1. The Scale of the Cliff: $236 Billion in Revenue at Risk Through 2030
The pharmaceutical industry is heading into the most consequential period of patent loss in its history. Between 2025 and 2030, an estimated $236 billion in annual branded drug sales face loss of exclusivity (LOE) in the U.S. market alone. GlobalData puts the figure at $230 billion; EY’s analysis, which includes international markets, reaches $356 billion. The precise number depends on which LOE dates one uses and how one treats partial-year exposure, but every estimate lands in the same catastrophic neighborhood.
The headline number, though striking, understates the disruption. Roughly 190 drugs will lose exclusivity during this window. Sixty-nine qualify as blockbusters, meaning they each generate more than $1 billion in annual revenue. Among them are drugs that define entire therapeutic categories: Merck’s Keytruda (pembrolizumab), the world’s best-selling oncology drug at roughly $25 billion in 2024 sales; Bristol-Myers Squibb and Pfizer’s Eliquis (apixaban), the leading oral anticoagulant at around $12 billion annually; and Johnson & Johnson’s Stelara (ustekinumab), whose global revenue peaked near $10 billion before biosimilar entry began eroding it in 2023.
Saying these drugs are ‘at risk’ requires qualification. Not every LOE event produces the same financial outcome. The depth and speed of revenue erosion depend on at least six variables that analysts must model independently: the number of generic or biosimilar competitors that actually enter, whether the molecule qualifies for automatic pharmacy substitution, the formulary behavior of the three largest PBMs (CVS Caremark, Express Scripts, and OptumRx), whether the innovator has erected a patent thicket requiring years of litigation to clear, the availability of an authorized generic, and whether the innovator has successor products ready to absorb displaced revenue.
The Humira cliff illustrated all six variables playing out simultaneously and in contradictory directions. The first U.S. biosimilar launched in January 2023 at an 85% discount to list price. By the end of that year, biosimilar market share was 2%. The rebate wall held for twelve months before CVS Caremark’s formulary decision in 2024 broke it open. Pfizer’s Lipitor cliff, by contrast, unfolded over four years, not one, because Pfizer deployed an authorized generic, PBM co-pay deals, and direct-to-patient coupons that slowed volume loss enough to justify continued branded promotion. Generic manufacturers face a fundamentally different problem with biologics than they do with small molecules, and investors who model the two the same way will consistently mis-price the LOE event.
Key Takeaways: Section 1
The 2025-2030 patent cliff is the largest in pharmaceutical history by revenue volume, but revenue-at-risk figures mask variance in actual erosion rates. LOE modeling requires six independent variables: competitor count, substitutability, PBM formulary positioning, patent thicket depth, authorized generic presence, and successor pipeline readiness. Small molecule and biologic LOE events require separate analytical frameworks.
2. The Anatomy of Market Exclusivity: Decoding Overlapping Protection Timelines
A drug’s effective period of exclusivity is not a single number. It is the terminal date of the last relevant protection to expire, drawn from two entirely separate legal systems: the patent system administered by the U.S. Patent and Trademark Office (USPTO) and the regulatory exclusivity system administered by the FDA. These two systems run concurrently, sequentially, or entirely independently, and confusing them is one of the most common analytical errors in pharmaceutical market research.
2a. Standard Patent Term and Its Real Commercial Meaning
A U.S. patent has a statutory term of 20 years from its earliest effective filing date. For pharmaceutical compounds, that filing date typically occurs during or shortly after the discovery phase, years before the molecule has proven clinical utility. The drug then spends years in preclinical testing and multiple phases of clinical trials before reaching the FDA for review. The FDA’s review itself consumes additional time. By the time a drug reaches the market, the original composition-of-matter patent, which is the strongest and broadest form of pharmaceutical protection, may have only 7 to 12 years of remaining life.
That 7-to-12-year window is the ‘effective patent life,’ and it is the single most important figure for revenue forecasting, not the nominal 20-year term. The gap between nominal and effective patent life can exceed a decade for complex biologics that require lengthy development programs.
2b. Patent Term Extension Under Hatch-Waxman
Congress partially addressed effective patent life erosion through the patent term extension (PTE) provision of the Hatch-Waxman Act. An innovator may apply for an extension of up to five years to compensate for time lost during FDA regulatory review, subject to a ceiling that the post-extension effective patent life cannot exceed 14 years from the date of first FDA approval. The PTE applies only to one patent per approved product, selected by the patentee. Companies routinely select the patent with the latest natural expiry to maximize the terminal protection date.
PTE applications are filed with the USPTO but require coordination with the FDA to establish the regulatory review period. The calculation is mechanical but not simple: the extension equals one-half the testing phase plus the full regulatory review phase, minus 14 years if the post-extension term would otherwise exceed that threshold. Biotech companies frequently file PTE on method-of-use patents rather than composition-of-matter patents when the latter are already near expiry, a tactic that can add years of protection for specific approved indications even after the molecule itself is in the public domain.
2c. Regulatory Exclusivity: The FDA’s Parallel System
Regulatory exclusivity runs on an entirely separate clock from patent protection and can block generic or biosimilar entry even after every relevant patent has expired. The FDA administers five primary exclusivity types, each with distinct durations and triggering conditions.
New Chemical Entity (NCE) exclusivity lasts five years from the date of NDA approval for a drug containing an active moiety never previously approved. During the first four years of this period, the FDA will not accept an ANDA for the generic. In year five, an ANDA with a Paragraph IV certification challenging the brand’s patents can be submitted, but the 30-month automatic stay triggered by brand litigation does not begin until year five, effectively compressing the available challenge window.
New Clinical Investigation exclusivity provides three years of protection for approved supplements to an existing NDA, provided that new clinical studies were essential to the approval. This exclusivity covers the change, not the underlying active ingredient, which means a generic can still reference the original active ingredient’s safety and efficacy data after the NCE exclusivity expires. The three-year exclusivity covers only the new indication, dosage form, or route of administration.
Orphan Drug Exclusivity (ODE) grants seven years of market protection for a drug designated to treat a rare disease affecting fewer than 200,000 U.S. patients. ODE is the most powerful exclusivity in the FDA’s toolkit because it cannot be broken by a Paragraph IV certification. No ANDA or 505(b)(2) application for the same drug in the same indication can be approved during the seven-year window, regardless of patent status. The Humira pattern of layering orphan designations for specific inflammatory conditions onto a broad-label biologic is one of the more sophisticated exclusivity strategies in the industry.
Pediatric Exclusivity adds six months to all existing patents and regulatory exclusivities, not just one. It attaches to the drug as a whole rather than to a specific patent. This means a drug with four patents and an NCE exclusivity that receives a pediatric study request, completes those studies, and earns the exclusivity extends all of those protections by six months simultaneously. For a blockbuster drug, the revenue generated during those six months can easily exceed $1 billion, making pediatric exclusivity one of the highest-return regulatory investments available to an innovator.
The first-filer 180-day generic exclusivity does not protect the brand; it protects the first ANDA applicant to file a Paragraph IV certification. The FDA cannot approve any subsequent generic ANDA until 180 days after the first generic commercially markets its product or a court enters final judgment of patent invalidity or non-infringement, whichever comes first. Forfeiture provisions added by the Medicare Modernization Act (MMA) of 2003 can strip this exclusivity from a first-filer that fails to market within 75 days of a court decision or within 30 months of the first filing date.
Exclusivity Type Reference Table
Protection Type
Governing Body
Duration
Trigger
Breakable by Para. IV?
Composition-of-Matter Patent
USPTO
20 yrs from filing (+ PTE up to 5 yrs)
Patent grant
Yes, via litigation
New Chemical Entity Exclusivity
FDA
5 years
NDA approval (new active moiety)
After year 4
New Clinical Investigation Exclusivity
FDA
3 years
NDA supplement approval requiring new trials
Yes
Orphan Drug Exclusivity
FDA
7 years
Approval for rare disease indication
No
Pediatric Exclusivity
FDA
6 months added to all protections
Completion of pediatric studies
N/A
180-Day Generic Exclusivity
FDA
180 days post-commercial marketing
First Paragraph IV ANDA filing
N/A
Biologic Reference Product Exclusivity
FDA
12 years
BLA approval
No
Key Takeaways: Section 2
Effective patent life averages 7 to 12 years, not 20. Patent protection and FDA regulatory exclusivity are legally distinct systems that must be tracked separately and that can interact in non-obvious ways. Orphan Drug Exclusivity is the most durable form of market protection because it cannot be challenged via a Paragraph IV filing. Pediatric Exclusivity extends all protections simultaneously and routinely generates more than $1 billion in additional branded revenue.
3. IP Valuation Framework: How to Price a Patent Portfolio as a Core Asset
For pharma IP teams, M&A analysts, and institutional investors, a drug’s patent portfolio is not background legal paperwork. It is the primary determinant of a product’s revenue duration and, by extension, its net present value. Rigorous IP valuation requires a purpose-built framework that goes well beyond checking the Orange Book expiry date.
3a. The Revenue-Duration Model
The most widely used approach to pharmaceutical patent valuation is a discounted cash flow (DCF) model that incorporates probability-weighted revenue duration. The core inputs are: peak branded revenue, the probability that each individual patent survives a Paragraph IV challenge, the probability of regulatory exclusivity expiring on schedule, the expected number of generic entrants upon LOE, and the projected price erosion curve under different competitor counts.
A composition-of-matter patent on a branded drug with no pending Paragraph IV challenges and no issued inter partes review (IPR) petitions at the Patent Trial and Appeal Board (PTAB) generally carries a high survival probability, often above 80%. A formulation patent or method-of-use patent filed after NDA approval, which covers incremental modifications rather than the core molecule, carries a materially lower probability, often in the 30-to-55% range, because these patents are more vulnerable to invalidity challenges based on obviousness. Courts and the PTAB have been increasingly willing to find secondary pharmaceutical patents obvious in view of the prior art, particularly when the improvement, such as a once-daily dosing schedule, flows predictably from known pharmacokinetics.
A realistic DCF for a drug with a composition-of-matter patent expiring in 2028 and seven secondary formulation patents expiring between 2029 and 2033 must separately probability-weight each layer. If the secondary patents collectively carry a 40% survival probability in Paragraph IV litigation and the composition-of-matter patent carries a 75% probability, then the expected terminal LOE date differs substantially from the most conservative reading of the patent thicket.
3b. Patent Thicket Density as an IP Asset Multiplier
Analysts often treat patent thicket density as a qualitative factor. It belongs in quantitative models. The correlation between the number of Orange Book-listed patents and the delay in generic entry is measurable. EvaluatePharma and academic researchers have documented that drugs with 10 or more Orange Book-listed patents delay generic entry by an average of 3.4 years beyond the composition-of-matter expiry date. Drugs with more than 20 listed patents see average delays exceeding five years.
