are marketed, but uptake—particularly for drugs where the reference product manufacturer offers aggressive rebates for formulary preference—continues to be slower than the European experience.
The Stelara (ustekinumab) biosimilar market that opened in 2024 is an instructive current case. J&J’s primary patents on Stelara expired following settlement agreements that allowed biosimilar entry beginning in January 2025. Multiple biosimilar manufacturers—including Amgen, Sandoz, Teva, and others—received FDA approval and began commercially launching. The price discount offered by Stelara biosimilars at launch was modest initially, and J&J responded with rebate offers designed to maintain formulary position with major PBMs.
Keytruda presents a more complex upcoming scenario. With composition-of-matter patents expiring in 2028 and a commercial profile involving complex oncology regimens, the biosimilar entry path will require navigating both the BPCIA patent dance and a market where physician and payer relationships with Merck are deeply entrenched. The first biosimilar approvals for pembrolizumab are expected in the 2028-2030 window, with commercial uptake likely to be gradual even after launch.
PBM Reform and What It Actually Changes
Congressional attention to PBM practices has increased substantially since the FTC’s 2024 report, which documented that the three major vertically integrated PBMs—Express Scripts (Cigna), CVS Caremark, and OptumRx (UnitedHealth)—engage in practices that inflate drug costs and disadvantage independent pharmacies. The report’s subtitle left nothing implicit: “The Powerful Middlemen Inflating Drug Costs and Squeezing Main Street Pharmacies.”
Multiple PBM reform bills have been introduced in Congress. A National Community Pharmacists Association 2024 survey found that 99% of independent pharmacy owners had experienced reduced PBM reimbursements in the prior year, and 32% said they were considering closing within 12 months. State-level action has been faster: multiple states passed PBM transparency and spread pricing prohibition legislation between 2023 and 2025.
If meaningful PBM reform passes—requiring disclosure of rebate retention rates, prohibiting spread pricing, or mandating pass-through of rebates to patients—the gross-to-net bubble will deflate faster and more broadly than the IRA alone can accomplish. That would change the competitive dynamics for both brand and generic manufacturers. Brand manufacturers who built their pricing strategy around high-list/large-rebate would need to rethink list price architecture. PBMs would lose a major revenue stream. Health plans would see more direct price pressure from manufacturers.
None of this changes the fundamental structure of the branded drug market: a patent-protected manufacturer retains pricing power that no distributor model can override. But it does change how that power is exercised and how much of the gross-to-net spread is captured by intermediaries versus passed through to patients and payers.
Where Cuban’s Model Goes Next
Cuban has been explicit that Cost Plus Drugs wants to expand into branded and specialty drugs. The company is “working with trade name manufacturers to add both single source brands and specialty biologics” to its pharmacy, according to its own website. In practice, this means negotiating with manufacturers for access at net prices competitive with what major PBMs receive—a task that requires scale and leverage that Cost Plus Drugs is still building.
The Humana partnership targeting employer groups is a step toward that scale. Direct-to-employer programs that bypass traditional PBMs give Cost Plus Drugs the kind of covered-lives count that manufacturers require before they take net price negotiations seriously. If Cost Plus Drugs can aggregate several million covered lives across its employer group program, it begins to have the leverage to demand net prices comparable to what OptumRx or CVS Caremark receives for their largest accounts.
But that path is long, requires capital Cost Plus Drugs is not currently generating (Cuban acknowledged the company was not profitable as of early 2024), and competes against incumbents with decades of manufacturer relationships and formulary contracts. The disruptive potential is real; the timeline is uncertain.
Cuban’s push to get the Trump administration to waive FDA generic drug fees—the hundreds of thousands of dollars in regulatory fees that accompany ANDA approvals—reflects a pragmatic understanding of where his model has leverage. Lower the barriers to generic manufacturing entry, reduce the friction of bringing new generic drugs to market, and the commodity economics that make cost-plus pricing viable for generics become even more favorable. It is not a strategy for the branded drug problem; it is a smart expansion of a strategy that already works for the generic problem.
