The $236 Billion Race Most Procurement Teams Are Losing
Between 2025 and 2030, more than $236 billion in branded pharmaceutical revenue will lose patent protection. Nearly 200 drugs will cross into generic territory in that window — roughly 70 of them blockbusters clearing over $1 billion a year in sales. Merck’s Januvia and Janumet lose exclusivity in 2026. Eliquis (apixaban) follows. Keytruda, the world’s best-selling drug at $29 billion in 2024 sales, hits its cliff in 2028.
For branded manufacturers watching that revenue evaporate, it is a reckoning. For generic drug companies and their API procurement teams, it is the most concentrated commercial opportunity in a generation.
But here’s where most teams get it wrong: they treat API supplier qualification as a compliance task — something that happens downstream, after the market opportunity has been confirmed, after the ANDA has been scoped, sometimes almost as an afterthought before filing. The companies that capture the real value in a patent expiry cycle treat API supplier qualification as a strategic intelligence exercise that begins years before a molecule goes off-patent.
Getting that discipline right requires knowing how to read three interconnected bodies of intelligence: the patent landscape around the active pharmaceutical ingredient and its manufacturing processes, the regulatory compliance record of candidate suppliers, and the Drug Master File (DMF) ecosystem that reveals who is already positioned to supply the market and who is not. Each layer eliminates candidates, surfaces risks, and narrows the field to a set of genuinely qualified partners.
This guide walks through that process in full — from Orange Book analysis to DMF status assessment, from ICH Q7 audit frameworks to the BIOSECURE Act’s structural effects on the global supplier map. It is written for regulatory affairs directors, procurement leads, business development managers, and anyone responsible for getting an ANDA across the finish line without a supplier-induced delay.
Why API Supplier Qualification Determines Launch Timing — Not Formulation
The common assumption is that formulation development and bioequivalence testing determine when a generic product reaches market. In practice, API supplier qualification is the variable most likely to delay, derail, or permanently foreclose a launch opportunity.
Consider the mechanics. A generic drug applicant filing an Abbreviated New Drug Application (ANDA) must specify one or more qualified API suppliers. Each supplier must hold a valid Type II Drug Master File with the FDA, or the ANDA must include full chemistry, manufacturing, and controls (CMC) data for the drug substance. If that DMF is deficient, incomplete, or has been placed on clinical hold, the ANDA review process stalls — regardless of the quality of the formulation work.
The choice of an API supplier and the comprehensive qualification of their material must begin years in advance of the planned market launch to align with the demanding timelines of regulatory submission and review. A failure in API sourcing — such as a supplier failing a Good Manufacturing Practice (GMP) audit, an unexpected impurity issue arising late in development, or a delay in the submission of a Drug Master File — can derail a product launch entirely.
That is not hyperbole. It is the operational reality documented in ANDA approval timelines across the industry. The FDA’s review clock for an ANDA currently averages around 12 months for a substantially complete application, but DMF deficiencies can add 6 to 18 months of back-and-forth before the underlying application ever gets substantive review. For drugs with a 180-day first-filer exclusivity window — where the entire commercial thesis depends on being first to market — an 18-month API-driven delay does not just reduce the opportunity. It eliminates it entirely.
The corollary is equally important. A company that identifies a qualified, regulatory-clean, DMF-ready API supplier two to three years before a patent expiry has a structural competitive advantage over every competitor sourcing reactively. That advantage compounds: the early mover can begin bioequivalence studies earlier, encounter and resolve impurity profile issues on a longer timeline, and file a more complete ANDA. The late mover inherits compressed timelines, higher development costs, and sometimes discovers — too late — that no qualified supplier exists for the specific polymorph or particle size specification required.
This is precisely why leading generic companies have organized their business development, regulatory affairs, and procurement functions around a shared intelligence infrastructure. The input that drives all three functions is the same: systematic analysis of the patent and regulatory data that surrounds every approaching patent expiry.
The Patent Landscape: What You’re Actually Reading (and What Most Teams Miss)
When a procurement team says they are ‘monitoring the patent landscape’ for an off-patent drug, they usually mean they have checked the FDA Orange Book. That is necessary but not sufficient. The Orange Book lists patents that the branded manufacturer has declared protect the listed drug product — it does not capture the full universe of intellectual property that can constrain a generic entrant’s API sourcing choices.
The Orange Book Is a Starting Point, Not the Answer
The Orange Book contains three categories of listed patents: drug substance patents (protecting the active ingredient), drug product patents (protecting the formulation), and method-of-use patents. For generic drug applicants, substance patents are the most directly relevant to API sourcing.
But the Orange Book has structural limitations. It only includes patents that innovators choose to list — and it excludes process patents entirely. A branded manufacturer that holds a patent on the synthetic route used to produce an API does not list that patent in the Orange Book, because process patents do not qualify for listing. Yet a generic API supplier using that same synthetic route without a license is infringing that patent, which means the finished drug product manufacturer who sources from that supplier inherits the infringement risk.
This is not a theoretical concern. Process patent litigation has been a recurring feature of generic drug competition for decades, and several well-documented cases have resulted in injunctions that prevented commercially launched generics from continuing to source APIs from their original suppliers. The supplier who cleared regulatory review did not clear IP review — and the finished drug manufacturer paid the price.
The correct protocol is to run parallel analyses:
A full Freedom-to-Operate (FTO) analysis covering the drug substance structure, known polymorphic forms, co-crystals, salts, particle size specifications, and published synthesis routes
A process patent landscape search covering every claimed synthesis pathway for the target molecule, including intermediate compounds that might be patented independently
A review of any supplemental protection certificates or patent term extensions that extend effective exclusivity beyond the nominal expiry date
Each of these searches should reference the USPTO database, the European Patent Register, and ideally Espacenet for international coverage. Platforms like DrugPatentWatch consolidate Orange Book data, patent expiry dates, Paragraph IV certification histories, and exclusivity status in a single interface — making it substantially faster to build the initial landscape without manual searches across multiple databases.
Process Patents: The Hidden Constraint on API Supplier Selection
The pharmaceutical industry has spent decades building process patent estates around molecules that were already off-patent on the composition-of-matter side. This is not a gray area of practice — it is standard lifecycle management. Branded manufacturers whose core compound patent is expiring often have engineering teams working in parallel to develop and patent improved synthesis routes. The goal is partly to create genuinely better manufacturing processes, and partly to establish a patent thicket that forces generic API manufacturers to develop non-infringing alternative routes at significant cost.
The practical effect for generic procurement teams is that a candidate API supplier cannot simply demonstrate that they manufacture the correct compound in specification. They must demonstrate — in documented form — that their synthesis route does not infringe any valid, in-force process patent held by the originator or any third party.
This analysis is the joint responsibility of the finished drug manufacturer’s IP team and the API supplier’s chemistry team. A supplier who cannot provide a route development report and FTO opinion for their specific synthesis pathway is not a qualified supplier, regardless of their GMP compliance record. Any procurement framework that approves an API supplier without this documentation has a material IP risk exposure that may not manifest until the product is commercially launched.
The documentation requirements are specific:
A written process description covering the synthesis route from defined starting materials through the final API
An impurity profile linked to the specific route, identifying route-specific impurities at characterization levels
A Freedom-to-Operate opinion from qualified patent counsel addressing identified relevant patents
A clear statement of the starting material definition per ICH Q11, which determines where the supplier’s regulatory responsibility begins and which intermediates fall within their DMF
Platforms like DrugPatentWatch are useful here for building the initial patent landscape, identifying process patent holders, and reviewing the litigation history around a molecule before commissioning a full FTO opinion. The Paragraph IV certification data available on such platforms also shows which generic challengers have already engaged with the innovator’s patent estate — signaling which synthesis routes have been commercially validated or legally contested.
Reading Paragraph IV History as Competitive Intelligence
A Paragraph IV certification is a formal declaration by an ANDA applicant that one or more listed patents is invalid, unenforceable, or will not be infringed by the proposed generic product. When a branded manufacturer receives a Paragraph IV certification, they typically file suit within 45 days, triggering an automatic 30-month stay on FDA approval.
For API procurement teams, this litigation history is a rich source of competitive intelligence. Each Paragraph IV challenge that has been resolved — either through settlement, court judgment, or consent decree — reveals something about which aspects of the originator’s patent estate are commercially contested and which are considered defensible. A molecule that has faced multiple Paragraph IV challenges, with most settling on terms that allowed generic entry, typically has a more navigable patent landscape than one that has never been challenged.