For Humira, AbbVie listed more than 130 patents in the FDA’s Purple Book equivalent and executed settlement agreements with all major biosimilar challengers that deferred U.S. commercial launch until January 2023, seven years after the core adalimumab composition-of-matter patents expired in 2016. That seven-year delay, at peak Humira U.S. revenues of $18.6 billion in 2022, represents over $100 billion in protected branded revenue. The patent thicket was not merely a defensive legal strategy; it was AbbVie’s primary asset worth more than most mid-cap pharma pipelines.
3c. PTAB Inter Partes Review: The Litigation Discount
The creation of the Patent Trial and Appeal Board in 2012 under the America Invents Act (AIA) introduced a new variable into pharmaceutical patent valuation: the IPR petition. A generic or biosimilar challenger can petition the PTAB to invalidate an Orange Book patent on grounds of prior art, bypassing district court litigation for a faster, cheaper administrative proceeding. The PTAB institution rate for pharmaceutical patents has historically ranged between 60% and 70%, and petitions that are instituted are invalidated in whole or in part roughly 75% of the time in final written decisions.
The practical consequence is that pharmaceutical patents, particularly secondary patents covering formulations and methods of use, face a credible annihilation risk that must be priced into IP valuation models. A formulation patent that appears to extend exclusivity to 2031 carries a materially lower expected value if a PTAB petition has been instituted and briefing is underway. Conversely, if the PTAB has declined to institute review, the patent’s effective value increases because the administrative challenge channel is closed for that proceeding.
3d. The 505(b)(2) Pathway and Its Impact on IP Asset Value
The 505(b)(2) pathway, which allows an NDA applicant to rely on previously published literature or an FDA finding of safety and effectiveness for an already-approved product, complicates standard IP valuation because it creates a competitor class that is neither a traditional generic nor a full innovator. A 505(b)(2) applicant must certify against Orange Book patents, just as an ANDA filer must, which means listed patents retain blocking power against this pathway. However, 505(b)(2) applicants can introduce incremental improvements that earn their own new regulatory exclusivity protections, effectively creating a new layer of competition that can erode the branded franchise while simultaneously accumulating its own IP.
For IP teams, the 505(b)(2) landscape around a given molecule is a critical monitoring priority. A competitor that files a 505(b)(2) for a next-generation formulation of a drug nearing LOE can legitimately challenge the innovator’s lifecycle management strategy while building its own protected position.
Investment Strategy: IP Valuation
Institutional investors should model pharmaceutical patent portfolios using three separate scenarios for each major product: (1) a ‘thicket holds’ scenario in which secondary patents survive litigation and delay entry to the last listed expiry date; (2) a ‘core patent only’ scenario in which secondary patents are stripped by PTAB or district court decisions and LOE occurs at the composition-of-matter expiry; (3) a ‘compound failure’ scenario in which an at-risk generic launch occurs before any patent expires, triggering injunction proceedings and damages. Probability-weight each scenario using publicly available PTAB petition data, district court docket records, and Orange Book patent certification history. The delta between the ‘thicket holds’ and ‘core patent only’ NPV scenarios is the maximum valuation benefit that incumbent management can claim from their patent strategy, and it should be scrutinized for realism at every earnings call.
Key Takeaways: Section 3
IP valuation requires probability-weighted DCF modeling, not simple LOE date lookups. Patent thicket density measurably delays generic entry and represents billions in protected revenue. PTAB IPR petitions impose a litigation discount on secondary pharmaceutical patents that must appear explicitly in valuation models. The 505(b)(2) pathway creates a third competitor archetype that standard generic entry models do not capture.
4. The Hatch-Waxman Act: Architecture of the U.S. Generic Drug Market
The Drug Price Competition and Patent Term Restoration Act of 1984, enacted as a compromise brokered by Senator Orrin Hatch and Representative Henry Waxman, established the entire legal framework within which pharmaceutical patent expiry operates in the United States. Forty-two years after its passage, the Act’s basic architecture remains intact, though it has been amended multiple times and interpreted by decades of litigation.
4a. The Problem Hatch-Waxman Solved
Before 1984, a generic drug manufacturer could not begin development work on a branded drug until after its patents expired, because doing so constituted patent infringement. Even after expiry, the generic manufacturer had to conduct its own full clinical trials to prove safety and efficacy, duplicating the innovator’s work at significant expense. The combined effect was a de facto monopoly extension of many years beyond the nominal patent term. Generic drugs constituted less than 20% of U.S. prescription volume in 1984. By 2024, they account for roughly 92% of dispensed prescriptions, though only about 20% of drug spending, which captures precisely the pricing dynamic Hatch-Waxman was designed to create.
4b. The ANDA Pathway: Abbreviated New Drug Application
The central mechanism for generic drug approval under Hatch-Waxman is the Abbreviated New Drug Application. An ANDA does not require the generic applicant to duplicate the innovator’s clinical trials. The generic manufacturer must demonstrate bioequivalence, meaning the generic delivers the same active ingredient to the same site of action at the same rate and to the same extent as the reference listed drug (RLD). Bioequivalence is typically established through pharmacokinetic studies measuring plasma concentration of the active ingredient over time in healthy volunteers. The 90% confidence interval for the ratio of AUC (area under the curve) and Cmax (peak plasma concentration) between the generic and the reference product must fall within 80% to 125%.
The ANDA pathway reduced generic drug development costs from hundreds of millions of dollars to a range of $1 million to $4 million and compressed development timelines from a decade or more to approximately 18 to 36 months. This cost reduction is what enables dozens of generic manufacturers to enter a single market simultaneously, producing the competitive pricing dynamics observed after LOE.
4c. The Orange Book: The Competitive Intelligence Registry
The FDA publishes ‘Approved Drug Products with Therapeutic Equivalence Evaluations,’ universally known as the Orange Book, which lists every FDA-approved drug along with any patents that the brand manufacturer has certified cover the product. Brand manufacturers are legally required to submit patent information for listing within 30 days of approval or patent issuance, whichever is later. The Orange Book is the definitive public registry of patent protection for small molecule drugs. Its biosimilar counterpart, the Purple Book, lists reference biological products and their biosimilars but operates under a different disclosure framework that does not include patent information directly.
Orange Book patent listings are not passive disclosures. They are strategic weapons. An innovator that lists a new formulation patent in the Orange Book immediately activates the 30-month stay mechanism against any subsequently filed ANDA with a Paragraph IV certification against that patent. Generic manufacturers and their lawyers monitor Orange Book submissions with the same intensity that equity analysts monitor SEC filings.
4d. The 30-Month Automatic Stay
When a brand manufacturer receives notice of an ANDA with a Paragraph IV certification and sues the generic applicant for patent infringement within 45 days, the FDA is automatically barred from approving the ANDA for 30 months. The stay does not require a court order or a judicial finding of any kind. The brand company’s decision to file suit is sufficient. This automatic stay is the innovator’s most powerful immediate tool in the post-filing period, because it provides 30 months of continued market exclusivity while litigation proceeds. If the court resolves the case within 30 months, the stay terminates at the court’s decision. If the case is unresolved after 30 months, the FDA may proceed with approval, and the generic can launch at risk if it chooses.
The 30-month stay has been criticized as an automatic reward for filing suit regardless of the merits of the patent claims. Congress partially addressed this criticism in the MMA of 2003 by limiting the stay to one 30-month period per product, regardless of how many patents the brand subsequently lists in the Orange Book against the same ANDA. This change reduced the ability of brands to ‘stack’ stays by listing new patents after an ANDA was already on file.
Key Takeaways: Section 4
The Hatch-Waxman Act’s ANDA pathway reduced generic development costs by more than 95% compared to a full NDA, enabling the 92% generic dispensing rate seen today. The Orange Book is a strategic asset map, not merely a regulatory reference. The 30-month automatic stay is the brand’s most immediate post-filing legal tool and requires no judicial finding to take effect. Congress limited ‘stay stacking’ in 2003, but brands retain powerful tools through Orange Book listings of newly issued secondary patents.
5. Paragraph IV Certification: The Legal Engine Driving Generic Entry
The Paragraph IV certification is the mechanism by which a generic manufacturer asserts that a brand patent is invalid, unenforceable, or will not be infringed. It is, in effect, a formal legal challenge to the brand’s patent position, and it triggers one of the most distinctive legal regimes in U.S. commercial law: a lawsuit based on an ‘artificial act of infringement’ because the infringing product does not yet exist.
5a. The Four Certifications and Their Commercial Significance
When filing an ANDA, a generic applicant must submit a patent certification for each patent listed in the Orange Book for the reference listed drug. The four certification options are:
Paragraph I certifies that no relevant patent information has been submitted to the FDA. Paragraph II certifies that the patent has already expired. Paragraph III certifies that the applicant will not market until the patent expires. Each of these three avoids litigation but waives any competitive advantage from early entry.
Paragraph IV certification, by contrast, asserts that the patent is either invalid or would not be infringed. It is the only path to generic entry before patent expiry. The Paragraph IV filer must send formal notice to the brand manufacturer and NDA holder within 20 days of FDA acknowledgment of the ANDA’s receipt. The notice must include a detailed statement of the legal and factual basis for the invalidity or non-infringement claim. This notice letter, often dozens of pages of technical and legal argument, initiates the 45-day window in which the brand company can file suit to trigger the 30-month stay.
5b. The 180-Day Exclusivity Race
The first generic applicant to file a Paragraph IV ANDA for a given drug earns a 180-day period during which no other generic can be approved. This exclusivity period is the most valuable prize in generic drug development. During those six months, the first-filer shares the market only with the brand (and any authorized generic the brand may launch). The duopoly pricing that results, typically 20% to 40% below the brand’s list price but dramatically above the price that materializes under full competition, generates revenues that can define a generic manufacturer’s fiscal year.
The race to achieve first-filer status is intense and commercially rational. For a brand drug with $5 billion in annual U.S. revenue, the 180-day exclusivity period, at a modest 40% market share for the generic and a 30% discount to brand pricing, generates approximately $420 million in revenue for the first-filer. Legal and development costs for the challenge typically run $5 million to $25 million. The expected return on investment is extraordinary if the Paragraph IV challenge succeeds.
5c. The MMA and Forfeiture Provisions
The Medicare Modernization Act of 2003 introduced forfeiture provisions that can strip 180-day exclusivity from a first-filer in specific circumstances. Forfeiture occurs if the first-filer fails to market within 75 days of a court decision finding the patent invalid or not infringed, fails to market within 30 months of its ANDA filing if no suit was filed, amends its Paragraph IV certification to a Paragraph III (i.e., agrees to wait for patent expiry), is acquired by the brand company, or enters into an agreement with the brand company that delays entry beyond the forfeiture trigger date.
The final forfeiture trigger, commercial agreements that delay entry, connects directly to the FTC’s ongoing scrutiny of reverse payment settlements. A ‘reverse payment’ or ‘pay-for-delay’ agreement involves the brand company paying the generic challenger to withdraw its Paragraph IV certification and delay market entry. In FTC v. Actavis (2013), the Supreme Court held that such agreements are not categorically immune from antitrust scrutiny and must be evaluated under the ‘rule of reason.’ The FTC has since pursued numerous enforcement actions against settlements it characterizes as anticompetitive.