Part VII: The Information Edge in a Two-Market System
What Investors Need to Track
For institutional investors in the pharmaceutical sector, the generics/brand divide has direct implications for portfolio construction and due diligence.
On the brand side, the key variables for any drug’s commercial longevity are: the breadth and defensibility of the patent estate, the pipeline of Paragraph IV challenges and their likely outcomes, the IRA negotiation timeline and expected magnitude of price reduction, and the biosimilar development landscape for biologics. A drug with a 2028 patent expiry and three active ANDA filers with strong invalidity arguments faces a very different commercial trajectory than a drug with a 2028 primary expiry backed by 50 secondary patents and no Paragraph IV filings to date.
On the generic side, the investment thesis for generic manufacturers increasingly depends on two factors: the ability to identify and win first-to-file Paragraph IV opportunities on high-value branded drugs, and the manufacturing quality and supply chain resilience to capture and hold market share when generic entry occurs. The commoditization of the generic market means that undifferentiated manufacturers face perpetual margin compression. First-filer exclusivity is a meaningful but temporary premium; after 180 days, the market reverts to commodity pricing.
Biosimilar development sits at the intersection of these two dynamics: the regulatory and IP complexity of the brand drug world combined with the eventual commodity economics of generic competition. The most valuable position in biosimilars is as first-mover with a high-volume drug, clean patent clearance, and formulary contracts secured before the reference product manufacturer can respond with defensive rebate offers.
What Health Systems and Employers Need to Track
For health systems, self-insured employers, and health plans managing drug spend, the practical intelligence needs are concrete. On the generic side: real-time pricing benchmarks, shortage intelligence, and immediate formulary switching protocols when new generics enter. On the brand side: IRA negotiation outcomes, generic entry timelines for high-spend drugs, biosimilar launch schedules, and PBM rebate pass-through transparency.
The specific branded drugs that will drive the most planning activity over the next three to five years are concentrated in a few categories. Immunology drugs facing biosimilar competition—Stelara, Enbrel, and the expanding adalimumab market—offer the most immediate large-scale switching opportunities. Oncology drugs approaching patent cliffs, including tyrosine kinase inhibitors and checkpoint inhibitors, will generate generic and biosimilar entry events with significant spend implications for oncology-heavy health systems. Cardiology and diabetes drugs subject to IRA negotiation in 2026 and 2027 will require formulary and contract updates as MFPs take effect.
Each of these planning activities requires accurate, timely patent data. A health system that plans a formulary transition to generic semaglutide based on an incorrect reading of Novo Nordisk’s patent estate—expecting entry in 2028 when the actual protected timeline extends to 2032—will find its projections badly wrong. A plan sponsor that misses a Stelara biosimilar transition opportunity because it didn’t know the settlement-based entry date will pay unnecessary brand prices for months. These are not trivial errors; on a large formulary with significant specialty drug spend, they represent millions of dollars in avoidable cost.
The Data Layer That Connects Both Markets
The pharmaceutical patent system is public, but navigating it is not simple. USPTO records, FDA Orange Book listings, ANDA first-filer databases, litigation dockets, ITC proceedings, and settlement agreement filings all exist in different repositories with different formats and update schedules. Synthesizing them into a coherent picture of when specific drugs will face competition—and how much confidence to place in that timeline—requires both technical data access and domain expertise.
DrugPatentWatch has built its intelligence platform around exactly this synthesis challenge. The service tracks patent portfolios, ANDA filings, 180-day exclusivity holders, litigation outcomes, and settlement terms for drugs across all therapeutic areas—providing both raw data access for sophisticated analysts and curated intelligence products for commercial and formulary planning teams. For a pharmaceutical business development team evaluating a licensing opportunity, a payer’s pharmacy leadership team building a five-year spend forecast, or a biosimilar manufacturer timing its launch strategy, the ability to answer specific questions—”When is the earliest credible generic entry date for Drug X, and what is the probability that date holds?”—has direct financial value.