The settlement terms themselves, while usually not fully public, are often partially disclosed in SEC filings or court records. Pay-for-delay agreements — where a branded manufacturer pays a generic challenger to delay market entry — are subject to FTC scrutiny and are partially disclosed as a result. A settlement that permits the generic entrant to launch several years before nominal patent expiry, but restricts certain API sourcing configurations, may be signaling that specific process patents are difficult to design around. <blockquote> “Pharmaceutical companies increasingly rely on structured patent intelligence to determine not just when a drug goes generic, but whether viable, non-infringing API supply chains actually exist for that molecule at commercial scale.” — DrugPatentWatch Industry Analysis, 2025 </blockquote>
DrugPatentWatch tracks the complete Paragraph IV filing history across thousands of drugs, including first-filer status, litigation outcomes, and authorized generic agreements — giving procurement and business development teams the context to assess whether an API sourcing strategy for a specific molecule is likely to face legal challenges before or after commercial launch.
Drug Master Files: The Regulatory Signal Layer
Once the patent landscape analysis has identified molecules worth pursuing and screened out IP-constrained synthesis routes, the next analytical layer is the DMF ecosystem. This is where the field of candidate suppliers narrows dramatically.
What a Type II DMF Actually Tells You
Drug master files are submissions to FDA used to provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of human drug products. They allow parties to reference material without disclosing DMF contents to those parties, are not required by statute or regulation, and are neither approved nor disapproved. Instead, FDA reviews the technical contents of DMFs in connection with the review of applications that reference them.
For API qualification purposes, the Type II DMF — covering drug substances — is the relevant document. Its existence alone tells you relatively little. What matters is its status, its completeness, its update history, and whether it has been successfully referenced in approved ANDAs. Each of these data points is accessible and tells a different part of the story.
A DMF that appears in the FDA’s DMF database but has never been referenced in an approved ANDA may be pristine — or it may have been reviewed and found deficient without the deficiency being publicly disclosed. A DMF that has been referenced successfully in multiple approved ANDAs has, by definition, survived regulatory scrutiny in a real commercial context. That is a materially different risk profile.
The Generic Drug User Fee Amendments (GDUFA) include provisions for DMF fees, a completeness assessment, and communications with DMF holders for Type II DMFs for drug substances used to support ANDAs. A completeness assessment is a series of questions that must be satisfied for a DMF to be made publicly available on FDA’s website. It does not replace the full scientific review to determine the adequacy of a DMF to support an ANDA regulatory action.
The GDUFA completeness assessment process, introduced to improve the quality of DMF submissions, created a two-stage filter. A DMF that has cleared completeness assessment is at least structurally adequate — it contains the required sections, covers the required topics, and meets basic format requirements. A DMF that has not cleared completeness assessment may have significant gaps that will become apparent only when the FDA reviews it in the context of an ANDA referencing it.
The practical implication: when screening API suppliers, confirm not just that a DMF exists but that it has cleared completeness assessment and has been successfully referenced in at least one approved ANDA. Suppliers who meet these criteria have demonstrated regulatory accountability in a concrete way.
The ASMF and European Regulatory Equivalence
For companies operating in European markets, the equivalent to the FDA Type II DMF is the Active Substance Master File (ASMF), sometimes referred to as the European Drug Master File. The ASMF process operates differently — the document has a publicly accessible ‘Applicant’s Part’ (AP) and a confidential ‘Restricted Part’ (RP), which the regulatory authority reviews but does not share with the finished drug manufacturer.
The European Directorate for the Quality of Medicines and Healthcare (EDQM) also issues Certificates of Suitability (CEPs) for monographs published in the European Pharmacopoeia. A CEP from EDQM is a shortcut — it certifies that the API meets Pharmacopoeia monograph requirements and has been manufactured in compliance with GMP standards as assessed by an EDQM inspection. A supplier holding a valid CEP for the target molecule has already completed a substantial portion of the European regulatory qualification process, which is a significant practical advantage.
DDReg experts assist with Certificate of Suitability (CEP), referenced in Marketing Authorization Applications after European Directorate for the Quality of Medicines & Healthcare (EDQM) approval, an API dossier detailing ingredient properties and manufacturing processes ensuring that your product stands out in competitive European markets. The expertise extends to Active Substance Master File (ASMF) submissions, offering comprehensive US FDA DMF services, European regulatory support, and specialized assistance with CADIFA submissions in Brazil.
For companies with multi-market ambitions, a supplier holding both a valid FDA Type II DMF (GDUFA-cleared) and a current EDQM CEP for the target molecule represents the highest level of pre-qualified regulatory standing available. There are typically far fewer such suppliers than the total population of API manufacturers claiming to produce a given compound.
Using DrugPatentWatch and the FDA Database to Map the Supplier Field
The FDA publishes its complete DMF list publicly, searchable by drug name, DMF number, and holder name. This raw data is useful but requires significant work to make actionable. DrugPatentWatch aggregates FDA DMF data alongside Orange Book information, ANDA approval records, Paragraph IV filing history, and Warning Letter data — enabling a procurement analyst to map the entire supplier landscape for a target molecule in a structured, searchable format.
A systematic supplier landscape analysis using these tools should produce a tiered output:
Tier 1: Suppliers with a current, GDUFA-cleared Type II DMF, no open import alerts or Warning Letters for the relevant facility, and a documented history of successful ANDA references. These suppliers represent the lowest-risk sourcing option and should be the primary focus of commercial engagement.
Tier 2: Suppliers with a current DMF that has not yet been referenced in an approved ANDA, or suppliers with an expired DMF that has not been updated in more than 18 months. These require additional diligence before qualification and should not be assumed to represent a ready commercial supply option.
Tier 3: Suppliers who produce the compound and have regulatory filings in other markets (China’s NMPA, India’s CDSCO, WHO prequalification) but lack FDA regulatory documentation. For US market ANDAs, these suppliers require full development-stage qualification work — effectively starting from scratch on the DMF submission — which should be factored into the timeline and resource calculation.
This tiered framework is not about eliminating Tier 2 and Tier 3 suppliers from consideration. For some molecules, the Tier 1 supplier pool is very small or nonexistent, and a finished drug manufacturer may have no choice but to qualify a supplier from a lower tier. The framework’s value is in setting realistic expectations about the regulatory work required and the timeline implications of each sourcing choice.
GMP Compliance History: Reading the Warning Letter and Inspection Record
A supplier’s DMF status tells you about their regulatory positioning. Their compliance history tells you about their operational reality. These two things often diverge.
What the FDA’s Enforcement Database Reveals
The FDA maintains publicly accessible databases that contain the complete history of Warning Letters, import alerts, Form 483 observations, and establishment inspection reports for registered drug manufacturing facilities. For API manufacturers, the most relevant enforcement action is the Warning Letter — a formal finding by the FDA that a facility has significant deviations from Current Good Manufacturing Practice (CGMP) for APIs as defined by 21 CFR Part 211 and, for API-specific standards, ICH Q7.
According to FDA annual reports, the number of quality-related warning letters has been steadily rising since FY2020, reaching its highest level in FY2024. That year alone, the FDA issued 105 warning letters for human drug quality issues, an 11% jump from the previous year. The FDA is focusing on systemic control, data credibility, and global supply chain reliability.
The pattern in recent Warning Letters to API manufacturers is consistent. It is not about isolated equipment failures or single-batch contamination events. The FDA’s enforcement focus has shifted to systemic quality system failures: data integrity lapses, inadequate out-of-specification (OOS) investigation protocols, failure to qualify incoming raw materials, and quality unit oversight failures that allow manufacturing deviations to go undetected or undocumented.
Of 148 warning letters directed at regulated lab environments in 2025, the following were the top risk categories: failure to investigate OOS results (34 letters, 34% of total); missing or inadequate written procedures (43 letters, 29%); method or process validation failures (40 letters, 27%); and failure to qualify suppliers or components (34 letters, 23%).
The last category — failure to qualify suppliers or components — is particularly notable in the API context. An API manufacturer who cannot demonstrate systematic qualification of their own raw material and intermediate suppliers has a structural blind spot in their quality system. The FDA has become increasingly attentive to this issue, and a Warning Letter citing supplier qualification deficiencies at an API manufacturer should be treated as a significant disqualifier by finished drug manufacturers evaluating that supplier.
The India and China API Enforcement Picture
The global API supply chain is heavily concentrated in India and China. The U.S. accounts for 22% of FDA-registered API manufacturing sites, while other major sources include India (21%), China (20%), and the European Union (19%). This means that the majority of FDA enforcement actions against API manufacturers involve facilities in these two countries.
Recent enforcement actions confirm that the problem is both persistent and patterned. In early 2025, the FDA issued Warning Letters and import alerts against multiple Indian and Chinese API manufacturers in rapid succession. Jagsonpal Pharmaceuticals, based in India, received a Warning Letter following an inspection during which FDA inspectors were initially denied entry at the facility — a detail that itself signals a quality culture concern. Tyche Industries, also Indian, was cited for failure to record quality-related activities at the time of performance, equipment cleaning failures, and inadequate identity testing of incoming production materials.