Investment Strategy: Paragraph IV Landscape
Investors monitoring generic entry timing should track Paragraph IV certifications as a leading indicator, typically 18 to 30 months ahead of generic launch. The ANDA filing date, combined with the likelihood of 30-month stay initiation and the strength of the patent thicket, allows construction of a probabilistic launch timeline. When multiple generic companies have filed Paragraph IV certifications for the same drug, the first-filer exclusivity mechanism is still valuable, but it may indicate that the patent challenge has sufficient merit to attract multiple challengers, which itself increases the probability of patent invalidation. Investors in branded companies should treat simultaneous Paragraph IV filings from four or more applicants as a material signal that the patent thicket may be weaker than management represents.
Key Takeaways: Section 5
The Paragraph IV certification is the commercial and legal engine of pre-patent-expiry generic entry. The 180-day first-filer exclusivity represents the highest return-on-investment opportunity in generic drug development. Forfeiture provisions introduced in 2003 and the Actavis antitrust framework limit the use of ‘pay-for-delay’ settlements to forestall entry. Multiple simultaneous Paragraph IV filings signal credible patent vulnerability and should function as a bearish signal for the brand company’s near-term revenue durability.
6. Small Molecules vs. Biologics: Regulatory Pathways and Economic Outcomes
The distinction between small molecule drugs and biologics is not merely semantic. It determines the regulatory pathway to approval, the scientific challenge of product characterization, the cost and duration of development, the number of competitors that realistically enter the market, the pricing dynamics post-LOE, and the role of the FDA’s interchangeability designation in driving retail-level substitution.
6a. Small Molecule Generics: The Economics of Identity
A small molecule drug is a compound with a defined molecular structure produced through chemical synthesis. Because the synthesis is reproducible and the structure is fully characterizable, a generic small molecule is, at the molecular level, identical to the reference listed drug. The active pharmaceutical ingredient (API) is the same compound. It binds to the same receptor, produces the same pharmacological effect, and is cleared by the same metabolic pathways. The generic’s task is to demonstrate bioequivalence, not to prove clinical efficacy from scratch.
This identity allows the Hatch-Waxman bioequivalence standard to function as intended. If two products deliver the same API to systemic circulation at the same rate and extent, they will produce equivalent clinical outcomes, and a head-to-head trial is not scientifically necessary to prove it. The regulatory and scientific consensus on this point has held for four decades.
The economic consequence is straightforward. Development costs are low, the regulatory pathway is well understood, and multiple manufacturers can produce chemically equivalent products simultaneously. When a blockbuster small molecule loses its composition-of-matter patent, a dozen or more ANDAs may have been on file for years. FDA approval of multiple ANDAs on the same date, which occurred with clopidogrel’s May 2012 LOE, produces immediate, full-scale price competition from day one and the steepest price erosion curves observed in the pharmaceutical market.
6b. Biologics: The Economics of Similarity
A biologic is produced by living cells, typically mammalian, bacterial, or yeast systems. The manufacturing process is not merely a technical detail; it is inseparable from the product. Minor variations in fermentation conditions, purification protocols, or cell culture media can alter the protein’s three-dimensional folding, glycosylation pattern, or aggregation behavior, all of which affect its safety and efficacy profile. This is not a deficiency in manufacturing quality; it is an inherent property of biological systems. Even the original reference product exhibits batch-to-batch variation within the bounds specified in its manufacturing license.
A biosimilar manufacturer cannot produce an exact copy of the reference biologic. The goal is to produce a molecule that is ‘highly similar’ to the reference product with no clinically meaningful differences in safety, purity, and potency. Proving this requires a ‘totality of the evidence’ analytical package that includes state-of-the-art structural and functional characterization using techniques such as mass spectrometry, X-ray crystallography, and surface plasmon resonance binding assays; animal pharmacology and toxicology studies; and at least one comparative clinical pharmacokinetic study, typically supplemented by a confirmatory efficacy and safety trial.
The BPCIA pathway requires an investment of $100 million to $250 million and 7 to 9 years. This cost and timeline naturally limits the number of biosimilar manufacturers who can compete for any given reference product. For Humira, which generated $200 billion in cumulative global revenue before U.S. biosimilar entry, the economics supported nine approved biosimilar products entering the U.S. market. For a biologic with $500 million in annual revenue, the economics may support only one or two biosimilar challengers, preserving a semi-competitive market with modest price reductions rather than the devastation seen with small molecule LOE.
6c. Interchangeability: The Retail Substitution Standard
The BPCIA created two tiers of biosimilar approval. A standard biosimilar demonstrates no clinically meaningful difference from the reference product. An interchangeable biosimilar meets a higher standard: it must be expected to produce the same clinical result as the reference product in any given patient, and the risk of alternating between the reference and the biosimilar must be no greater than the risk of using the reference product alone, without switching.
Interchangeability matters at the pharmacy counter. State pharmacy practice acts generally allow, and some require, pharmacists to substitute a generic for a branded small molecule without prescriber notification. An interchangeable biosimilar earns the same automatic substitution right. A standard biosimilar does not. This means that even a biosimilar priced 40% below the reference product may fail to capture meaningful volume if it lacks an interchangeability designation, because every patient switch requires active prescriber engagement, which imposes friction that slows adoption.
The FDA has approved interchangeable designations for several adalimumab biosimilars, insulin biosimilars, and filgrastim biosimilars. The interchangeability requirement for switching studies has been a persistent competitive moat for reference product sponsors, because completing the switching studies adds 12 to 24 months to the development timeline and several tens of millions of dollars in additional cost.
Small Molecule vs. Biosimilar Comparison Table
Feature
Small Molecule Generic
Biosimilar
Molecule Size
< 1,000 daltons
10,000-150,000+ daltons
Manufacturing
Chemical synthesis (reproducible)
Biological expression systems (variable)
Reference Relationship
Chemically identical
Highly similar (not identical)
Regulatory Standard
Bioequivalence (80-125% CI)
Totality of evidence; no clinically meaningful difference
Clinical Trials Required
Generally none
Comparative PK study plus confirmatory trial
Development Cost
$1-4 million
$100-250 million
Development Timeline
18-36 months
7-9 years
Typical Competitors at LOE
8-15+
2-9
Initial Price Discount
80-95%
15-40%
Automatic Pharmacy Substitution
Yes (in most states)
Only if designated interchangeable
Key Takeaways: Section 6
Biosimilars and generic drugs are distinct categories with fundamentally different development economics, competitive dynamics, and pricing outcomes. Biosimilar development costs are 30-to-100 times higher than small molecule generic development, limiting competitor count and moderating post-LOE price erosion. The FDA’s interchangeability designation is a binary switch that determines whether retail-level pharmacy substitution is possible, making it a material determinant of biosimilar commercial success.
7. The Economics of Patent Cliff: Competitive Entry and Price Erosion Curves
Quantifying the magnitude and timing of drug price erosion after patent expiry is the core analytical challenge for anyone managing pharmaceutical revenue forecasting, healthcare budget modeling, or biopharmaceutical investment. The relationship between competitor count and price level is well established empirically.
7a. The Competition-Price Relationship: Quantifying the Cascade
Data from the FDA’s generic drug program and HHS ASPE analyses demonstrate a consistent, non-linear relationship between the number of generic manufacturers and market price. With a single generic entrant, prices drop approximately 20% to 30%. With two generic entrants, prices fall to roughly 40% to 55% of the branded price. At four to five entrants, prices typically reach 20% to 30% of brand. At eight or more entrants, prices compress to 5% to 15% of the pre-LOE branded price.
The non-linearity of this curve is important for modeling. The marginal price impact of each additional entrant diminishes as the market already has many competitors. The largest price drops occur at entry counts of two, three, and four. A generic manufacturer entering a market with already seven competitors faces a very different revenue opportunity than the first or second entrant, even if all are nominally competing in the same therapeutic space.
The relationship also varies by molecular complexity. For simple small molecules with well-characterized APIs, competition drives prices to near-commodity levels quickly. For complex drug products such as inhalation solutions, modified-release oral dosage forms, transdermal patches, or drug-device combination products, the regulatory and manufacturing barriers are higher, competition is less intense, and the price floor is higher. The FDA’s Drug Competition Action Plan, initiated in 2017, specifically targeted this dynamic by releasing product-specific guidance documents for complex generics to clarify the regulatory standards, thereby encouraging more ANDA filers.
7b. The ‘Generic Paradox’: Brand Price Inflation After LOE
The generic paradox, documented in economic literature by Grabowski and Vernon and subsequently confirmed in numerous market studies, describes the counterintuitive phenomenon in which the reference listed drug’s list price increases after generic entry. The empirical evidence is robust: branded drugs in markets with more than four generic competitors show average list price increases of 5% to 15% in the two years following generic entry.
The mechanism is market segmentation. Upon generic entry, the market bifurcates into two distinct demand segments. The price-elastic segment, which includes most health plan enrollees whose formularies mandate generic substitution, transitions to the generic immediately. The brand-loyal, price-inelastic segment consists of patients who pay out-of-pocket or whose benefits design creates minimal cost differential, physicians who have expressed a persistent brand preference, and institutional purchasers who have contractual reasons for continuing branded sourcing. The innovator maximizes revenue from this residual segment by abandoning price competition with the generic and instead optimizing for margin.
For revenue forecasting, the generic paradox means that branded product revenue does not go to zero at LOE. The terminal branded revenue stream, though much smaller than peak, can persist for years at elevated per-unit prices. Pfizer’s Lipitor revenues, which reached $12.9 billion in 2006, had declined to approximately $1.86 billion by 2015, four years after generic entry, and continued generating measurable branded revenue beyond that point.
7c. Time to Price Floor and Market Maturity
The rate at which generic prices reach their floor varies by market structure. In markets where multiple ANDAs receive simultaneous FDA approval (as with clopidogrel in 2012), price compression is immediate. In markets where FDA approves ANDAs sequentially over 12 to 24 months following the 180-day first-filer exclusivity period, price decline is more gradual. The FDA’s Generic Drug User Fee Amendments (GDUFA) program, first enacted in 2012 and reauthorized in 2017 and 2022, has progressively reduced ANDA review times, meaning more recent LOE events tend to feature faster competitive entry than those before 2015.
Investment Strategy: Modeling the Erosion Curve
Analysts should build LOE revenue models using four parameters: entry date (probability-weighted using patent thicket analysis), entry count (modeled from existing ANDA filings and FDA approval pipeline data), price floor by competitor count tier (calibrated to drug class and formulation complexity), and brand revenue persistence (based on market segmentation research for the specific therapeutic area and patient population). A model that treats LOE as a binary event, with revenue going to zero at a single date, materially understates both the long-tail branded revenue and the total generic market revenue in the 18 months following initial entry.