The fact that Cuban’s model works for generics is not a limitation of his vision; it is a function of where the legal structure of pharmaceutical markets creates pricing flexibility. In the patent-protected branded drug space, the structure does not create that flexibility. It creates, instead, a different set of opportunities: for litigation, for negotiation, for timing-based formulary management, and for the kind of patent intelligence that separates accurate commercial planning from expensive guesswork.
Part VIII: The Accountability Gap and What Comes Next
What Cuban Got Right That the Industry Ignored
Whatever the limitations of the Cost Plus Drugs model in the branded segment, Cuban diagnosed real pathologies in the pharmaceutical supply chain that the industry had successfully obscured for decades. The spread pricing practices of PBMs on generics were not a secret to industry insiders, but they were invisible to the patients, employers, and policymakers who were paying for them. The absence of price transparency was not an accident; it was a design feature that benefited every party in the supply chain except the end payer.
By publishing its full pricing stack—acquisition cost, markup, pharmacy fee, shipping—and demonstrating that imatinib could be sold for $14.40 when retail pharmacies were charging $2,500, Cost Plus Drugs provided empirical proof of the problem’s scale. That proof had political and commercial consequences. It contributed to increased regulatory scrutiny of PBM spread pricing, accelerated state-level PBM transparency legislation, and created competitive pressure on retail pharmacy chains to improve their generic pricing. The Interagency Prescription Drug Affordability Board data and the FTC’s PBM investigation both trace partly to the political climate that Cuban’s company helped create.
He also correctly identified that the U.S. was heavily dependent on overseas generic manufacturing with fragile supply chains and inconsistent quality oversight, and that domestic manufacturing capacity for essential generic drugs—particularly sterile injectables—was a national vulnerability. The COVID-19 pandemic had made this visible; Cost Plus Drugs’ Dallas facility was an early concrete private-sector response to it.
The Work That Transparency Cannot Do
Transparency is the right tool for markets where information asymmetry is the core problem. In the generic drug market, the drug itself is a commodity; the problem is that patients and employers cannot see what it actually costs to produce and distribute. Cost-plus transparency solves that directly.
In the branded drug market, the core problem is not information asymmetry. It is market power. A manufacturer with a valid patent has a legal monopoly, and that monopoly pricing power persists regardless of how much transparency the distribution layer achieves. You can publish AbbVie’s entire pricing stack for Humira, including every layer of rebates and gross-to-net adjustments, and none of that changes the fact that AbbVie is the only legal source of adalimumab until its patents are challenged and defeated or expire.
Solving the branded drug pricing problem therefore requires tools that affect market power directly: patent challenges, biosimilar development, government price negotiation (as the IRA now provides for Medicare), and transparency reforms that force the rebate system into the open so that cost-sharing and formulary decisions can be made on actual net prices rather than inflated list prices.
All of those tools are more legally complex, more capital-intensive, and more dependent on specific regulatory and IP outcomes than operating a transparent pharmacy. They require different kinds of expertise—patent attorneys, regulatory scientists, formulary economists, government affairs specialists—and produce outcomes on timescales measured in years or decades rather than days.
The industry is slowly developing those tools. The IRA’s Medicare negotiation program is the most consequential government-side instrument. The FTC’s expanded antitrust scrutiny of patent thickets and Orange Book gaming is a regulatory complement. The growth of sophisticated biosimilar developers willing to invest in the multi-year patent dance is a market-based complement. And resources like DrugPatentWatch provide the informational infrastructure that all of these efforts depend on.
The Long Game
Over the next decade, the U.S. pharmaceutical pricing landscape will change significantly. The IRA’s negotiation program will expand annually, putting increasing portions of high-spend branded drug spending under a government-negotiated price cap for Medicare. Generic and biosimilar competition will intensify as the 2024-2030 patent expiry wave plays out, removing exclusivity protections from drugs that currently represent $300 billion in annual revenue. PBM transparency reforms—federal or state—will make the gross-to-net bubble more visible and, gradually, harder to sustain.