The FDA issued warning letters to two Chinese active pharmaceutical ingredient manufacturers, Wuhu Nuowei Chemistry and Chengdu Innovation Pharmaceutical, for ‘significant’ deviations from current Good Manufacturing Practice. The agency placed both companies on Import Alert 66-40, blocking their products from entering the U.S. market. Following an inspection of Wuhu Nuowei Chemistry’s facility in September 2024, the FDA found that the company used impurity testing methods from the Chinese Pharmacopeia without proving they were equivalent to the U.S. Pharmacopeia (USP) standards. A batch of API imported into the U.S. exceeded USP impurity limits, rendering the drug adulterated under the Federal Food, Drug, and Cosmetic Act.
This last point — using domestic pharmacopeial methods without establishing USP equivalence — is a recurring and preventable failure. Chinese API manufacturers working primarily for their domestic market often have quality systems calibrated to Chinese Pharmacopeia (ChP) standards. When those manufacturers begin exporting to the US market without adapting their analytical methodology, the gap between ChP and USP impurity limits can result in adulterated product reaching US supply chains.
For procurement teams, the practical lesson is that a supplier’s compliance history with their domestic regulatory authority is not a substitute for FDA compliance data. A manufacturer with an excellent standing with China’s National Medical Products Administration (NMPA) may simultaneously be on FDA Import Alert 66-40. These are not equivalent credentials, and they should not be treated as such.
The Import Alert Hierarchy
Import alerts are the mechanism by which FDA blocks the importation of products from specific manufacturers without examining each individual shipment. Import Alert 66-40, ‘Detention Without Physical Examination of Drugs From Firms Which Have Not Met Drug GMPs,’ is the most commonly applied alert for API manufacturers with CGMP deficiencies.
A manufacturer on Import Alert 66-40 cannot legally supply APIs for FDA-regulated products until the alert is lifted — a process that requires a demonstrated correction of the deficiencies identified in the original Warning Letter, followed by a re-inspection and FDA determination that the facility is in compliance. This process typically takes 12 to 24 months at minimum, and there is no guarantee that a re-inspection will result in removal from the alert.
The implication for procurement teams is that import alert status must be checked actively and recently for any supplier under consideration. Import alerts are added and removed continuously, and a supplier who passed a preliminary screening six months ago may have been placed on import alert since then. This is not a one-time check — it is a recurring monitoring requirement throughout the supplier relationship.
The FDA’s public databases, combined with aggregation tools like DrugPatentWatch, enable this kind of ongoing monitoring at scale. A company managing dozens of active API supplier relationships across multiple product lines cannot manually check FDA enforcement databases for each supplier on a regular basis; a systematic monitoring protocol using these tools is both a practical necessity and a regulatory compliance expectation.
Data Integrity: The Dominant Quality Theme of the Current Enforcement Cycle
Data integrity has been the FDA’s dominant enforcement focus for API manufacturers since approximately 2012, when a series of inspections at Indian manufacturers revealed systematic falsification of laboratory records — including backdated HPLC runs, deleted out-of-specification results, and manufactured audit trails. The problem has not gone away.
The FDA has warned an active pharmaceutical ingredient maker in India for falsifying manufacturing records and issued a warning letter to a contract testing lab in China for its lax testing approach.
Data integrity failures are categorically different from other CGMP violations. A facility that consistently struggles with equipment calibration or cleaning validation can often be remediated with targeted investment and procedural changes. A facility where laboratory data has been systematically falsified has a quality culture problem that cannot be fixed by buying new equipment or writing new SOPs. The FDA recognizes this distinction, which is why data integrity findings are treated as particularly serious and often result in consent decrees or extended import alert periods rather than the more routine remediation expected for other types of deficiencies.
When auditing a potential API supplier, data integrity assessment should be a primary focus rather than a supplementary check. The specific areas to probe include:
The completeness and integrity of HPLC raw data files, including audit trails showing when runs were performed and whether any results were deleted
The documentation of out-of-specification (OOS) investigations, including whether initial OOS results are recorded before subsequent retesting
The traceability between batch production records and analytical results
The management of electronic data systems, including access controls that prevent unauthorized modification of records
The handling of laboratory instruments, particularly whether analysts have the ability to generate ‘practice’ or ‘test’ injections that are not captured in the official data system
An API supplier who cannot demonstrate robust controls in these areas should not be qualified regardless of their DMF status, GMP certificate, or commercial capabilities.
The Audit Process: What On-Site Qualification Actually Requires
DMF analysis and regulatory database screening narrow the field of candidate suppliers to a manageable shortlist. What confirms or disqualifies a supplier from that shortlist is the on-site audit.
ICH Q7 as the Audit Framework
The International Council for Harmonisation (ICH) Q7 guideline, ‘Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients,’ is the globally recognized standard for API GMP and forms the basis for regulatory inspections and audits. The on-site audit remains the gold standard. It allows a qualified audit team to thoroughly evaluate a facility’s quality systems, equipment maintenance and calibration programs, personnel training, data integrity practices, and the overall culture of quality and compliance.
ICH Q7 organizes the requirements for API manufacturing GMP into 19 sections covering everything from personnel qualifications through production operations, quality management systems, materials management, and documentation requirements. An audit team evaluating an API supplier against ICH Q7 should have qualified professionals with direct API manufacturing experience — not just finished drug product auditors, because the GMP requirements for API manufacture differ from those for finished dosage forms in important ways.
The key ICH Q7 requirements that most commonly drive audit findings at API manufacturers include:
Section 7 — Materials Management: The requirement to qualify all suppliers of critical raw materials and intermediates, to test the identity of each lot of incoming materials, and to maintain traceability from raw material through to final API. This is an area where deficiencies are both common and consequential.
Section 11 — Laboratory Controls: Requirements for documented test methods, method validation, stability testing programs, OOS investigation procedures, and data integrity. This section is the primary focus of FDA inspection activity at API manufacturers in the current enforcement environment.
Section 12 — Validation: Requirements for process validation at commercial scale, analytical method validation, and cleaning validation. Suppliers who have validated their process only at development scale and extrapolated to commercial manufacture without formal revalidation are a common source of impurity profile surprises during development.
Section 15 — Complaints and Recalls: The supplier’s quality system for receiving, investigating, and reporting product quality complaints — and their process for managing potential recalls. A supplier with no documented complaint history is not necessarily a high-quality supplier; it may simply mean they have an inadequate complaint receipt and documentation system.
Section 16 — Contract Manufacturers and Laboratories: If the supplier uses any contract facilities for specific steps in the synthesis or for analytical testing, those contract facilities fall within the scope of the audit. API manufacturers who outsource specific synthesis steps to contract manufacturers without adequate oversight of those facilities have a supply chain blind spot that finished drug manufacturers inherit.
Remote and Desktop Audits: What They Can and Cannot Do
The COVID-19 pandemic accelerated the adoption of remote audit methodologies across the pharmaceutical industry, and remote audits are now an established tool in the supplier qualification toolkit. They are faster, cheaper, and logistically more practical than on-site audits — especially for suppliers in geographically remote locations or countries with complex visa requirements.
But remote audits have hard limitations. They cannot assess facility conditions, equipment state, or the operational environment. A remote audit that reviews documentation packages and conducts interviews with quality staff can confirm that the written systems are adequate; it cannot confirm that those systems are actually implemented as written. The gap between documented procedures and operational reality is precisely where the most serious GMP deficiencies reside.
The appropriate protocol is to use desktop and remote audits for initial screening and for ongoing monitoring of established suppliers, while requiring at minimum one on-site audit before approving a new supplier for commercial use. For API suppliers manufacturing complex or high-potency compounds, or for suppliers where the preliminary screening has identified any areas of concern, on-site audits are non-negotiable.
More than 60% of import alerts added in FY2024 were linked directly to records requests. FDA oversight no longer depends solely on in-person inspections. Remote regulatory assessments became part of the new normal, and ignoring or delaying a records request practically invites enforcement action.
This point applies equally to finished drug manufacturers’ own audit programs. A supplier who delays providing records in response to an audit preparation request, who is selective about which documents they make available for review, or who provides documents that have clearly been prepared specifically for the audit rather than generated in the ordinary course of manufacturing operations is exhibiting the same red flags that regulators identify during inspections.
Geopolitical and Supply Chain Dimensions of API Sourcing
The market for off-patent APIs is global, and the decision about where to source has expanded well beyond a pure cost-and-quality calculation. Geopolitical factors now affect supplier qualification in ways that were not present even five years ago.