Key Takeaways: Section 7
The price-competition relationship is non-linear: the largest price drops occur at two-to-four generic entrants, and marginal price impact diminishes with each additional competitor. The generic paradox is empirically well supported; branded drugs frequently raise list prices after LOE to maximize revenue from price-inelastic loyalists. Generic price floors are higher for complex formulations than simple small molecules, and the FDA’s product-specific guidance program directly targets this gap. LOE revenue models should treat entry count, entry timing, and branded price trajectory as independent variables.
8. The Innovator’s Playbook: Evergreening, Patent Thickets, and Authorized Generics
The strategic response to an approaching patent cliff is one of the most studied problems in pharmaceutical management. The tools available to innovators fall into three broad categories: legal extension of the patent protection perimeter, commercial strategies to retain revenue after generic entry, and scientific investment in successor molecules. The most effective LOE management programs combine all three.
8a. Evergreening: What the Term Actually Covers
‘Evergreening’ is a term applied loosely to a wide range of secondary patenting strategies. In its most straightforward form, it covers the filing of patents on modifications to a previously approved drug molecule or formulation. The modifications may include new salt forms, new polymorphic crystalline structures of the API, new hydrates or solvates, new enantiomers from a racemic mixture (chiral switches), new prodrugs that metabolize to the active compound, new dosage forms with altered release profiles, and new delivery systems such as autoinjectors or prefilled syringes.
Each of these strategies has scientific merit in isolation. Extended-release formulations genuinely improve patient adherence for drugs that require twice-daily or more frequent dosing. Chiral switches can produce an enantiomerically pure compound with a better tolerability profile than the racemic mixture. New salt forms can improve API stability or solubility. The controversy around evergreening is not about whether these modifications are real innovations; it is about whether the patent protection they earn is proportionate to the incremental benefit they deliver relative to the original drug, and whether that protection is being sought primarily to extend monopoly or to serve patients.
From an IP strategy standpoint, the efficacy of secondary patenting depends critically on execution timing and claim breadth. A secondary patent filed and issued well before the composition-of-matter expiry, with claims broad enough to cover the product as it is actually marketed, can extend effective exclusivity by three to seven years. A secondary patent filed late, with narrow claims that a generic can readily design around, provides limited value.
8b. Patent Thickets: Quantitative Assessment
AbbVie’s Humira patent thicket is the most analyzed example in the literature. The company held more than 250 patents related to adalimumab, with approximately 90% filed after the drug received its initial FDA approval in 2002. The thicket covered the molecule, specific formulation concentrations, manufacturing processes, methods of use for individual inflammatory conditions, dosing intervals, and delivery device components. The breadth and density of this coverage, combined with AbbVie’s willingness to litigate aggressively, produced settlement agreements with every major U.S. biosimilar developer, each of which included a deferred launch date of January 2023.
Other notable patent thickets include Johnson & Johnson’s Stelara portfolio, which lists 145 patents in the Purple Book; AstraZeneca’s Nexium (esomeprazole) strategy, which extended esomeprazole’s market life through chiral switch patenting after omeprazole’s composition-of-matter patent expired; and Cephalon’s modafinil (Provigil) portfolio, which combined multiple formulation and method patents with reverse payment settlements to delay generic entry for years. The FTC’s 2008 complaint against Cephalon for its settlement agreements with generic applicants was among the first major enforcement actions in the pay-for-delay area.
8c. Authorized Generics: The Brand’s Most Effective Commercial Weapon
An authorized generic (AG) is the reference listed drug’s exact formulation, produced by the brand manufacturer or its licensee, marketed without the brand name under a generic applicant’s ANDA. The AG is bioequivalent to the RLD by definition, because it is the RLD. It requires no separate bioequivalence testing and no ANDA filing by the brand, because the brand already holds the NDA.
The commercial logic of an AG is that it allows the innovator to participate directly in the generic revenue stream rather than surrendering all post-LOE market share to independent competitors. The AG typically prices below the brand but above full generic competition levels, capturing a portion of the price-sensitive market while maintaining a higher-margin branded product for the price-inelastic segment.
The AG’s most strategically important feature is that it is not blocked by the first-filer’s 180-day exclusivity. The exclusivity provision applies only to ANDA approvals. Because the AG is marketed under the existing NDA, not an ANDA, it can enter the market on day one of the 180-day exclusivity period, converting the first-filer’s intended duopoly into a three-way competition and compressing the first-filer’s revenue opportunity materially. Published research by the FTC estimated that an AG launched during the exclusivity period reduces the first-filer’s revenue by 40% to 52% compared to what it would earn in a true duopoly.
8d. Product Hopping: The Next Formulation
Product hopping involves transitioning patients from an approved drug approaching LOE to a next-generation formulation of the same molecule that carries fresh patent protection. The classic example is AstraZeneca’s shift from omeprazole (Prilosec) to esomeprazole (Nexium). Omeprazole’s composition-of-matter patent expired; AstraZeneca patented the S-enantiomer of omeprazole, obtained NDA approval for it as a separate drug (Nexium), and launched an aggressive marketing campaign to move prescribers to the new product before generic omeprazole could erode the franchise. Generic omeprazole was available, but AstraZeneca had already redirected the prescribing base to a product it protected for several additional years.
Courts have scrutinized product hopping when it involves product withdrawal rather than mere promotion. If a brand company reformulates a drug and then withdraws the original formulation from the market, it prevents generic substitution at the pharmacy for patients already on the brand, because pharmacists can only substitute generics for drugs that are currently on the approved reference listed drug list. The Second Circuit Court of Appeals, in New York v. Actavis (2015), held that reformulation combined with market withdrawal can constitute an exclusionary practice under antitrust law if the primary purpose is to prevent generic substitution.
Key Takeaways: Section 8
Evergreening through secondary patents is commercially rational and legally permissible but is increasingly scrutinized by courts, the PTAB, and antitrust regulators. Patent thicket density measurably delays generic entry and is quantifiable using Orange Book and Purple Book data. Authorized generics reduce the first-filer’s 180-day exclusivity revenue by 40% to 52%, which both captures generic revenue for the brand and deters future Paragraph IV challenges. Product hopping paired with market withdrawal is legally risky under post-Actavis antitrust doctrine.
Biologic lifecycle management is a distinct discipline from small molecule patent strategy. The tools available are different, the timelines are longer, the manufacturing complexity is higher, and the regulatory interactions are more intricate. An innovator managing a biologic approaching its 12-year reference product exclusivity window must execute across at least five strategic dimensions simultaneously.
9a. Formulation Innovation: Concentration, Stability, and Delivery
The first and most accessible lifecycle extension tactic for a biologic is formulation development. The reference product is typically approved at a specific concentration, pH, excipient composition, and presentation (vial or prefilled syringe). Secondary patents covering alternative concentrations, stabilization systems, or delivery device integrations are commercially valuable because they enable clinical differentiation and regulatory exclusivity without requiring new efficacy data.
Humira’s transition from an intravenous to a subcutaneous formulation, and subsequent concentration increases from 40 mg per 0.8 mL to 40 mg per 0.4 mL (high-concentration, low-volume formulation), generated new patents covering the high-concentration formulation and the citrate-free excipient system that reduced injection site pain. AbbVie listed these formulation patents separately from the core adalimumab patents, adding them to the thicket and requiring biosimilar developers to either match the formulation (and potentially infringe) or develop an alternative formulation (and potentially show clinical non-inferiority to the new standard of care rather than the original).
9b. Device Integration and Combination Product Patents
Biologics administered by injection increasingly rely on auto-injector pens or prefilled syringe systems that qualify as drug-device combination products under FDA regulation. A novel auto-injector design can receive its own patents through the USPTO independently of any drug-related patent and is listed in the Purple Book as a device component of the biologic’s approval. A biosimilar developer must either use an interchangeable device or submit its own device data demonstrating equivalent human factors performance.
AbbVie’s Humira Pen auto-injector had numerous patents covering the plunger mechanism, needle shield, and dosing indicator. Biosimilar developers had to design their own auto-injectors, which required separate human factors studies and usability engineering documentation. This added cost and development time to the biosimilar program and created a basis for product differentiation that AbbVie could market to physicians as a convenience or tolerability advantage.
9c. Indication Expansion and the 3-Year New Clinical Investigation Exclusivity
Each FDA-approved indication for a biologic can generate its own three-year new clinical investigation exclusivity if the approval required new clinical studies. An innovator that earns approval for a new indication in year 10 of its biologic’s market life gains three additional years of exclusivity for that indication, even after the 12-year reference product exclusivity has expired. Biosimilar developers pursuing approval for that indication must wait until the three-year exclusivity expires before referencing the innovator’s data.
The strategic implication is that an innovator should pursue indication expansion aggressively and time new indication approvals to maximize terminal exclusivity. A new indication approved in year 11 of the reference product’s life provides exclusivity for that indication until year 14, extending the effective protection window for a significant patient population by two years beyond what the baseline 12-year exclusivity would provide.
9d. Second-Generation Molecule Development: The ‘Biobetter’ Strategy
A ‘biobetter’ is a next-generation biologic that has the same therapeutic target or mechanism of action as the reference product but incorporates molecular engineering improvements that deliver superior clinical performance, a differentiated safety profile, or a more convenient dosing schedule. A biobetter is not a biosimilar; it is a new molecular entity that requires a full BLA submission and clinical development program. However, it can leverage the mechanistic and clinical precedent established by the reference product to design more efficient trials.
AbbVie’s Skyrizi (risankizumab) and Rinvoq (upadacitinib) function as biobetters in the immunology space, not in the molecular sense but in the commercial and clinical sense. They address the same patient populations as Humira (plaque psoriasis, Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis) with more selective mechanisms, improved efficacy data, and different dosing profiles. AbbVie began their clinical development well before Humira’s biosimilar entry, specifically to ensure they would be commercially available as the Humira revenue stream declined. Skyrizi and Rinvoq combined are projected to exceed $30 billion in annual revenue by 2027, more than offsetting Humira’s erosion.
9e. Subcutaneous vs. Intravenous Reformulation
Several biologics that are administered intravenously (typically in clinic or infusion center settings) have been reformulated for subcutaneous (SC) self-administration. The SC formulation requires co-formulation with hyaluronidase (a recombinant enzyme that increases subcutaneous tissue permeability and allows larger injection volumes) or development of a concentrated formulation that fits within an acceptable SC injection volume.
Johnson & Johnson reformulated Darzalex (daratumumab) from an IV infusion requiring several hours of administration to a subcutaneous formulation (Darzalex Faspro) with an 8-minute administration time. This reformulation generated new patent protection, improved patient convenience substantially, changed the site of care from infusion centers to home settings (reducing healthcare system cost), and created a differentiated product that biosimilar developers of the original IV formulation cannot automatically substitute for. The SC formulation’s clinical and commercial advantages made it the standard of care before IV biosimilars entered the market, effectively pre-empting the biosimilar opportunity for the IV formulation.