In that environment, the generic drug market will continue to work roughly as Cost Plus Drugs and academic analyses suggest it should: competitive, commodity-priced, with ongoing opportunities to reduce costs further through manufacturing efficiency and supply chain transparency. The Cuban model, or variations of it, will become more mainstream as employer groups adopt direct purchasing approaches and as Amazon and other entrants continue building transparent pharmacy models.
The branded drug market will remain the real financial battleground. The fights over patent validity, biosimilar formulary positioning, IRA negotiation scope, and PBM reform will determine how much of that $300 billion in annual revenue at risk actually translates to savings for payers and patients—and how much is recaptured by manufacturers through patent extensions, lifecycle management, and next-generation products that reset the exclusivity clock.
Winning in that battleground requires understanding the rules. The patent system, the Paragraph IV litigation process, the IRA’s negotiation mechanics, the PBM rebate architecture, and the biosimilar launch dynamics are complex systems that interact in ways that produce non-intuitive outcomes. The Humira story—a drug whose primary patent expired in 2016 but whose manufacturer continued collecting royalties from competitors who entered the market seven years later—is not an anomaly. It is the system working exactly as sophisticated brand IP management intends.
Cuban disrupted the easy half. The harder half is still being fought.
Key Takeaways
The generic/brand split defines where the money is: Generic drugs fill roughly 90% of U.S. prescriptions but account for only about 20% of drug expenditure. Brand-name drugs fill the remaining 10% of prescriptions and consume roughly 80% of expenditure. Any drug pricing strategy that focuses only on generics leaves the majority of the financial problem unaddressed.
Cost Plus Drugs works within specific structural conditions: The model succeeds because generic drugs are commodities—multiple manufacturers, no exclusivity, transparent acquisition costs. Those conditions do not exist for patent-protected branded drugs, where the manufacturer is the sole legal source and sets list prices without competitive constraint.
Patent expiration dates are misleading: The nominal patent expiry printed in an FDA database is not the date competition arrives. Patent thickets, secondary patent filings, serial litigation, settlement agreements, and product hopping strategies routinely extend effective exclusivity years beyond the primary patent. Real-time patent intelligence—tracking the full IP estate, ANDA filings, litigation outcomes, and settlement terms—is necessary for accurate forecasting of generic entry timelines.
The IRA is the most significant structural change to branded drug pricing in 40 years: The Medicare Drug Price Negotiation Program directly targets what Cost Plus Drugs cannot—patent-protected, high-spend branded drugs with no generic competition. The first round of negotiated prices takes effect January 1, 2026. The program expands annually through 2029 and beyond, eventually covering all of the highest-spending Medicare drugs.
The gross-to-net bubble obscures branded drug economics: The $356 billion gap between brand drug list prices and net prices after rebates and discounts is not visible to patients and creates systematic distortions in formulary management. PBM reform—federal or state—that mandates rebate pass-through or disclosure would change the incentive structure for branded drug pricing more broadly than any distributor-side transparency model can achieve.
The manufacturing bet may be Cuban’s most durable contribution: Cost Plus Drugs’ $11 million manufacturing facility targeting generic drug shortages addresses a real supply chain failure that retail pricing transparency cannot fix. Domestic manufacturing capacity for essential sterile injectables is a national vulnerability; private investment in that capacity has strategic value independent of the company’s pharmacy business.
The next decade’s pharmaceutical financial battles are already mapped: The 2024-2030 patent expiry wave puts roughly $300 billion in annual brand drug revenues at risk. GLP-1 pricing, Keytruda’s 2028 patent cliff, biosimilar competition for Stelara and Enbrel, and IRA negotiation expansion are all in motion. Organizations that invest in patent and competitive intelligence now will have the planning foundation to capitalize on those transitions when they arrive.