The BIOSECURE Act and What It Actually Changes
The BIOSECURE Act was included in the FY 2026 National Defense Authorization Act, which the President signed on December 18, 2025. A primary implication of the BIOSECURE Act is that using equipment or services — such as outsourcing the manufacturing of a drug substance or active pharmaceutical ingredient for a pharmaceutical product — from a Biotechnology Company of Concern could render that product ineligible for sale to U.S. executive agencies. The Act also has implications for pharmaceutical companies relying on U.S. federal grants to support drug research and development.
The BIOSECURE Act is now law. Its most direct effect on API sourcing is that any API manufacturer who is subsequently designated as a ‘Biotechnology Company of Concern’ under the Act’s designation process (administered by the Office of Management and Budget) cannot supply APIs used in products sold to US executive agencies or manufactured with federal research funding, without a waiver.
For pharmaceutical companies, the most significant consequence may not be the immediate prohibition on sourcing from listed Chinese entities, but the gradual decoupling of federal biomedical programs from globally integrated supply chains.
The implications vary by company type. For pharmaceutical manufacturers whose primary customers are commercial health plans and retail pharmacies, the BIOSECURE Act has limited immediate direct effect on most API sourcing decisions. For manufacturers with significant government contract revenue — including through Medicaid, VA formularies, or DoD medical supply programs — the Act’s restrictions apply more broadly.
The more important effect may be structural and medium-term. The BIOSECURE Act accelerates a ‘China+1’ sourcing strategy that many pharmaceutical manufacturers had already begun developing in response to pandemic-era supply chain disruptions. Manufacturers who have been slow to diversify away from Chinese API sources now face a regulatory and legislative environment that adds compliance risk to the existing geopolitical and logistics risk of single-source China dependence.
China leads the world in chemical manufacturing facilities for APIs and is also a powerhouse in generic drug manufacturing. It is estimated that the US sources approximately 80% of its API imports from China and India. There are also concerns growing among US policymakers that China could use this dependency as an economic weapon to hurt US businesses.
India depends on China for approximately 70% of its bulk drug and intermediate imports. This indirect reliance means many pharmaceutical products originating from India still contain Chinese raw materials, making true diversification an ongoing challenge. Long-term supply chain resilience will require a more strategic and diversified approach.
This is the key structural vulnerability that the ‘India as alternative to China’ strategy does not fully address. An API manufactured in India using Chinese-sourced intermediates has China dependency one step removed — but still present. For pharmaceutical manufacturers building a BIOSECURE Act compliance program or a supply chain resilience strategy, a genuine China+1 or China-independent sourcing configuration requires tracing the supply chain back to defined starting materials and confirming the origin of those materials, not just the country of final manufacture.
Tariff Exposure and the Cost Recalculation
In 2018, the U.S. government initiated Section 301 tariffs, imposing up to 25% duties on various Chinese imports, including certain pharmaceutical chemicals, active pharmaceutical ingredients, raw materials, and intermediates. In 2019, the U.S. government expanded these tariffs to cover additional products, further impacting pharmaceutical raw materials and finished drugs imported from China.
For off-patent API procurement, tariff exposure can materially shift the cost calculus that previously made Chinese API sourcing the default choice. A 25% duty applied to an API that already faces commodity-level pricing pressure in the generics market can eliminate the cost advantage that justified sourcing from China in the first place.
The tariff situation remains fluid. Companies building long-term API sourcing strategies for off-patent drugs should model supply chain costs under multiple tariff scenarios — including the current tariff levels, a scenario of further escalation, and a scenario of partial relief — and select suppliers whose cost profiles are acceptable across that range rather than optimizing exclusively for the lowest current-cost option.
India: Regulatory Maturation and HPAPI Growth
India’s pharmaceutical manufacturing sector has spent the last decade in a largely adversarial relationship with the FDA over CGMP compliance. The wave of Warning Letters, import alerts, and consent decrees that swept through Indian API manufacturers between 2014 and 2018 forced a substantial quality systems investment across the industry. The leading Indian API manufacturers — Sun Pharmaceutical, Dr. Reddy’s Laboratories, Aurobindo Pharma, Divi’s Laboratories, and a cluster of specialized manufacturers — have built quality organizations that are materially more sophisticated than those of a decade ago.
This matters for supplier qualification because the risk profile of a major, FDA-inspected Indian API manufacturer with a multi-year clean compliance record is fundamentally different from that of a mid-size Indian manufacturer with a recent Warning Letter or an unreinspected facility. The category ‘Indian API manufacturer’ is far too broad to be analytically useful. What matters is the specific facility’s compliance history, the recency of its most recent FDA inspection, and the nature of any observations that inspection generated.
The LOE wave covering major branded oncology compounds — including multiple kinase inhibitors, PARP inhibitors, and immunotherapy checkpoint inhibitors with patents expiring from 2025-2030 — creates the conditions for significant generic API demand growth in a product category where API synthesis complexity has historically limited the number of qualified global manufacturers. The technical difficulty of oncology API synthesis, including high potency active pharmaceutical ingredient (HPAPI) handling requirements at occupational exposure limits below 1 microgram per cubic meter, creates regulatory and engineering requirements for containment that further limit competitive entry.
For the wave of oncology API opportunities opening up in the late 2020s, HPAPI synthesis capability is a genuine barrier to entry that further narrows the qualified supplier pool. Not every API manufacturer with adequate GMP for conventional APIs has the containment infrastructure required to safely manufacture a cytotoxic compound. Qualification of HPAPI suppliers requires specific assessment of the occupational safety management systems, containment engineering controls, and dedicated synthesis equipment that these molecules require.
European API Manufacturing: Regulatory Premium and Cost Reality
European API manufacturers — in Germany, Italy, Ireland, and a few other centers — operate under EMA oversight and typically hold both EDQM CEPs and FDA DMF filings. Their regulatory standing is the highest in the global API supply market. Their costs are commensurately higher.
For off-patent APIs in therapeutic areas where pricing competition is intense — oral solid dosage generics, for instance — European API sourcing is often economically unviable at commercial scale. The regulatory premium commanded by European manufacturers does not translate into a corresponding price premium in highly commoditized generic drug markets.
Where European API sourcing makes economic sense is for regulated markets willing to pay for regulatory certainty (hospital formulary products, government tenders with strict source-of-origin requirements), for complex APIs where the technical sophistication of European manufacturers delivers genuine quality advantages, and for HPAPI synthesis where European manufacturers often hold technical and regulatory leadership.
The practical framework is market-segment specific. For commoditized oral generics, the commercially viable API sourcing options are primarily Indian manufacturers with clean compliance records. For injectable generics, oncology APIs, and products destined for hospital or government contract markets, European manufacturers and the leading Indian manufacturers with sterile or HPAPI capabilities represent the practical supplier set.
Building the Supplier Qualification Framework: From Intelligence to Contracts
The intelligence-gathering work described above — patent landscape analysis, DMF status assessment, compliance history review, geopolitical risk evaluation — produces a shortlist of candidate suppliers who are qualified candidates on paper. Converting that shortlist to a contractually engaged, operationally ready supply partner requires a structured qualification process with specific milestones.
The Four-Stage Qualification Process
Stage 1: Desk-Based Pre-Qualification
Before any commercial engagement, the desk-based pre-qualification establishes a baseline eligibility threshold. A supplier who does not clear this stage does not advance to resource-intensive audit or development activities.
Pre-qualification criteria include:
Active Type II DMF for the target API, with GDUFA completeness assessment clearance
No current import alert status for the relevant manufacturing facility
No unresolved Warning Letters for the relevant facility (a Warning Letter that has been formally closed by FDA with an acceptance of the firm’s corrective action plan is different from an open Warning Letter with no FDA response)
Documented FTO position for their synthesis route, or written confirmation that an FTO analysis is underway and will be completed before samples are supplied
Current GMP certificate issued by a recognized authority (FDA, EMA, TGA, PMDA, or equivalent) within the last two years
This pre-qualification stage can be completed almost entirely using public databases, supplemented by direct requests to the supplier for their current GMP certificate and FTO documentation. The time requirement is typically one to four weeks for a single supplier assessment by a trained regulatory analyst.
Stage 2: Technical Evaluation
Suppliers clearing pre-qualification move to technical evaluation, which involves obtaining and analyzing API samples against a defined specification. The specification should cover, at minimum:
Identity confirmation by multiple orthogonal methods (IR, NMR, mass spectrometry)
Assay by validated HPLC or equivalent method
Impurity profile (known impurities specified individually, unknown impurities reported per ICH Q3A limits)
Residual solvents per ICH Q3C, with specific attention to solvents known to be used in the supplier’s synthesis route
Physical characterization: particle size distribution (D10/D50/D90), polymorphic form, and bulk/tapped density where relevant to the dosage form
Microbiological testing per pharmacopoeial requirements
Stability data: at minimum, one batch of accelerated stability data (40°C/75%RH for 6 months or 25°C/60%RH for 12 months) demonstrating the API is within specification at the proposed retest date
The impurity profile deserves particular attention. Route-specific impurities from the supplier’s synthesis pathway may be present at levels that are acceptable against the current USP monograph but that require characterization and qualification per ICH Q3A if they are not listed in the monograph. Identifying these impurities early in the qualification process is critical — it takes two to three years to generate the genotoxicity and repeat-dose toxicity data required to qualify a novel impurity above ICH threshold limits. Discovering a new impurity after ANDA filing is a serious development setback.