Key Takeaways: Section 9
Biologic lifecycle extension operates across five technical dimensions: formulation optimization, device integration patents, indication expansion, biobetter development, and subcutaneous reformulation. The subcutaneous reformulation strategy (exemplified by Darzalex Faspro) is particularly powerful because it changes the site of care, improves patient experience, generates new patents, and creates a product standard that biosimilar developers of the original formulation cannot directly substitute for. AbbVie’s Skyrizi/Rinvoq launch cadence is the canonical example of next-generation succession planning that transforms a patent cliff into a managed revenue transition.
10. The Challenger’s Gambit: Generic and Biosimilar Market Entry Strategies
Generic and biosimilar manufacturers approach the LOE event from the opposite direction, seeking to maximize revenue capture during the entry window. The strategic toolkit has become substantially more sophisticated over the past decade, particularly in the biosimilar space, where straightforward price competition has proven insufficient to drive rapid market penetration.
10a. The Paragraph IV Race and Pre-Filing Strategy
For small molecule generics, the Paragraph IV race is the primary offensive strategy. Generic manufacturers maintain dedicated teams that monitor Orange Book patent additions, track NDA supplement approvals, conduct freedom-to-operate analyses on newly issued patents, and prepare ANDA packages well in advance of targeted filing dates. The goal is to file on the first eligible day, typically the day the first year of NCE exclusivity ends (day 1460 of the 5-year NCE period), to maximize the probability of first-filer status and the associated 180-day exclusivity.
The pre-filing period involves extensive patent invalidity research, prior art searches, claim construction analysis, and often retention of expert witnesses who will testify in the anticipated Paragraph IV litigation. Generic manufacturers that are serial first-filers, such as Teva, Sandoz, and Mylan (now Viatris), maintain institutional expertise in this process that functions as a competitive moat within the generic industry.
10b. At-Risk Launch: The Financial Calculus
An at-risk launch is the decision by a generic or biosimilar manufacturer to begin commercial marketing before resolution of ongoing patent litigation. The expected value of an at-risk launch depends on the probability of winning the litigation (or having the stay expire), the revenue available during the at-risk period, the magnitude of damages if the court ultimately upholds the brand’s patents, and the cost of reversing the distribution if an injunction issues.
The Apotex clopidogrel at-risk launch in 2006 is the case study most frequently cited as a cautionary lesson. Apotex launched generic clopidogrel while Plavix’s patents were still under litigation, captured approximately $1 billion in revenue over the several months before the court issued an injunction, and ultimately paid $442.2 million in damages to Bristol-Myers Squibb and Sanofi. The net outcome was negative because the patent was upheld. The case is instructive precisely because the revenue captured during the at-risk period was substantial but insufficient to overcome the damages award.
The calculus differs when a court has already found a patent invalid or not infringed at the district court level but an appeal is pending. In that scenario, a launch carries lower litigation risk because a reversal on appeal is required to produce an infringement finding. The damages exposure is also structured differently when the infringement decision does not yet exist. Generic manufacturers typically conduct at-risk launches in this posture rather than against an unreviewed patent.
10c. Biosimilar Commercial Strategy: Beyond List Price
The early experience of Humira biosimilars in the U.S. demonstrated definitively that a lower list price does not guarantee market penetration when PBM rebate structures favor the incumbent brand. This led biosimilar manufacturers to develop commercial strategies that operate at the payer and PBM level rather than at the patient or physician level.
Dual pricing, pioneered by several adalimumab biosimilar manufacturers, involves launching two distinct commercial offerings simultaneously. The high-list/high-rebate version has a list price within 5% to 10% of Humira’s list price and offers a large confidential rebate to PBMs, allowing the PBM’s formulary economics to remain competitive with Humira’s rebate. The low-list/low-rebate version has a list price 80% to 85% below Humira’s and targets cash-paying patients, high-deductible plan members, and self-insured employers who prefer transparent net pricing over rebate-dependent economics. This dual-track approach acknowledges that the U.S. pharmaceutical market contains two structurally different demand segments with incompatible pricing preferences.
The private-label partnership strategy represents an even more direct approach to formulary capture. Sandoz licensed its Hyrimoz (adalimumab-aqvh) biosimilar to CVS’s Cordavis subsidiary, which markets it under a separate label and receives preferential placement on CVS Caremark’s formularies. When CVS removed brand-name Humira from its main commercial formularies in 2024, it effectively transferred Humira’s entire formulary-captive patient population to Cordavis/Hyrimoz. The biosimilar’s market share climbed from low single digits in 2023 to above 20% within the following year. The Sandoz-Cordavis model is now the template that other biosimilar manufacturers are pursuing with Express Scripts and OptumRx.
Key Takeaways: Section 10
At-risk launches carry asymmetric risk profiles: the revenue opportunity during the at-risk period must materially exceed the probability-weighted damages award, which requires a credible assessment of patent vulnerability before launch. Biosimilar commercial success depends less on list price than on formulary positioning, which requires either PBM partnership, dual-pricing strategy, or direct-to-employer contracting outside the traditional PBM channel. The Sandoz-Cordavis private-label model set the standard for biologic LOE market entry.
11. PBMs and Formulary Gatekeeping: Who Actually Controls Market Access
The most important variable in post-LOE market dynamics is not the price of the generic or biosimilar, nor the patent strength of the brand. It is the formulary decision of the three PBMs that collectively process approximately 80% of U.S. prescription drug claims: CVS Caremark, Express Scripts (Cigna), and OptumRx (UnitedHealth).
11a. Formulary Architecture: How Tiers Determine Volume
A drug formulary is a tiered list of covered medications in which tier placement determines the patient’s cost-sharing obligation. Tier 1 (preferred generic) carries the lowest co-pay, often $0 to $10. Tier 2 (preferred brand) carries a moderate co-pay, typically $30 to $60. Tier 3 (non-preferred brand) carries a higher co-pay, often $70 to $100 or higher. The specialty tier, which covers most biologics, requires co-insurance rather than a flat co-pay, and the patient may pay 20% to 33% of a drug’s list price.
A drug’s tier assignment drives volume through patient cost-sharing incentives. A biosimilar placed on Tier 1 while the reference product sits on Tier 3 creates a $60 to $100 monthly cost differential for the patient at point-of-sale. Most patients will accept a therapeutically equivalent option at that cost differential. A biosimilar placed on Tier 3 while the reference biologic remains on Tier 2, which is what many PBMs initially did with Humira biosimilars due to the rebate economics, produces negligible volume for the biosimilar regardless of its clinical equivalence or list price discount.
11b. The Rebate Wall: How Incumbent Brands Defend Formulary Position
Brand manufacturers pay PBMs confidential rebates, calculated as a percentage of the drug’s WAC (wholesale acquisition cost), in exchange for preferred formulary placement. A brand biologic with a $60,000 annual WAC might pay a 50% rebate, meaning the PBM receives $30,000 per patient per year in rebate income. A biosimilar launching at a 30% discount to WAC ($42,000 annual WAC) might offer a 20% rebate ($8,400 per patient per year), because its lower list price constrains the absolute dollar rebate it can offer even at a higher rebate percentage.
From the PBM’s perspective, the financial incentive runs counter to biosimilar adoption. Switching 10,000 patients from the brand biologic to the biosimilar at the above numbers would reduce the PBM’s gross rebate income from $300 million to $84 million annually, a loss of $216 million, even though the biosimilar has a lower net price for the health plan. This is the rebate wall: the PBM’s financial interest in maintaining the high-WAC incumbent conflicts directly with the healthcare system’s interest in lower net drug costs.
The rebate wall is not absolute. Sophisticated employers who self-insure and who demand transparency in PBM contracting have increasingly structured their agreements to eliminate or reduce rebate pass-through, aligning PBM incentives with low net cost rather than high gross rebate. The Consolidated Appropriations Act of 2021 attempted to ban rebates in Medicare Part D (the provision was subsequently delayed, then modified), and the Biden and Trump administrations each explored rebate reform at different levels of ambition. The trajectory of rebate reform, more than any other policy variable, will determine how quickly biosimilar adoption accelerates across the U.S. market.
11c. Step Therapy and Prior Authorization: Utilization Management as Competitive Weapon
Step therapy protocols require patients to try a preferred drug before the plan will cover a non-preferred alternative. When a PBM implements step therapy requiring a trial of a biosimilar before it will cover the reference biologic, it effectively mandates biosimilar use for new patients, producing rapid market share transition in the affected populations. Conversely, step therapy requiring a trial of the reference biologic before the biosimilar is covered, which some plans initially implemented to earn brand rebates, suppresses biosimilar uptake.
Prior authorization (PA) is a utilization management tool that requires prescriber justification before a drug is covered. PA is typically applied to high-cost specialty drugs. A PA protocol that requires documentation of biosimilar failure before approving the brand biologic implements a de facto first-line biosimilar policy. A PA protocol that applies only to the biosimilar and not the brand creates a procedural barrier to biosimilar adoption by imposing administrative burden on prescribers.
The direction of utilization management policy, as determined by PBMs in response to formulary economics, is a leading indicator of biosimilar market share trajectory. Analysts tracking the Keytruda (pembrolizumab) LOE event, expected to begin with biosimilar entry in 2028-2029, should monitor PBM formulary policy positions well before that date.
Investment Strategy: PBM Policy as LOE Trigger
Formulary decisions by the top three PBMs function as discrete, identifiable events that produce step-changes in biosimilar market share. CVS Caremark’s 2024 Humira formulary decision produced a measurable inflection in adalimumab biosimilar share within two quarters of announcement. Investors should monitor PBM formulary policy updates, contract renewals between brand manufacturers and PBMs (frequently disclosed in quarterly earnings calls), and CMS Part D formulary submissions (which are public) for advance signals of formulary position changes that will drive volume shifts.
Key Takeaways: Section 11
PBMs control formulary placement, and formulary placement determines biosimilar market penetration more reliably than list price. The rebate wall arises from a structural misalignment between PBM rebate income and net drug cost minimization. Rebate reform, whether regulatory or contractual, is the most powerful policy lever for accelerating biosimilar adoption. Step therapy and prior authorization protocols function as volume switches that analysts can track as leading indicators of market share transitions.
12. Case Study: Lipitor (Atorvastatin) and Its IP Asset Valuation
12a. The Asset Profile
Atorvastatin calcium, sold by Pfizer as Lipitor, reached peak global annual sales of $12.9 billion in 2006. At that point, Lipitor was the single best-selling drug in the history of the pharmaceutical industry, a distinction that held until Humira eclipsed it. Its composition-of-matter U.S. patent (U.S. Patent No. 4,681,893) expired November 30, 2011. Pfizer had secured a patent term extension that pushed the effective expiry from an earlier date to November 2011, and had aggressively listed secondary formulation and salt patents to complicate the generic entry landscape.