FAQ
1. Can Cost Plus Drugs ever offer patented brand-name drugs at lower prices?
Not through the same mechanism it uses for generics. For off-patent drugs, Cost Plus Drugs negotiates directly with multiple competing manufacturers, purchases at market-clearing acquisition cost, and applies its 15% markup. There is no equivalent purchasing leverage for a patented drug where a single manufacturer controls supply. What Cost Plus Drugs can do is include branded drugs at the net price it negotiates with the manufacturer directly—functionally similar to what large PBMs do, but with the pricing visible to the patient rather than buried in rebate contracts. The company has stated it is working with trade name manufacturers on exactly this kind of arrangement, but the commercial and financial terms depend on scale that Cost Plus Drugs is still building. Until it has enough covered lives to matter to a manufacturer’s formulary strategy, its negotiating position for branded drugs is weaker than that of OptumRx or CVS Caremark, which together manage pharmacy benefits for hundreds of millions of Americans.
2. Why does a drug like Humira cost $7,000 per month in the U.S. but far less in Europe, if the same patent system applies?
The patents are similar, but the negotiating structures are different. In the U.S., Medicare was legally prohibited from negotiating drug prices until the IRA in 2022, and the commercial insurance market negotiates through fragmented PBMs whose rebate-based incentives actually reward high list prices. In Europe, national health systems negotiate directly with manufacturers on a country-by-country basis, typically using a cost-effectiveness threshold (the UK’s NICE uses £20,000 to £30,000 per quality-adjusted life year) to determine the maximum they will pay. A manufacturer that cannot meet that threshold does not sell its drug through the national formulary. That market exclusion threat is a negotiating lever the U.S. did not use until the IRA, and even now the IRA applies only to Medicare, covers a limited number of drugs annually, and faces ongoing political and legal challenges. European biosimilar competition for Humira began in 2018; U.S. competition was delayed until 2023 by AbbVie’s patent thicket strategy, which U.S. courts declined to challenge as anticompetitive even though it clearly delayed access and inflated costs.
3. What does the IRA actually mean for a self-insured employer’s drug benefit in 2026 and beyond?
The IRA’s Medicare drug price negotiation directly affects only Medicare Part D and Part B. Self-insured employers are not Medicare, so they do not automatically receive the negotiated Maximum Fair Prices for the 10 drugs whose prices take effect January 1, 2026. However, there are indirect commercial effects. When Medicare negotiates a significantly lower net price for Eliquis, for example, and that price becomes public, it provides employers and their PBMs with a benchmark for their own negotiations with Bristol-Myers Squibb and Pfizer. Manufacturers who accepted IRA negotiated prices at substantial discounts have less defensible ground for maintaining much higher prices for commercial payers covering similar populations. The IRA also changes the dynamics of the gross-to-net bubble for drugs subject to negotiation, which can affect how PBMs structure rebate agreements for those drugs in commercial accounts. The practical impact on employer plans will unfold over 2026 and 2027 as the first negotiated prices take effect and manufacturers respond to the new commercial environment.
4. If a drug has 30 patents listed in the Orange Book, does that mean it will have exclusivity for 30 patent terms?
No, though brand manufacturers sometimes structure their portfolios to create that impression. The patents listed in the Orange Book vary considerably in strength, scope, and expiration date. A primary “composition of matter” patent covering the active molecule itself is typically the strongest and most difficult to challenge—but it is also the first filed and therefore often the first to expire. Secondary patents on formulations, delivery devices, manufacturing processes, and methods of use expire later but are generally narrower in scope and more vulnerable to challenge. A generic manufacturer filing a Paragraph IV certification typically must certify against all listed patents, but the practical barrier is not challenging all 30 simultaneously; it is challenging the handful of patents that would actually block the generic product as formulated. The Humira case shows what happens when a manufacturer files enough patents that the sheer cost of challenging all of them—even if each individual patent is vulnerable—exceeds what any single competitor can rationally spend. AbbVie assembled over 130 patents not because each was a strong independent barrier but because the collective cost of litigating them all was prohibitive for any single biosimilar developer.
5. How should a pharmaceutical investor use patent expiry data when evaluating a branded drug company’s long-term revenue trajectory?