Stage 3: On-Site Quality Audit
The on-site audit, conducted by a qualified team against ICH Q7 and FDA CGMP requirements, evaluates the operational reality behind the documentation reviewed during pre-qualification. The audit should be conducted by personnel with direct API manufacturing experience, include unannounced or short-notice elements where regulatory and commercial relationships permit, and cover the full scope of manufacturing operations relevant to the target API — not just a facility-level tour.
Audit report findings should be classified by severity using a consistent scale: critical (immediate risk to product quality or patient safety), major (systemic deficiencies requiring documented corrective actions with completion timelines), and minor (isolated observations not indicating systemic failure). A supplier with unresolved critical or major audit findings should not be qualified until those findings have been closed to the auditor’s satisfaction.
Stage 4: Quality Agreement and Commercial Qualification
The final stage converts the technical qualification into a contractually governed supply relationship. The quality agreement (QA) is the central document — it defines the respective quality responsibilities of the finished drug manufacturer and the API supplier, the change management and notification requirements, the agreed specification and the method used to test against it, the documentation that accompanies each batch, and the escalation process for quality events.
Quality assurance and documentation requirements include a full Certificate of Analysis (COA), traceable raw material sources, validated test methods, impurity profile, and retention of samples. Risk management for supply chain vulnerabilities includes single sourcing, raw material shortages, geopolitical risks, regulatory changes, and intellectual property and patent issues.
The quality agreement should specifically address:
Change control: the supplier’s obligation to notify the finished drug manufacturer before implementing any change to the synthesis route, raw material sources, manufacturing site, or analytical methods. This notification obligation is critical because changes at the API supplier level may require ANDA supplements or prior approval supplements, which can take 12 months or more to obtain.
Audit rights: the finished drug manufacturer’s right to conduct announced and unannounced audits of the API manufacturing facility, including access to all relevant quality records
Regulatory inspection notification: the supplier’s obligation to notify the finished drug manufacturer immediately upon receipt of a regulatory inspection notice, and to share inspection observations (Form 483 equivalents) and Warning Letters within a defined timeframe
Exclusivity provisions where applicable: for first-to-file ANDA strategies, a supplier agreement that limits or restricts the API manufacturer’s ability to supply competing generic manufacturers for the target molecule during the 180-day exclusivity window
Maintaining Qualification: Ongoing Monitoring and Change Management
Supplier qualification is not a one-time event. The compliance landscape changes continuously, and suppliers who were qualified at one point can subsequently develop problems that affect supply quality, regulatory standing, or commercial reliability.
The Ongoing Monitoring Protocol
A structured ongoing monitoring protocol should include, at minimum:
Batch-Level Review: The Certificate of Analysis (COA) for every commercial batch received should be reviewed against specification before the batch is approved for use in finished product manufacturing. Trends in key parameters — assay, specific impurities, residual solvents, particle size — should be tracked across batches and investigated if they show directional drift toward specification limits.
Regulatory Intelligence Monitoring: The FDA Warning Letter database, import alert list, and Form 483 observation database should be checked for all qualified suppliers at least quarterly. Changes in regulatory status can happen rapidly — a Warning Letter issued in one month can result in an import alert within weeks. A systematic monitoring program prevents the situation where a manufacturer discovers mid-batch that their API supplier has been placed on import alert.
Annual Audit or Desktop Review: At minimum, an annual desktop review of the supplier’s regulatory filings, DMF update history, and quality metrics should be conducted. Suppliers with any identified areas of concern from the previous year’s review or audit should receive an on-site audit rather than a desktop review.
Change Management: Any notification received from the supplier regarding changes to their manufacturing process, site, personnel, raw material suppliers, or analytical methods should be assessed for its impact on the finished drug product’s regulatory filings and initiated through a formal change management process.
When to Dual-Source
The pandemic-era supply chain experience demonstrated the cost of single-source API dependence at scale. When a single API manufacturer shuts down due to a regulatory action, natural disaster, or operational failure, every finished drug manufacturer relying on that supplier simultaneously faces the same crisis — and there is typically no short-term solution because qualifying a new API supplier takes 12 to 24 months even under ideal conditions.
The risk calculus for dual-sourcing varies by molecule and market. For high-volume, commoditized generic APIs — atorvastatin, metformin, lisinopril — the qualified supplier pool is large enough that rapid alternative sourcing is feasible in an emergency. For complex APIs, controlled substances, HPAPIs, or molecules where the total number of qualified global suppliers is small, dual-sourcing is a business-continuity imperative rather than a preference.
The cost of maintaining a qualified secondary supplier — audit costs, development costs, the commercial volume required to keep the secondary supplier’s DMF current and their manufacturing processes validated — is a known, manageable expense. The cost of a supply disruption on a commercially launched product, including back-order penalties, regulatory notification obligations, and patient harm from drug shortages, is an uncertain, potentially catastrophic outcome. The calculation typically favors dual-sourcing for any API representing more than $5 million in annual cost exposure.
The First-to-File Race: Aligning API Qualification with ANDA Strategy
For generic drug companies, the highest-value opportunity in the patent expiry cycle is the Paragraph IV first-to-file position — the first ANDA filed containing a Paragraph IV certification challenging one or more Orange Book-listed patents. The first filer who successfully navigates the resulting patent litigation (or settles on terms that allow early market entry) receives 180 days of generic exclusivity during which no other generic can be approved. For blockbuster drugs, that exclusivity period can generate hundreds of millions of dollars in gross profit.
The 180-day exclusivity does not just reward the first filer — it punishes every competitor who is even a day behind. A generic company whose ANDA was filed second receives no exclusivity benefit and must wait until the first-filer launches and either exhausts or forfeits the exclusivity period before their own product can be approved. The difference between first and second in filing order can be worth tens to hundreds of millions of dollars.
To win the ‘First-to-File’ race, procurement must secure a viable API source years before the patent expires. The API supplier must be technically capable (able to produce the API to the required specifications), document ready (able to provide a Drug Master File to the FDA to support the ANDA), and non-infringing (capable of synthesizing the API via a route that does not infringe on the innovator’s process patents). This often requires deep collaboration between the generic company’s R&D team and the API supplier to validate the synthesis pathway.
The timeline requirements for first-to-file positioning are more demanding than most procurement teams appreciate. Filing a Paragraph IV ANDA requires a substantially complete ANDA — including a complete CMC section covering the API supplier, the finished drug formulation, and bioequivalence data. The bioequivalence study alone typically requires 12 to 18 months from API receipt to study completion. Before that, the API supplier must be qualified, samples must clear technical evaluation, and the supplier’s DMF must be in order.
Working backwards from a target Paragraph IV filing date two or three years after a patent’s nominal expiry date leaves very little time for anything to go wrong at the API sourcing stage. Working backwards from a target filing date one to two years after patent expiry — the timeframe required for genuine first-to-file competition on a major molecule — means that API sourcing work must begin before the patent expiry date, sometimes years before it.
Utilizing platforms like DrugPatentWatch to monitor Paragraph IV certification windows and ‘skinny label’ opportunities allows firms to lock in API supply for ‘First-to-File’ exclusivity, a critical revenue driver.
‘Skinny labeling’ — also called Section viii carve-outs — allows a generic manufacturer to launch for unpatented indications while omitting indications still covered by method-of-use patents. For drugs with complex multi-indication profiles, this strategy can accelerate market entry significantly. But it requires API sourcing decisions that account for the full indications ultimately intended, because post-launch label amendments to add carved-out indications require supplemental ANDAs that reference the existing qualified API supplier configuration. Changing API suppliers after launch is possible but requires either Prior Approval Supplements (PAS) or Changes Being Effected (CBE) filings depending on the nature of the change — additional time and regulatory risk that should be anticipated during the original sourcing decision.
Practical Case Studies: What Intelligent Sourcing Looks Like in Action
Case Study 1: Apixaban (Eliquis) — The Complexity of a Crowded Cliff
Apixaban exemplifies the challenge of a high-value, intensely competitive patent expiry in which IP complexity has delayed generic entry well beyond the nominal patent expiry date. Bristol Myers Squibb and Pfizer hold multiple patents on apixaban across the Orange Book, including composition-of-matter, polymorphic form, and formulation patents, with a litigation campaign that has kept most generic challengers at bay.