12b. IP Asset Valuation at Peak
At peak 2006 revenues, atorvastatin’s IP portfolio had a net present value estimable by standard DCF methodology. With 2006 revenues of $12.9 billion, a conservative post-exclusivity erosion assumption of 80% in year one and a terminal branded revenue of roughly 15% of peak persisting indefinitely, the NPV of the atorvastatin patent portfolio (using a 10% discount rate and the then-estimated 2011 expiry) was in the range of $30 billion to $40 billion at 2006 pricing. This was Pfizer’s single most valuable asset in its entire portfolio and larger than the market capitalization of most mid-cap pharmaceutical companies.
By 2009, with the expiry date now two years closer and some analyst downgrades of peak revenue forecasts, the NPV of the remaining patent life had compressed to approximately $15 billion to $20 billion. The erosion of the patent portfolio’s NPV in advance of LOE is itself an investable signal.
12c. Pfizer’s Defensive Playbook in Execution
Pfizer’s response to the Lipitor patent cliff was the most heavily documented single-drug lifecycle management campaign ever executed. The core elements were: a negotiated agreement with the largest PBMs to maintain preferred formulary status for branded Lipitor at a rebated price that competed with the first-generic’s expected price during the 180-day exclusivity period; the launch of the ‘Lipitor for You’ co-pay assistance program, which reduced patient out-of-pocket costs to as low as $4 per month, eliminating the financial incentive for patients to accept generic substitution; a partnership with Watson Pharmaceuticals (now Allergan/AbbVie’s Allergan Generics) to launch an authorized generic on day one of generic entry, denying Watson’s generic division a clear first-mover advantage and capturing generic market revenue directly; and continued direct-to-consumer advertising, which is unusual for a drug facing patent expiry but was justified by the scale of the branded revenue at stake.
The result was that Pfizer retained approximately 18% of atorvastatin prescription volume with the branded product as of mid-2012, far above the 2% to 5% brands typically retain. Revenue declined from $9.6 billion in 2011 to $3.9 billion in 2012 (a 59% decline), which was severe but considerably less catastrophic than the 90%+ declines seen when brands are entirely passive at LOE. By 2015, Pfizer’s Lipitor revenues had stabilized at approximately $1.86 billion globally, supported primarily by international markets where generic penetration was lower and pricing structures different.
Lipitor Revenue Cliff Table
Year
Global Lipitor Revenue ($M)
YoY Change
Key LOE Events
2006
$12,900
—
Peak sales year
2010
$10,700
—
Pre-LOE high
2011
$9,577
-10.5%
U.S. patent expires Nov. 30
2012
$3,947
-58.8%
Full generic competition; first-filer exclusivity period ends
2013
$2,315
-41.3%
Continued erosion with 10+ generics
2014
$2,058
-11.1%
Market approaching price floor
2015
$1,860
-9.6%
Stable branded tail
Key Takeaways: Section 12
Lipitor’s IP portfolio was worth $30-40 billion at peak NPV in 2006, making it the most valuable pharmaceutical patent asset in history at that time. Pfizer’s active defensive strategy retained 18% branded prescription volume, substantially above the passive-brand baseline. The authorized generic partnership with Watson was the most commercially consequential single tactic, as it captured generic revenue for Pfizer while simultaneously suppressing the first-filer’s 180-day windfall. Global market fragmentation (slower generic penetration in Europe and Japan) created a revenue tail that persisted for years beyond U.S. LOE.
13. Case Study: Plavix (Clopidogrel) and the Cost of At-Risk Launch
13a. The Asset Profile
Clopidogrel bisulfate, co-marketed by Bristol-Myers Squibb and Sanofi as Plavix, was the world’s second-best-selling drug at its peak, generating more than $7 billion in combined annual sales. BMS’s share of U.S. Plavix revenue represented approximately 46% of its total U.S. net revenue at the drug’s peak, creating exceptional earnings concentration risk that every sell-side analyst tracked obsessively. The core U.S. patents were scheduled to expire May 17, 2012.
13b. The Apotex at-Risk Launch: A Quantitative Post-Mortem
In August 2006, Canadian generic manufacturer Apotex launched clopidogrel bisulfate tablets at a discount to Plavix despite multiple patent infringement suits pending in the New Jersey federal district court. The company had obtained Paragraph IV certifications on the Plavix patents and concluded its challenge was strong enough to support commercial launch before resolution. The revenue opportunity was compelling: several months of market penetration in a drug generating $7 billion annually, even at a 40% to 50% discount, would generate revenues in the hundreds of millions.
For approximately three months, Apotex’s generic clopidogrel circulated in the market. Pharmacies dispensed it; mail-order PBMs routed prescriptions to it. BMS and Sanofi obtained a preliminary injunction, and Apotex withdrew the product, but an estimated 65% of dispensed clopidogrel prescriptions had shifted to the generic during those months.
The financial outcome for Apotex: approximately $1.1 billion in gross revenue from the at-risk period, before product withdrawal costs. The ultimate damages payment: $442.2 million to BMS and Sanofi, plus Apotex’s own legal costs through years of litigation. The net economic outcome for the at-risk launch was negative, and the precedent cost the entire generic industry a cautionary case study taught in every pharmaceutical law curriculum.
For BMS, the impact was different in character but equally significant. Plavix revenues fell sharply during the three months of generic availability, the markets registered the first concrete proof of the drug’s LOE vulnerability, and BMS’s share price declined substantially. Even after Apotex’s withdrawal and the resumption of brand-only sales, some portion of the converted patient population stayed on the generic (or was re-prescribed it by physicians who had observed no efficacy difference), creating a persistent base of converted patients that reduced Plavix’s market share even before the 2012 LOE.
13c. The 2012 LOE Event: Full Competition from Day One
When Plavix’s patents finally expired in May 2012, the market structure differed fundamentally from Lipitor’s more orderly post-LOE environment. The litigation history around Plavix’s patents involved multiple generic applicants, and the patent challenges by some applicants had been resolved through court judgments (some in the brand’s favor, some not) rather than settlements. This left no clean first-filer exclusivity situation. The FDA approved ANDAs from several generic manufacturers simultaneously on the expiry date.
The result was immediate, full-scale price competition from day one of commercial availability. Within weeks of the May 2012 expiry, clopidogrel tablets were available from multiple suppliers at prices ranging from $10 to $50 per month’s supply, versus the brand’s $180 to $200 per month. The price of clopidogrel at wholesale collapsed in some instances to less than $5 for a 30-day supply within months. BMS’s Plavix U.S. revenues fell from approximately $5.4 billion in 2011 to approximately $1.4 billion in 2012, a 74% single-year decline that stands as one of the steepest annual LOE drops in pharmaceutical history.
The Plavix case demonstrates that the litigation history surrounding a patent thicket determines the day-one competitive structure of the post-LOE market. If multiple generic challengers have filed Paragraph IV certifications and the brand has settled with none of them (or has lost in court), investors should model day-one full competition rather than a 180-day exclusivity period followed by a gradual ramp of additional entrants. The absence of a clear first-filer exclusivity situation is a bearish indicator for the brand’s post-LOE revenue trajectory.
Key Takeaways: Section 13
The Apotex at-risk clopidogrel launch produced a negative net economic outcome for the generic challenger and demonstrated the asymmetric downside of launching against upheld patents. BMS’s Plavix experienced a 74% single-year U.S. revenue decline in 2012 because multiple generic manufacturers entered simultaneously without a 180-day exclusivity buffer. Pre-LOE litigation history determines day-one market structure and must be incorporated into revenue forecasting models.
14. Case Study: Humira (Adalimumab) and the Rebate Wall
14a. The Asset Profile
AbbVie’s adalimumab (Humira) is the best-selling non-vaccine drug in pharmaceutical history, generating approximately $200 billion in cumulative global revenues between its 2002 approval and 2024. It peaked at $21.2 billion in global revenue in 2022. Its adalimumab composition-of-matter patents in the U.S. expired in 2016. AbbVie then held the market for an additional seven years through its patent thicket strategy, securing settlement agreements from all major U.S. biosimilar developers that deferred commercial launch until January 2023.
14b. AbbVie’s IP Asset Valuation: The Thicket as Core Asset
At the time of Humira’s composition-of-matter patent expiry in 2016, AbbVie’s patent thicket represented approximately $80 billion to $100 billion in NPV of protected future revenues, assuming the thicket successfully delayed biosimilar entry to 2023. AbbVie’s strategy to build and defend that thicket through 250+ patents and aggressive settlement negotiations was the most commercially consequential patent strategy in pharmaceutical history by absolute dollar value of protected revenue.
The thicket’s value can be measured by what happened without it. European Humira biosimilars launched in 2018, five years earlier than the U.S., and European biosimilar market share reached 60% within 18 months of first availability. AbbVie’s European Humira revenues declined approximately 75% between 2018 and 2021. Applying that erosion rate to U.S. Humira revenues in the counterfactual scenario where the thicket failed and biosimilars entered in 2018 rather than 2023 yields an estimated $60 billion to $80 billion in protected U.S. revenues attributed solely to the patent thicket strategy. This is the quantifiable value of AbbVie’s IP legal strategy, dwarfing the legal cost of maintaining and enforcing the patents.
14c. The Rebate Wall: Market Share Data
When Amgen’s Amjevita (adalimumab-apat) launched January 31, 2023, at a list price approximately 55% below Humira’s list price, market observers expected substantial and rapid biosimilar penetration. Instead, the first full year of U.S. biosimilar competition produced a biosimilar market share of approximately 2% by the end of 2023. AbbVie lost 32% of total Humira revenues in 2023 ($14.4 billion versus $21.2 billion in 2022), primarily from accelerating non-U.S. erosion and a modest U.S. volume loss, but the U.S. biosimilar market share figure confirmed that the rebate wall was fully intact.
The key data points: AbbVie disclosed that it had retained formulary coverage for Humira on approximately 90% of commercial plan lives in 2023. This meant that for 90% of insured patients, Humira remained the formulary-preferred adalimumab product. PBMs maintained this preference because AbbVie’s rebates (estimated at 40-50% of WAC) made Humira economically competitive with biosimilars on a net-cost basis despite the much higher list price.
14d. The CVS Caremark Formulary Decision: The Rebate Wall Breaks
CVS Caremark’s decision in early 2024 to remove brand-name Humira from its main commercial and Medicare Part D formularies, effective July 1, 2024, was the event that broke the adalimumab rebate wall. CVS Caremark covers approximately 100 million U.S. lives. Removing Humira from its commercial formularies and giving preferred status to Cordavis (Hyrimoz) and other biosimilars produced a formulary-driven patient transition that no pricing or marketing program could counter.
The financial impact was immediate and measurable. AbbVie’s U.S. Humira revenues, which had been approximately $12.2 billion in 2023 (full year), declined to approximately $7.1 billion in 2024. Q3 2024 biosimilar market share exceeded 20%, versus approximately 2% at the end of 2023.