Patent expiry data is necessary but not sufficient for revenue forecasting. The first step is understanding the structure of the patent estate: which patents cover the primary molecule, which cover formulations or devices, when each expires, and which have been listed in settlement agreements with known generic entry dates. A drug whose primary composition patent expires in 2027 but whose only formulation patent expires in 2032 may have negotiated generic entry in 2027 as part of a settlement—or may not have faced a Paragraph IV challenge at all. The second step is understanding the litigation landscape: which ANDAs have been filed, which have survived initial 30-month stays, and what the likely litigation outcomes are. Resources like DrugPatentWatch provide this patent and litigation data systematically, allowing investors to distinguish between a drug with a theoretical 2032 patent expiry and no ANDA filings—meaning full exclusivity through 2032—and a drug with a theoretical 2032 expiry but active Paragraph IV litigation and a settlement-based generic entry date of 2028. That four-year difference in effective exclusivity can represent billions of dollars in revenue, and the information is knowable with proper patent intelligence. The third step is understanding how the IRA negotiation timeline interacts with the patent estate: a drug that becomes negotiation-eligible in 2027 faces a different revenue trajectory than an identical drug that won’t reach eligibility until 2030, even if their patent expiry dates are similar.
Sources
Sheehan, M. (2022, July 28). Mark Cuban pharmacy Cost Plus Drugs: Trying to solve brand-name drugs. CNBC. https://www.cnbc.com/2022/07/28/mark-cuban-pharmacy-cost-plus-drugs-struggling-with-brand-name-drugs.html
Emanuel, E. J., & Connolly, J. (2024, July 29). Mark Cuban Cost Plus Drugs: More impact on shortages than costs. STAT News. https://www.statnews.com/2024/07/29/mark-cuban-cost-plus-drug-company-bigger-impact-drug-shortages-not-drug-costs/
Mark Cuban Cost Plus Drug Company. (2024). About MCCPDC. https://www.markcubancostplusdrugcompany.com/
Saraceno, N. (2026, February 15). Mark Cuban pushes for generic drug fee waivers as Cost Plus Drugs eyes US manufacturing expansion. Pharmaceutical Commerce. https://www.pharmaceuticalcommerce.com/view/mark-cuban-pushes-for-generic-drug-fee-waivers
Landi, H. (2024, March 5). Mark Cuban says Cost Plus Drugs targeting generic meds in short supply as it opens manufacturing facility. Fierce Healthcare. https://www.fiercehealthcare.com/health-tech/mark-cuban-says-cost-plus-drugs-has-couple-million-patients-2500-generic-drugs-it-moves
Osei-Frimpong, K., Agbata, R., Osafo-Mensah, B., & Boateng, P. K. (2023). Generic cardiology drug prices: The potential benefits of the Mark Cuban Cost Plus Drug Company model. PMC/Frontiers in Pharmacology. https://pmc.ncbi.nlm.nih.gov/articles/PMC10506155/
DrugPatentWatch. (2026, March 20). The complete architecture of pharmaceutical drug pricing: Patents, power, and the post-IRA market. https://www.drugpatentwatch.com/blog/deconstructing-drug-pricing-strategy-for-big-pharma/
Evernorth. (2025, June 4). How drugmakers exploit the patent system to delay competition and inflate prices. https://www.evernorth.com/articles/how-drugmakers-exploit-patent-system-delay-competition-and-inflate-prices
DrugPatentWatch. (2025, July 31). The global patent thicket: A comparative analysis of pharmaceutical monopoly strategies in the U.S., Europe, and emerging markets. https://www.drugpatentwatch.com/blog/how-do-patent-thickets-vary-across-different-countries/
DrugPatentWatch. (2025, November 4). The thicket maze: A strategic guide to navigating and dismantling drug patent fortresses. https://www.drugpatentwatch.com/blog/the-thicket-maze-a-strategic-guide-to-navigating-and-dismantling-drug-patent-fortresses/