BMS reported over $13 billion in 2024 Eliquis revenue. Key patents expire in 2026, though BMS and Pfizer have mounted an aggressive litigation campaign against generic challengers. BMS has announced cost restructuring programs explicitly linked to anticipated Eliquis revenue decline. Eliquis represents the largest single near-term patent cliff event in pure revenue terms.
For API suppliers and generic manufacturers entering the apixaban market, the immediate challenge is synthesis route differentiation. Apixaban’s synthesis involves multiple stereochemically complex steps, and several of the efficient routes have been patented by the originators or by third parties. API suppliers offering apixaban must demonstrate non-infringement through routes that are either novel or clearly outside the scope of existing process patents — a technical and IP challenge that has limited the number of fully qualified, IP-cleared API suppliers in the early phase of generic competition.
The DMF landscape for apixaban reflects this complexity. DrugPatentWatch data shows multiple DMF filings for apixaban from Indian and Chinese manufacturers, but the subset of those filings that are both GDUFA-cleared and accompanied by documented FTO opinions for the specific synthesis route is substantially smaller. Procurement teams who accepted DMF existence as a proxy for supplier readiness, without confirming the FTO position, faced the possibility of sourcing from a supplier whose synthesis route became the subject of litigation post-launch.
Case Study 2: Sitagliptin (Januvia) — First-Filer Economics at Scale
Sitagliptin, Merck’s DPP-4 inhibitor marketed as Januvia, generated over $2 billion annually at peak before generic competition began. The patent expiry in 2026, combined with a Paragraph IV landscape that multiple generic companies had been building positions on for several years, created an unusually well-anticipated competitive entry.
For API procurement teams, the sitagliptin case illustrates the value of early landscape intelligence. Companies that began evaluating sitagliptin API sourcing three to four years before patent expiry had time to identify the synthesis route constraints, qualify Indian suppliers who had developed non-infringing routes, and file ANDAs in positions that qualified for first-to-file exclusivity consideration. Companies that began evaluation one to two years before expiry found a more crowded DMF landscape, higher development service costs from API suppliers already managing multiple qualification projects for the same molecule, and reduced likelihood of achieving a first-filer position.
The sitagliptin phosphomonohydrate salt form adds a layer of complexity: Merck held patents on the specific salt form used in Januvia, which required generic API suppliers to demonstrate either non-infringement or invalidity of those salt patents in addition to the core compound patents. Several early Paragraph IV filers settled with Merck on terms that allowed authorized generic supply prior to full generic exclusivity expiry — a commercial outcome that effectively transferred a portion of the patent-expiry value from independent generic companies to Merck through authorized generic fees.
Case Study 3: Atorvastatin — The Commoditization Trajectory
Atorvastatin (Lipitor, Pfizer) reached generic entry in 2011-2012 and is now among the most commoditized APIs in global supply chains. Dozens of qualified suppliers hold active FDA DMFs, and the API price has declined to levels that make meaningful margin recovery difficult for any individual generic manufacturer.
The atorvastatin case is instructive for what happens to API supplier qualification dynamics at the far end of the commoditization cycle. When a molecule has been off-patent for more than a decade, the qualified supplier pool is large enough that procurement decisions are primarily driven by price, delivery reliability, and quality consistency — with patent IP risk effectively eliminated. The challenge shifts from identifying the right supplier to managing a competitive multi-supplier sourcing program and maintaining the quality monitoring required to catch the GMP degradation that sometimes occurs when suppliers de-prioritize products with minimal margin.
Recent FDA enforcement actions have included Warning Letters and import alerts against atorvastatin API manufacturers who had previously maintained acceptable compliance records. The pattern reflects a well-documented phenomenon: when an API reaches full commodity pricing, the economics of maintaining high-quality manufacturing become strained, and suppliers who cannot sustain quality investment at low margins either exit the market or cut corners. Ongoing monitoring of atorvastatin API suppliers — despite the molecule’s maturity — continues to identify quality management issues that require active management.
How to Use DrugPatentWatch in the API Qualification Intelligence Process
DrugPatentWatch is a commercial pharmaceutical intelligence platform that aggregates FDA Orange Book data, patent expiry dates, DMF filing records, Paragraph IV certification histories, ANDA approval records, and a range of other regulatory data into a searchable, structured format. For API procurement teams and generic drug business development professionals, it is the most comprehensive single source of integrated patent and regulatory intelligence currently available.
The platform’s utility in API supplier qualification spans multiple stages of the process described throughout this guide:
Patent Landscape Screening: DrugPatentWatch’s Orange Book search returns all listed patents for a target drug, including expiry dates, patent type classifications, and any Paragraph IV certification history. This provides the baseline patent intelligence needed to frame the FTO analysis and identify the scope of process patent research required.
DMF Landscape Mapping: The platform’s DMF database allows users to search by drug substance name and identify all DMF holders with filings for a target API. The DMF status information — including holder name, facility location, filing date, and amendment history — supports the Tier 1/2/3 classification exercise described earlier.
Competitive Intelligence on First-to-File: Paragraph IV filing records and first-to-file status information on DrugPatentWatch enable business development teams to assess the competitive landscape for a target molecule before committing resources to ANDA development.
Compliance Monitoring: Cross-referencing DMF holder names against FDA Warning Letter and import alert databases, supported by DrugPatentWatch’s aggregated data, enables the kind of ongoing supplier compliance monitoring that a systematic quality program requires.
Authorized Generic Tracking: DrugPatentWatch tracks authorized generic agreements, which are commercially important for understanding the effective competitive environment a generic ANDA filer will face upon market entry. An authorized generic launched by the originator on day one of generic exclusivity materially dilutes the first-filer’s revenue advantage.
The platform is used by generic pharmaceutical companies, branded manufacturers monitoring their own competitive exposure, investment analysts valuing pharmaceutical patent estates, and litigation teams building patent challenges. In the API qualification context, it operates as the intelligence layer that converts raw regulatory and patent data into actionable supplier screening output.
Building Internal Capabilities: The Patent-Regulatory Intelligence Function
Most pharmaceutical companies that have a sophisticated API qualification process have, either formally or informally, built an internal function responsible for integrating patent intelligence with regulatory intelligence. In companies with more formal structures, this may be organized as a ‘portfolio intelligence’ or ‘business intelligence’ function within the generic pharmaceutical business development or regulatory affairs organizations.
The core capabilities this function needs include:
Patent Analysis: The ability to read and interpret patent claims — not to the level of conducting full freedom-to-operate opinions (that requires qualified patent counsel), but to the level of identifying which patents in a landscape are potentially relevant to API sourcing, which have been challenged or invalidated, and which remain untested. This is a learnable analytical skill that does not require a law degree.
Regulatory Database Literacy: Fluency with FDA databases — the Orange Book, the DMF database, the Warning Letter database, the Import Alert database, the Establishment Inspection Report database — and the ability to synthesize information across these databases into a coherent supplier risk picture. This is a skill that can be developed with training and structured practice.
Commercial API Market Knowledge: Understanding of the global API manufacturing landscape — which suppliers are active in which therapeutic areas, which manufacturers have invested in specific capabilities like HPAPI synthesis or sterile API production, which are growing their regulatory footprints and which are retrenching. This knowledge comes from industry experience and from platforms like DrugPatentWatch that make the regulatory footprint of manufacturers visible.
Geopolitical Risk Assessment: The ability to evaluate how macroeconomic and legislative developments — tariffs, BIOSECURE Act designations, logistics disruptions — affect the risk profile of suppliers in specific geographies. This does not require geopolitical expertise, but it does require active monitoring of the relevant policy developments.
For smaller companies that cannot build these capabilities in-house, regulatory consulting firms and specialized intelligence platforms offer access to comparable capabilities on a project or subscription basis. The important thing is that the function exists in some form — either internally or through reliable external partners — before the company begins committing resources to ANDA development for a target molecule.
The Regulatory Filing: How API Supplier Qualification Appears in the ANDA
The work described throughout this guide ultimately surfaces in the drug substance section (Module 3.2.S) of the ANDA submission. This section must contain:
The drug substance name, structure, molecular formula, and physicochemical properties
A complete description of the manufacturer(s), including facility name, address, and DMF number
A description of the manufacturing process and process controls, either in full detail or by reference to a DMF
A characterization section covering the impurity profile, polymorphic form, and any relevant physical properties
Reference standards and their sources
A specification, with justification for all limits
Analytical procedures, with validation data
Stability data supporting the proposed retest date
The quality of this section — and the underlying supplier qualification work that supports it — directly affects both the completeness of the ANDA as submitted and the ease with which the FDA can review and approve it. ANDAs with incomplete or inconsistent drug substance sections are among the most common sources of Complete Response Letters (CRLs), which reset the review clock and delay approval.