Humira LOE Revenue Table
Period
Global Humira Revenue ($M)
U.S. Humira Revenue ($M)
Key Events
Full Year 2022
$21,237
$18,619
Peak year; no U.S. biosimilars
Full Year 2023
$14,404
$12,160
First U.S. biosimilars launch; rebate wall holds; biosimilar share ~2%
50% revenue decline; AbbVie total revenue resilient via Skyrizi/Rinvoq
Investment Strategy: Humira Lessons for Keytruda
Humira’s LOE trajectory carries direct implications for Keytruda (pembrolizumab), whose U.S. biosimilar entry is projected to begin in late 2028 or 2029. Merck has been aggressively building its own Keytruda patent thicket and developing formulation and device innovations. Investors should expect: (1) a delay between Keytruda’s nominal patent expiry and actual biosimilar market share accumulation, driven by both the patent thicket and PBM rebate dynamics; (2) a first year of biosimilar availability with low single-digit market share if PBM formulary economics favor Keytruda’s rebate; (3) a sharp inflection in biosimilar share at the point when a major PBM shifts formulary position, analogous to CVS Caremark’s 2024 decision for Humira; (4) a long Keytruda revenue tail supported by indications, particularly combination therapies and tumor mutation burden (TMB) applications, where biosimilar interchangeability may be more complex to demonstrate.
Key Takeaways: Section 14
AbbVie’s Humira patent thicket protected an estimated $60-80 billion in U.S. revenues beyond the composition-of-matter patent expiry. The rebate wall held biosimilar market share at approximately 2% for the entire first year of biosimilar competition. A single formulary decision by CVS Caremark (covering 100 million lives) produced a market share inflection from 2% to above 20% within two quarters. Skyrizi and Rinvoq demonstrate that next-generation successor planning is the most durable LOE mitigation strategy.
15. The Next Cliff: Keytruda, Eliquis, Stelara, and What Comes After
15a. Keytruda (Pembrolizumab): The $25 Billion Looming Event
Merck’s Keytruda (pembrolizumab) generated approximately $25 billion in global revenue in 2024, making it the world’s best-selling drug and the largest single patent-cliff event in pharmaceutical history. Its core U.S. composition-of-matter patents begin expiring in 2028, with the effective U.S. LOE window for biosimilar entry expected in the 2028-2029 timeframe.
Keytruda’s LOE event differs from Humira’s in several clinically important respects. Pembrolizumab is administered intravenously, meaning pharmacy-level interchangeability is less relevant than for subcutaneous products; the substitution decision occurs at the infusion center or hospital level rather than the retail pharmacy. Oncology prescribing behavior tends to be more conservative than rheumatology or dermatology prescribing; oncologists may resist biosimilar substitution for patients on active treatment more vigorously than rheumatologists or dermatologists, whose patients often have stable, long-term disease control. And pembrolizumab’s approval covers more than 16 tumor types and more than 30 individual indications, each of which may generate its own method-of-use patent protection and three-year new clinical investigation exclusivity.
The biosimilar development programs for pembrolizumab have been initiated by several companies, including Samsung Bioepis, Celltrion, and others. The development cost for a PD-1 antibody biosimilar, given the complexity of the reference product and the requirement for comparative clinical trials in oncology patients (which takes longer to enroll and generate data than comparative trials in inflammatory disease), is likely to run toward the high end of the $100-250 million range.
15b. Eliquis (Apixaban): The Anticoagulant Cliff
BMS and Pfizer’s Eliquis (apixaban) generated approximately $12 billion in U.S. revenues in 2024. Its primary U.S. patents are expected to expire in 2026, though litigation and secondary patent challenges could shift that timeline. The FTC has raised concerns about reverse payment settlements in the apixaban patent challenge cases, which have attracted scrutiny given the drug’s importance to stroke prevention and the scale of revenues at stake.
Apixaban is a small molecule, meaning generic entry, once it arrives, will likely follow the classic Lipitor-style cascade with rapid price erosion and high generic penetration. Unlike Humira, there is no rebate wall structural barrier for small molecule generics; pharmacy substitution is automatic upon generic availability if the FDA has established therapeutic equivalence ratings. The key variable for Eliquis is the timing and patent strength analysis of its secondary formulation and method-of-use patents, which Pfizer and BMS have used to maintain Orange Book listings beyond the core composition-of-matter expiry.
For Pfizer, which also owns the rights to Eliquis’s successor and faces simultaneous LOE pressure from several other products, the Eliquis cliff arrives at a particularly complex moment in its portfolio management cycle.
15c. Stelara (Ustekinumab): Biosimilar Market Already Open
J&J’s Stelara (ustekinumab), an IL-12/23 inhibitor approved for plaque psoriasis, psoriatic arthritis, Crohn’s disease, and ulcerative colitis, began its U.S. biosimilar competition in early 2024, making it the second major biologic to face U.S. biosimilar entry after Humira. Stelara’s global revenues peaked near $10 billion. Unlike Humira, Stelara’s patent thicket was substantially thinner, and biosimilar developers reached market in the U.S. more quickly after the core patent expiry.
The Stelara case will be the second data point for biosimilar LOE market dynamics in immunology, testing whether the Humira patterns around rebate wall, formulary decision lag, and PBM-driven inflection repeat themselves or differ based on prescribing specialty, patient heterogeneity, and the number of biosimilar entrants.
Key Takeaways: Section 15
Keytruda’s LOE event, expected in 2028-2029, represents the largest single patent cliff in pharmaceutical history by revenue at approximately $25 billion in peak annual sales. Oncology prescribing conservatism and IV administration complicate biosimilar penetration dynamics relative to subcutaneous immunology biologics. Eliquis’s small-molecule status means its LOE will produce rapid price erosion more closely resembling Lipitor than Humira. Stelara’s ongoing biosimilar competition is generating the second major data set for biologic LOE market dynamics, with results that analysts should track closely as a Keytruda preview.
16. Global Comparative Frameworks: U.S., EU, and Japan
16a. The European Union: Predictable Timelines, Faster Biosimilar Uptake
The EU operates on the ‘8+2+1’ market protection framework. An innovator receives eight years of data exclusivity, during which generic or biosimilar applications referencing the innovator’s data cannot be submitted. After the eight-year data exclusivity period expires, applications can be submitted but the product cannot launch for another two years, providing 10 total years of market protection. One additional year of market exclusivity can be earned if the innovator obtains approval of a significant new therapeutic indication within the first eight years, extending total market protection to 11 years.
This framework’s clarity produces two important commercial outcomes. Generic and biosimilar developers can plan their development timelines with greater precision than in the U.S. system, where litigation outcomes can shift LOE dates by multiple years. National health systems in the EU, which are the primary payers for most medicines, can plan their budget accommodations for incoming generics and biosimilars with corresponding accuracy. The combination of planning certainty and national health system cost-containment pressure has produced faster and deeper biosimilar penetration in most EU markets than in the U.S. Humira biosimilars captured more than 50% of the adalimumab market in Germany, France, and the UK within 12 to 18 months of their 2018 EU launch, years before the U.S. even allowed biosimilar entry.
EU member states also use centralized tendering and reference pricing mechanisms that accelerate price erosion in ways the U.S. system does not replicate. Germany’s AMNOG process, which subjects new drug approvals to a benefit assessment and negotiated price within 12 months of launch, applies continuous cost-effectiveness pressure throughout the product lifecycle. In this environment, an innovator approaching LOE faces not merely patent expiry but also potential price renegotiation based on the comparator now available in the form of a biosimilar.
16b. Japan: Re-Examination Period and Administrative Patent Linkage
Japan’s pharmaceutical regulatory framework, overseen jointly by the Pharmaceuticals and Medical Devices Agency (PMDA) and the Ministry of Health, Labour, and Welfare (MHLW), uses the re-examination period as its functional equivalent of data exclusivity. New drugs receive a re-examination period of 8 to 10 years (6 to 10 years for orphan drugs) during which the MHLW evaluates post-marketing safety and efficacy data before the drug can be submitted for re-examination. Generic applications cannot reference the innovator’s data during this period.
Japan’s ‘administrative patent linkage’ is informal but effective. The MHLW, as a matter of practice, will not approve a generic application for a drug that it knows to be covered by a valid patent. Generic manufacturers must resolve relevant patents either through litigation or settlement before the MHLW will proceed with their approval. This creates a de facto Hatch-Waxman-style linkage without the formal statutory framework.
The NHI (National Health Insurance) price list is Japan’s central pricing mechanism. Every drug, brand and generic, must be listed on the NHI price to receive reimbursement in Japan’s universal healthcare system. NHI prices for generics are set as a percentage of the brand’s NHI price, with sliding scale reductions as market share increases. This creates a predictable pricing trajectory for generic entry that differs from the competitive market pricing dynamics in the U.S.
Japan has also been aggressive in promoting generic use as a matter of national health policy. The government has set explicit targets for generic penetration as a percentage of total drug volume, and has imposed regulatory and reimbursement incentives to drive physician and pharmacist behavior toward generic dispensing. Generic penetration rates have risen substantially over the past decade as a result of these policies.
Global Regulatory Comparison Table
Feature
United States
European Union
Japan
Primary Regulatory Body
FDA
EMA
PMDA / MHLW
Patent Term Extension
Up to 5 years (Hatch-Waxman)
Up to 5 years (Supplementary Protection Certificates)
Up to 5 years
Data Exclusivity
5 years (NCE); 12 years (biologics)
8 years
Re-examination period: 6-10 years
Market Exclusivity
Varies by exclusivity type
10 years (8+2), +1 for new indication
Tied to re-examination period
Patent Linkage
Formal (Orange Book / Paragraph IV)
None (national courts handle)
Informal / administrative
Primary Pricing Mechanism
PBM formulary / rebate
National tender / reference pricing
NHI price list
Biosimilar Uptake Speed
Slow initially (rebate wall)
Fast (national tendering)
Moderate (improving with policy)
Key Takeaways: Section 16
The EU’s ‘8+2+1’ framework provides greater LOE date predictability than the U.S. system, and national tendering produces faster, deeper biosimilar penetration. Japan’s re-examination period functions similarly to data exclusivity, and administrative patent linkage produces Hatch-Waxman-like blocking effects without formal statute. Global LOE strategy must account for these regulatory differences, because European and Japanese LOE events frequently occur earlier and with steeper price trajectories than U.S. LOE events for the same drug.
17. Investment Strategy: Patent Expiry as Alpha Signal
Patent expiry data is among the most systematically underutilized quantitative inputs in pharmaceutical equity analysis. Most of the information that drives LOE events is public, filed with the USPTO or the FDA, visible in PTAB petition dockets, and disclosed in quarterly earnings materials. Yet the analytical depth applied to patent timelines by most buy-side teams remains shallow compared to the depth applied to clinical trial readouts or regulatory decisions.