DrugPatentWatch. (2025, December 4). The biosimilar launch window: A predictive framework for navigating patents, payers, and litigation. https://www.drugpatentwatch.com/blog/the-biosimilar-launch-window-a-predictive-framework-for-navigating-patents-payers-and-litigation/
BioSpace. (2024, December 18). Lessons from Humira on how to tackle unjust extensions of drug monopolies with policy. https://www.biospace.com/policy/opinion-lessons-from-humira-on-how-to-tackle-unjust-extensions-of-drug-monopolies-with-policy
Congress.gov / Congressional Research Service. (2024). Medicare Drug Price Negotiation Under the Inflation Reduction Act: Industry responses and potential effects. https://www.congress.gov/crs-product/R47872
Medicare Rights Center. (2025, October 9). Negotiated prices take effect for ten drugs in 2026. https://www.medicarerights.org/medicare-watch/2025/10/09/negotiated-prices-take-effect-for-ten-drugs-in-2026
Rodwin, M. A. (2025). Negotiating Medicare drug prices: A new attempt to control purchase prices. Journal of Law and Medicine & Ethics, 53(1), 147–157. https://pmc.ncbi.nlm.nih.gov/articles/PMC12179530/
Vizient Inc. (2025, December 18). Early impacts of the IRA’s Medicare Drug Price Negotiation Program: Pricing trends for Medicare Parts B and D. https://www.vizientinc.com/insights/all/2025/early-impacts-of-the-iras-medicare-drug-price-negotiation-program-pricing-trends
DrugPatentWatch. (2026, February 23). Top Paragraph IV litigation trends and what they mean for pharma. https://www.drugpatentwatch.com/blog/top-paragraph-iv-litigation-trends-and-what-they-mean-for-pharma/
DrugPatentWatch. (2026, March 22). Drug patent strategy: The definitive guide for pharmaceutical IP teams, R&D leads, and institutional investors. https://www.drugpatentwatch.com/blog/optimizing-your-drug-patent-strategy-a-comprehensive-guide-for-pharmaceutical-companies/
Fischer, M. A., et al. (2021). Serial patent litigation: An emerging strategy to delay entry of generic competition. PMC. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12757684/
Congress.gov / Congressional Research Service. (2024). ‘Skinny labels’ for generic drugs under Hatch-Waxman. https://www.congress.gov/crs-product/IF12700
Association for Accessible Medicines. (2025, June 4). Patent settlements are necessary to help combat patent thickets. https://accessiblemeds.org/resources/blog/patent-settlements-are-necessary-to-help-combat-patent-thickets/
Epstein Becker Green. (2025, May 2). Medicare Drug Price Negotiation Program: The IRA “Pill Penalty” and other IRA reforms on the horizon for 2026. https://www.healthlawadvisor.com/medicare-drug-price-negotiation-program-the-inflation-reduction-act-pill-penalty-and-other-ira-reforms-on-the-horizon-for-2026
ASPE / HHS. (2023). Inflation Reduction Act research series: Understanding development and trends in utilization and spending for drugs selected under the Medicare Drug Price Negotiation Program. https://aspe.hhs.gov/reports/ira-research-series-medicare-drug-price-negotiation-program
The Hospitalist. (2025, May 27). Understanding the Inflation Reduction Act’s impact on prescription drug costs. https://www.the-hospitalist.org/hospitalist/article/38496/critical-care/understanding-the-inflation-reduction-acts/
MUSC Graduate Studies. (2024, September). Mark Cuban is making medication costs an easier pill to swallow. https://gradstudies.musc.edu/about/blog/2024/09/easier-pill-to-swallow
DrugPatentWatch. (2025, April). Mark Cuban: Unexpected leader in orphan drug development reform. https://www.drugpatentwatch.com/blog/mark-cuban-unexpected-leader-in-orphan-drug-development-reform/
DrugPatentWatch. (2026, March 30). How are prescription drug prices determined? https://www.drugpatentwatch.com/blog/how-are-prescription-drug-prices-determined/
DrugPatentWatch. (2025, November 20). A strategic guide to biologic patent exclusivity and competitive advantage. https://www.drugpatentwatch.com/blog/a-strategic-guide-to-biologic-patent-exclusivity-and-competitive-advantage/















")

")