The GDUFA program has introduced specific commitments from the FDA regarding review timelines for ANDAs — currently a goal of reviewing 90% of standard ANDAs within 10 months of receipt. But GDUFA goal dates apply to substantially complete applications. An ANDA with drug substance section deficiencies will receive a Refuse to Receive (RTR) action or a Complete Response Letter, neither of which is covered by the GDUFA timeline commitment.
Invest the quality assurance work upfront. A well-qualified API supplier with a current, complete DMF and a clean compliance record is not just a regulatory necessity — it is the foundation of a complete, approvable ANDA.
Sector-Specific Considerations: Oncology, Cardiovascular, and Controlled Substances
The general framework described throughout this guide applies across therapeutic areas, but each therapeutic area has specific considerations that affect API supplier qualification.
Oncology APIs
The oncology API opportunity in the late 2020s is substantial and complex. The LOE wave covering major branded oncology compounds — including multiple kinase inhibitors, PARP inhibitors, and immunotherapy checkpoint inhibitors with patents expiring from 2025-2030 — creates the conditions for significant generic API demand growth. The technical difficulty of oncology API synthesis, including HPAPI handling requirements at occupational exposure limits below 1 microgram per cubic meter, creates regulatory and engineering requirements for containment that further limit competitive entry.
For oncology API qualification, the additional requirements beyond standard ICH Q7 compliance include: validated contained manufacturing systems meeting the required occupational exposure band (OEB) for the specific molecule, operator health surveillance programs, validated cleaning methods specific to the potent compound, and cross-contamination risk assessments for all other products manufactured at the same facility. A supplier who produces conventional small-molecule APIs in shared facilities with a potent oncology API without validated segregation and containment is not a qualified supplier for the potent product, regardless of their general GMP standing.
Cardiovascular APIs
The cardiovascular portfolio is one of the most commoditized in the global generic drug market — statins, ACE inhibitors, beta blockers, and several anticoagulants have been off-patent long enough that the supplier pool is large and price competition is intense. The apixaban case is somewhat exceptional in this category because its synthesis complexity and patent estate have maintained a more constrained supplier landscape through the early years of generic entry.
For cardiovascular APIs being sourced from established, commodity-stage molecules, the primary qualification concerns are quality consistency over time (given the commoditization economics described above), supply reliability (given the high volume and just-in-time purchasing patterns in this segment), and ongoing GMP compliance monitoring.
Controlled Substances
API sourcing for controlled substance drugs — opioids, benzodiazepines, CNS stimulants, and related classes — involves an additional layer of regulatory complexity beyond CGMP compliance. DEA registration and scheduling requirements apply both to the API manufacturer and to the finished drug manufacturer, and the DEA’s Aggregate Production Quota (APQ) system limits the total US production of each controlled substance.
For controlled substance APIs, supplier qualification must include confirmation of the supplier’s DEA Schedule I or II manufacturing registration (as applicable), their DEA Form 3 authorization for export, and their history of compliance with DEA quota requirements. The DEA maintains its own inspection program for Schedule I and II registrants, and a supplier with DEA compliance issues creates a supply security risk that has no simple regulatory alternative.
Key Takeaways
Patent intelligence precedes supplier selection. For every target API, a systematic analysis of the Orange Book, process patent landscape, and Paragraph IV history must precede any commercial engagement with candidate suppliers. IP risk discovered after commercial qualification is exponentially more expensive to manage than IP risk identified before commitment.
DMF status is a screening criterion, not a qualification standard. The existence of a Type II DMF establishes that a supplier has engaged with the FDA regulatory process. GDUFA completeness clearance and a history of successful ANDA references establish substantially more — that the DMF has survived at least some level of regulatory scrutiny in a commercial context.
The compliance record of the specific facility matters. Warning Letters, import alerts, and inspection observations are facility-specific. A corporate parent with a clean compliance record does not guarantee that a specific manufacturing site within that organization operates to the same standard.
Data integrity is a disqualifying risk. A supplier with documented data integrity issues — falsified records, deleted OOS results, manufactured audit trails — presents a risk that cannot be remediated with process changes alone. These should be treated as disqualifying findings pending demonstrated and sustained cultural remediation.
Geopolitical exposure requires explicit management. The BIOSECURE Act is now law, Section 301 tariffs remain in force, and India’s indirect dependence on Chinese intermediates means that ‘diversification’ from China that only addresses the final manufacturing step does not achieve meaningful supply chain independence.
First-to-file economics require API qualification to begin years before patent expiry. The window for first-filer positioning on a major molecule is determined by the time required to qualify an API supplier, complete bioequivalence studies, and file a complete ANDA — a timeline of three to five years from decision to filing for complex molecules.
DrugPatentWatch and regulatory databases enable systematic, scalable intelligence. No team can manually track the patent and regulatory status of dozens of molecules and hundreds of suppliers. Systematic use of integrated intelligence platforms converts that complexity into manageable, actionable analysis.
Dual sourcing is not a luxury. For any API that represents meaningful commercial exposure and is sourced from a limited number of qualified global suppliers, maintaining a qualified second source is a business continuity requirement, not a procurement preference.
FAQ
Q1: Can I use a foreign regulatory approval — such as an EDQM Certificate of Suitability or an Indian CDSCO GMP certificate — as a substitute for an FDA Type II DMF when filing a US ANDA?
No. For an Abbreviated New Drug Application filed with the FDA, the drug substance must be supported by either a complete CMC section within the ANDA itself or a Type II Drug Master File filed with FDA by the API manufacturer. An EDQM CEP or a domestic regulatory certificate from another authority does not satisfy the FDA’s regulatory requirements, regardless of the scientific quality of those documents. That said, an EDQM CEP or WHO prequalification status is useful supporting evidence that a supplier’s manufacturing process has received regulatory scrutiny — it informs your risk assessment, but it does not substitute for the FDA-specific filing.
Q2: What happens if my qualified API supplier receives a Warning Letter after my ANDA is filed but before it is approved?
This situation requires immediate action on multiple fronts. If the Warning Letter indicates that the API supplier’s products are adulterated or that their manufacturing does not conform to CGMP, the FDA may issue a Complete Response Letter to your ANDA citing the supplier’s non-compliance as a basis for non-approval. You should notify your regulatory affairs team immediately, assess whether the Warning Letter observations affect the specific API lot or manufacturing process covered by your ANDA, and evaluate whether filing an ANDA supplement to add a second qualified API supplier is feasible within the review timeline. This scenario is precisely why ongoing compliance monitoring of qualified suppliers is not optional.
Q3: How should I evaluate an API supplier who has a current DMF but has never been referenced in an approved ANDA?
A DMF that has never been successfully referenced in an approved ANDA has not been fully tested against FDA review standards. The GDUFA completeness assessment confirms structural adequacy, but it explicitly does not replace the scientific review that occurs when the DMF is assessed in the context of an actual ANDA review. An unreferenced DMF may be scientifically adequate — particularly if it has been prepared by an experienced regulatory team and follows current guidance. But you bear the risk that a deficiency identified during the first ANDA review will affect your application’s approval timeline. For high-priority molecules where approval timing matters commercially, the risk-adjusted preference is for suppliers with proven DMF histories.
Q4: How does the BIOSECURE Act specifically affect API sourcing decisions for a generic drug company with no federal government contracts?
For a generic drug company whose revenue comes entirely from commercial health plans and retail pharmacies, the direct regulatory effect of the BIOSECURE Act on API sourcing is currently limited. The Act’s restrictions apply specifically to U.S. executive agency procurement and to entities receiving federal research funding. Commercial generic pharmaceutical sales to non-government payers are not directly covered. However, three indirect effects are worth managing: first, the Act accelerates a general de-risking of Chinese supply chain exposure that institutional investors, strategic partners, and large commercial customers may apply to supplier scorecards even in the absence of legal requirement; second, any future expansion of the Act’s scope (or state-level equivalents) could extend restrictions to Medicaid and other government health program reimbursements; third, maintaining a supply chain that is heavily dependent on entities that may be designated as biotechnology companies of concern creates a binary transition risk if circumstances change.
Q5: Is it possible to qualify a new API supplier after an ANDA is already approved and the product is commercially launched, and what does that process entail?