17a. LOE as a Catalyst Calendar
Patent expiry events are datable. The Orange Book lists every relevant U.S. patent with its expiry date. PTAB dockets record IPR petition dates and institution decisions. District court dockets track Paragraph IV litigation milestones. From these public sources, an analyst can construct a probabilistic LOE calendar for any major pharmaceutical product that is more granular and more accurate than the summary disclosures companies provide in their annual reports.
The calendar should include: the composition-of-matter patent expiry date, any PTE extension applied and its remaining term, secondary patent expiry dates with individual probability weights based on PTAB and litigation history, regulatory exclusivity expiry dates (all applicable types), the number and identity of Paragraph IV filers, the litigation status of each filed challenge, and the dates of any settlement agreements disclosed in litigation documents or earnings calls.
The pharmaceutical market has a persistent tendency to overvalue branded drugs in the three-to-five years before LOE. Analysts habitually extend revenue forecasts at or near peak levels for longer than the underlying patent portfolio warrants, partly because management guidance is optimistic, partly because the exact LOE date is genuinely uncertain, and partly because the structural complexity of the patent thicket makes precise analysis difficult.
Short-selling or put-option strategies structured to profit from branded revenue decline post-LOE have historically generated strong risk-adjusted returns when initiated 18 to 24 months before the expected effective LOE date, with position sizing weighted by the probability of the thicket holding versus failing. The Humira case offered a short-side opportunity on AbbVie in 2022 that materialized more slowly than expected due to the rebate wall; investors who sized the position based on a 2023 rebate-wall collapse versus a 2024 collapse would have been stopped out or subject to significant mark-to-market loss before the thesis played out.
17c. Long-Side Opportunity: Generic and Biosimilar Entry Events
The 180-day first-filer exclusivity period for a major drug LOE is a dateable, high-probability revenue event for the winning generic manufacturer. A generic company that wins first-filer status for a $5 billion drug generates $400 to $700 million in incremental revenue during those six months. For a company with a $2 billion annual revenue base, a single successful first-filer exclusivity event can represent 20-35% of annual revenues in a single period. Generic company stock prices frequently show measurable underreaction to first-filer status confirmations in district court decisions, creating a buying opportunity.
For biosimilar companies, the investable event is not the initial approval but the formulary transition. The CVS Caremark Humira formulary decision was publicly announced months before the effective date, providing investors with a clear signal to increase exposure to Sandoz (Cordavis), Amgen (Amjevita with preferred status at Express Scripts), and other favorably positioned biosimilar manufacturers.
17d. Patent Expiry Data as Macro Signal
At the portfolio level, aggregate patent cliff data is a reliable leading indicator of sector-level capital allocation trends. In the three to five years before a major patent cliff cycle, innovator companies accelerate M&A activity as they seek to replace the pipeline capacity that will be eroded by LOE. Valuations for pipeline-stage companies rise as large-cap pharma pays premium prices for late-stage assets to fill the gap. Conversely, during a patent cliff period itself, large-cap pharma free cash flow that was previously allocated to dividends and buybacks gets redirected to business development, constraining shareholder return programs.
The 2025-2030 patent cliff, at $236 billion in at-risk revenues, is already driving measurable M&A behavior. AstraZeneca, Merck, BMS, and Pfizer have each executed multi-billion-dollar acquisitions since 2022 that are explicitly linked to building pipeline depth ahead of major LOE events. Tracking deal flow and target selection against the patent cliff calendar provides a structured framework for anticipating M&A targets and acquirers.
Key Takeaways: Section 17
Patent expiry data is public, systematic, and analytically rich; most buy-side LOE analysis remains shallower than the data supports. Short-side opportunities exist in pre-LOE overvaluation of branded drugs, calibrated against patent thicket probability weights. Long-side opportunities exist in first-filer generic exclusivity events and formulary-transition inflection points for biosimilars. The $236 billion 2025-2030 patent cliff is a macro signal for accelerated M&A, rising pipeline valuations, and constrained shareholder return programs at the major innovators facing the largest LOE events.
18. Master Key Takeaways
Effective patent life is 7 to 12 years, not 20. The 20-year nominal patent term is commercially misleading. By the time a drug reaches the market, its composition-of-matter patent may have less than half its nominal life remaining, and that remaining period must generate sufficient revenue to recoup a development investment that commonly exceeds $1 billion.
Patent protection and FDA regulatory exclusivity are separate systems requiring separate tracking. A composition-of-matter patent expiry does not equal LOE. Orphan Drug Exclusivity can extend market protection for seven years post-approval regardless of patent status and cannot be broken by a Paragraph IV challenge.
Patent thicket density measurably delays generic and biosimilar entry. Drugs with 10 or more Orange Book-listed patents experience average generic entry delays of 3.4 years beyond the composition-of-matter expiry. AbbVie’s Humira thicket produced a seven-year delay worth an estimated $60-80 billion in protected U.S. revenues.
The PTAB is a litigation discount factory for secondary pharmaceutical patents. IPR petition institution rates above 60% and final invalidation rates above 75% for instituted petitions impose material probability discounts on formulation, method-of-use, and device patents. These discounts must appear explicitly in IP valuation models.
Price erosion follows a predictable competitor-count curve. Two generic entrants produce 40-55% price reduction. Four to five entrants produce 70-80% reduction. Eight or more entrants compress prices to 5-15% of pre-LOE branded price. The curve is non-linear; the largest marginal price impact occurs at entrant counts of two through four.
Biosimilar and generic LOE events require separate analytical frameworks. Biosimilar development costs are 30-100 times higher than small molecule generics, limiting competitor count and moderating price erosion. Biosimilar interchangeability status is a binary determinant of retail-level substitution and must be tracked separately from biosimilar approval status.
PBMs control biosimilar market penetration more decisively than pricing does. The rebate wall can hold biosimilar market share at 2% for an entire year despite 85% list price discounts. A single formulary decision by a PBM covering 100 million lives can produce a market share inflection from 2% to 20% within two quarters.
Authorized generics reduce first-filer 180-day exclusivity revenue by 40-52%. This both captures generic revenue for the innovator and materially reduces the expected payoff from Paragraph IV challenges, functioning as a deterrent to future patent challenges.
The $236 billion 2025-2030 patent cliff is driving accelerated M&A. Keytruda, Eliquis, Stelara, and approximately 186 other drugs facing LOE are creating a strategic vacuum at the world’s largest pharmaceutical companies that can only be filled through internal R&D productivity or external acquisition.
Global LOE events are not synchronized. EU biosimilar penetration has consistently been faster and deeper than U.S. penetration for the same drugs, due to national tendering, absence of a rebate wall, and clearer exclusivity timelines. Japanese LOE dynamics are distinct from both. A global LOE revenue model must country-code its inputs.
Appendix: Glossary of Key Technical Terms
ANDA (Abbreviated New Drug Application): The regulatory submission used by generic drug manufacturers to obtain FDA approval for a generic drug by referencing the innovator’s safety and efficacy data.
Authorized Generic (AG): The reference listed drug’s formulation marketed without the brand name by the innovator or its licensee; not blocked by the first-filer’s 180-day exclusivity.
BPCIA (Biologics Price Competition and Innovation Act): The 2009 statute establishing the regulatory pathway for biosimilar approval in the U.S. and creating the 12-year reference product exclusivity period for biologics.
Biosimilar Interchangeability: A designation granted by the FDA to biosimilars that meet a higher standard of evidence on switching studies, enabling automatic pharmacy-level substitution in states that permit it.
Composition-of-Matter Patent: A patent claiming the chemical or biological structure of a new molecular entity; the broadest and most commercially valuable form of pharmaceutical patent protection.
Evergreening: The practice of obtaining secondary patents on formulations, methods of use, delivery devices, or other incremental modifications to extend effective market exclusivity beyond the composition-of-matter patent expiry.
Hatch-Waxman Act: The Drug Price Competition and Patent Term Restoration Act of 1984 that created the ANDA pathway, the Orange Book, the 30-month stay, and the 180-day first-filer exclusivity.
LOE (Loss of Exclusivity): The date on which the last relevant patent or regulatory exclusivity protecting a drug from generic or biosimilar competition expires, allowing market entry.
NCE Exclusivity (New Chemical Entity Exclusivity): Five years of FDA-granted market protection for drugs containing an active moiety never previously approved by the FDA.
ODE (Orphan Drug Exclusivity): Seven years of FDA-granted market protection for drugs designated to treat rare diseases; not breakable by Paragraph IV certification.
Orange Book: The FDA’s official publication listing approved drug products with therapeutic equivalence evaluations, including all patents that brand manufacturers have certified cover each listed drug.
Paragraph IV Certification: A certification filed by an ANDA applicant asserting that a brand patent is invalid, unenforceable, or will not be infringed; triggers the 30-month automatic stay if the brand sues within 45 days.
Patent Term Extension (PTE): An extension of up to five years on a single patent granted by the USPTO to compensate for regulatory review time consumed during FDA approval.
Patent Thicket: A dense, overlapping web of multiple patents surrounding a single drug product, covering the molecule, formulations, manufacturing processes, methods of use, and delivery devices.
PBM (Pharmacy Benefit Manager): An intermediary that administers prescription drug benefits for health plans, manages formularies, and negotiates rebates with drug manufacturers.
PTAB (Patent Trial and Appeal Board): The administrative body within the USPTO that adjudicates inter partes review (IPR) petitions challenging the validity of issued patents.
Purple Book: The FDA’s equivalent of the Orange Book for biological products; lists approved biologics and their biosimilars but does not include patent information directly.
Rebate Wall: The economic barrier to biosimilar adoption created by PBM financial dependence on large rebates from high-WAC branded biologics, which can exceed the cost savings from biosimilar net pricing.
Reference Listed Drug (RLD): The innovator drug product to which a generic ANDA or biosimilar application refers as the basis for approval.
Totality of the Evidence: The regulatory standard used by the FDA to evaluate biosimilar applications, requiring comprehensive analytical, nonclinical, and clinical data to demonstrate no clinically meaningful differences from the reference product.
30-Month Automatic Stay: The FDA’s automatic delay in approving an ANDA with a Paragraph IV certification if the brand manufacturer sues the generic applicant for patent infringement within 45 days of receiving notification of the filing.
WAC (Wholesale Acquisition Cost): The manufacturer’s list price to wholesalers; the starting point from which rebates are calculated and the reference price for most PBM contracting.
This analysis is intended for pharmaceutical IP professionals, R&D strategists, biotech investors, and healthcare policy analysts. It reflects data and public filings. Patent expiry dates and regulatory exclusivity periods are subject to change based on patent term extension grants, PTAB decisions, litigation outcomes, and FDA administrative actions. All revenue figures sourced from company quarterly and annual financial disclosures unless otherwise noted.
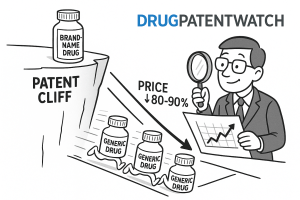





















")