Yes, but the process is more involved than pre-approval qualification. A post-approval API supplier change requires an ANDA supplement — either a Prior Approval Supplement (PAS) or a Changes Being Effected in 30 Days (CBE-30) supplement, depending on the nature of the change as defined in FDA’s guidance on pharmaceutical manufacturing changes. If the new supplier uses a materially different synthesis route, or if the impurity profile or physical characteristics of the API differ from what was approved, a PAS is likely required, which means waiting 12 months or more for FDA review before the new supplier can supply commercial product. For changes considered minor — same synthesis route, same facility type, no change in physical or chemical properties — a CBE-30 may be applicable. Get this classification confirmed by a qualified regulatory affairs professional before assuming the shorter path applies; misclassification of a supplement type is a significant regulatory compliance issue.
References
[1] DrugPatentWatch. (2025, December 8). The strategic core: A definitive guide to API sourcing for generic drug manufacturers. https://www.drugpatentwatch.com/blog/the-strategic-core-a-definitive-guide-to-api-sourcing-for-generic-drug-manufacturers/
[2] DrugPatentWatch. (2026, March 22). How to find a reputable active pharmaceutical ingredient (API) supplier: A comprehensive guide. https://www.drugpatentwatch.com/blog/how-to-find-a-reputable-api-supplier/
[3] DrugPatentWatch. (2026, March 22). Pharmaceutical procurement practice aspects: A comprehensive guide to market domination. https://www.drugpatentwatch.com/blog/pharmaceutical-procurement-practice-aspects/
[4] PharmInt. (2025, September 18). API sourcing: Definition and global procurement guide. https://pharmint.net/glossary/api-sourcing/
[5] U.S. Food and Drug Administration. (2025). Types of Drug Master Files (DMFs). https://www.fda.gov/drugs/drug-master-files-dmfs/types-drug-master-files-dmfs
[6] U.S. Food and Drug Administration. (2025). Drug Master Files (DMFs). https://www.fda.gov/drugs/forms-submission-requirements/drug-master-files-dmfs
[7] Celegence. (2025, July 11). Drug Master Files (DMFs): Types and global use. https://www.celegence.com/drug-master-file-types-global-submission-requirements/
[8] DDReg Pharma. (2025). DMF filing and submission services for API. https://www.ddregpharma.com/solutions/regulatory-affairs/api-dmf-services
[9] Fierce Pharma. (2025, February 20). FDA hits pair of Indian API makers with warning letters, import alerts. https://www.fiercepharma.com/manufacturing/fda-hits-pair-indian-api-drugmakers-warning-letters
[10] Pharmaceutical Technology. (2025, December 2). FDA issues warning letter to Aspen Biopharma Labs for API manufacturing deviations. https://www.pharmtech.com/view/fda-issues-warning-letter-aspen-biopharma-labs-api-manufacturing-deviations
[11] Pharma Manufacturing. (2025, February 12). FDA slaps two Chinese API manufacturers with warning letters over cGMP violations. https://www.pharmamanufacturing.com/industry-news/news/55267470/fda-slaps-two-chinese-api-manufacturers-with-warning-letters-over-cgmp-violations
[12] SciLife. (2026, April). FDA warning letters 2025: Trends, violations, and how to avoid them. https://www.scilife.io/blog/worst-fda-warning-letters-pharma
[13] QBench. (2026, March 12). Inside 470 FDA warning letters from 2025: What labs need to know. https://qbench.com/resources/inside-470-fda-warning-letters-from-2025-what-labs-need-to-know
[14] RAPS. (2025, February). FDA warns Indian API supplier for data integrity lapses, Chinese contract lab for inadequate testing. https://www.raps.org/news-and-articles/news-articles/2025/2/fda-warns-indian-api-supplier-or-data-integrity-la
[15] LGM Pharma. (2025, March 6). Tariffs and geopolitics: API supply chain resilience in 2025. https://lgmpharma.com/blog/tariffs-api-supply-chain-resilience-in-2025/
[16] Morrison Foerster. (2025, December 18). BIOSECURE Act update. https://www.mofo.com/resources/insights/251218-biosecure-act-update
[17] Greenberg Traurig. (2025, November 3). BIOSECURE Act advances in the U.S. Senate. https://www.gtlaw.com/en/insights/2025/11/biosecure-act-advances-in-the-us-senate
[18] Foley Hoag. (2025, December). Congress passes BIOSECURE Act: Here’s what you need to know. https://foleyhoag.com/news-and-insights/publications/alerts-and-updates/2025/december/congress-passes-biosecure-act-here-s-what-you-need-to-know/
[19] PharmaVoice. (2025, December 15). After years of debate, the Biosecure Act could be headed for Trump’s desk. https://www.pharmavoice.com/news/biosecure-act-china-us-pharma-drug-trump-congress/807812/
[20] DrugPatentWatch. (2026, March 10). The patent cliff playbook: Pharmaceutical IP valuation, generic entry timing, and biosimilar strategy. https://www.drugpatentwatch.com/blog/patent-expirations-seizing-opportunities-in-the-generic-drug-market/
[21] Drug Discovery News. (2026, February 24). Blockbuster drugs face a massive patent cliff in 2026. https://www.drugdiscoverynews.com/blockbuster-drugs-face-a-massive-patent-cliff-in-2026-17019
[22] Credevo. (2026, March 5). Trending molecules and generic drug opportunities: Blockbuster drugs with upcoming patent expirations (2026-2030). https://credevo.com/articles/2026/03/05/trending-molecules-and-generic-drug-opportunities-blockbuster-drugs-with-upcoming-patent-expirations-2026-2030/
[23] DrugPatentWatch. (2026, March 19). EU API manufacturing: The complete regulatory, IP, and supply chain resilience guide. https://www.drugpatentwatch.com/blog/regulatory-considerations-for-api-manufacturing-in-the-eu-ensuring-compliance-and-supply-chain-resilience/
[24] Pharmaceutical Technology. (2026, February 12). Managing supply chain challenges for CDMOs. https://www.pharmaceutical-technology.com/sponsored/managing-supply-chain-challenges-for-cdmos/
[25] GlobalData/Pharmaceutical Technology. (2025). State of the biopharmaceutical industry: 2025 mid-year update. GlobalData.
[26] MIAS Pharma. (2026, January 2). US BIOSECURE Act 2.0 and its impact on the pharma supply chain. https://miaspharma.com/us-biosecure-act-impact-pharma-supply-chain/
[27] BioSpace. (2024, October 3). BIOSECURE Act could signal a seismic shift for biopharma in US and China. https://www.biospace.com/policy/biosecure-act-could-signal-a-seismic-shift-for-biopharma-in-us-and-china
[28] Global Pricing Innovations. (2025, November 4). Patent cliff in pharma: Navigating disruption and creating opportunity. https://globalpricing.com/patent-cliff-in-pharma-navigating-disruption-and-creating-opportunity/
[29] ECA Academy/GMP Compliance. (2025, February 19). FDA warning letter: Chinese API manufacturer on import alert due to CGMP violations. https://www.gmp-compliance.org/gmp-news/fda-warning-letter-chinese-api-manufacturer-on-import-alert-due-to-cgmp-violations
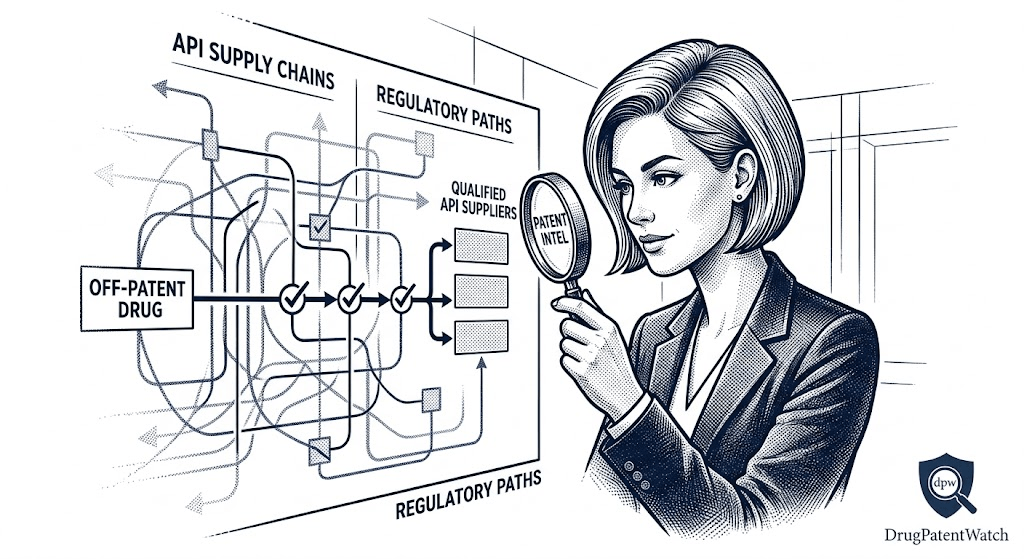



















")






