A strategic guide to patent expiry, lifecycle defense, and the economics of generic competition — for executives, investors, and analysts who need to move faster than the market
When Pfizer’s Lipitor lost patent protection in November 2011, the company’s U.S. revenue from the drug fell roughly 80% within two years [1]. Not gradually. Not in stages. Eighty percent. The drug had generated more than $13 billion annually at its peak, and within 24 months it was a commodity. Pfizer knew it was coming. They had an entire lifecycle management apparatus dedicated to slowing it down. It barely mattered.
This is what a patent cliff looks like from the inside: a precisely dated, mathematically certain, commercially catastrophic event that innovator pharmaceutical companies spend billions trying to soften and billions more pretending they can prevent.
For everyone else in the ecosystem — generic manufacturers, biosimilar developers, hospital purchasing departments, pharmacy benefit managers, investors shorting or longing biotech positions — a patent expiry is an opportunity. The same cliff that destroys a company’s revenue is the starting gun for an entirely different competitive race.
This guide covers what actually happens when a drug patent expires: the legal mechanics, the competitive dynamics, the financial fallout, the defensive strategies innovators use, and why many of those strategies fail. It draws on publicly available patent data, FDA regulatory records, court decisions, financial filings, and the kind of competitive intelligence that services like DrugPatentWatch compile to help professionals track exactly where and when the next cliff is coming.
Part One: What a Drug Patent Actually Protects
The Anatomy of Pharmaceutical Patent Protection
A drug patent does not protect a drug. That framing, while common, is imprecise enough to be dangerous for anyone making strategic decisions based on it.
A pharmaceutical patent protects a specific, narrowly claimed invention. That invention might be the chemical structure of an active pharmaceutical ingredient (API). It might be a particular salt or polymorph of that API. It might be the process used to manufacture the API, the formulation that delivers it (the capsule, the coating, the suspension medium), or the method of using it to treat a specific disease. Most blockbuster drugs are not protected by a single patent. They are protected by a portfolio of overlapping patents covering different aspects of the product, each with its own expiration date.
This layered architecture is not accidental. It is the product of decades of legal refinement by pharmaceutical companies seeking to maximize the period of market exclusivity — and the primary reason why analyzing “the patent on Drug X” without specifying which patent is nearly meaningless.
A compound patent, which covers the novel chemical structure itself, is typically the most important and earliest-expiring form of protection. These patents are usually filed during preclinical development, often a decade or more before a drug reaches market. Under current law, U.S. patents have a 20-year term from the filing date [2]. For pharmaceuticals, however, Congress created a partial offset through the Drug Price Competition and Patent Term Restoration Act of 1984 — commonly called the Hatch-Waxman Act — which allows patent holders to apply for patent term extensions (PTEs) of up to five years to compensate for time lost during FDA review, subject to a cap of 14 years of post-approval protection [3].
Even with a PTE, the math rarely works in favor of the innovator. By the time a compound patent expires, the drug behind it has typically been on the market for 10 to 12 years. The first several years of that commercial window are consumed by the slow ramp of physician adoption, payer negotiations, and market access challenges. The peak revenue years often land in the back half of the exclusivity window, right before the cliff hits.
The Six Types of Pharmaceutical Patents
Understanding the patent landscape requires knowing what types of protection companies actually seek:
Compound patents cover the active molecule itself — the core chemical structure, often called the new chemical entity (NCE). These are the foundational patents and typically the first to expire.
Formulation patents cover how the drug is delivered — the extended-release coating, the nanoparticle suspension, the lipid nanoparticle carrier. AstraZeneca’s extended-release formulation of omeprazole (Nexium) generated years of additional exclusivity after the base compound patent on omeprazole expired.
Process patents cover how the drug is manufactured at scale, including synthesis routes and purification methods. These matter most in markets where API manufacturing is dominated by a small number of third-party producers.
Method-of-use patents cover the specific therapeutic indications in which a drug is used. A compound whose compound patent has expired can retain meaningful protection if its primary approved use is covered by a separate, later-expiring method patent.
Dosing regimen patents cover specific administration schedules — once-daily dosing, fixed-dose combinations, particular titration protocols. These are among the most controversial patent types because they add minimal therapeutic innovation while extending commercial protection.
Metabolite and active moiety patents cover the biologically active form of a compound that is produced in the body after administration of a prodrug. AstraZeneca’s esomeprazole (the S-enantiomer of omeprazole) became the basis for Nexium partly because it could be claimed separately from the racemate.
What the FDA’s Orange Book Tells You
The FDA maintains a publication called “Approved Drug Products with Therapeutic Equivalence Evaluations,” universally known as the Orange Book. For every approved drug application (NDA), the Orange Book lists the patents the applicant has certified as covering the drug or its approved uses, along with their expiration dates.
The Orange Book is the legal battlefield on which patent cliff timing is determined. When a generic company files an Abbreviated New Drug Application (ANDA), it must certify its relationship to each Orange Book-listed patent. A Paragraph IV certification — the certification that a listed patent is invalid, unenforceable, or will not be infringed by the generic — triggers a potential 30-month stay of FDA approval while the innovator litigates [4].
This means the Orange Book is not simply a disclosure document. It is a strategic instrument. Innovator companies have strong incentives to list every arguably applicable patent, whether or not a court would actually find infringement. Generic companies have strong incentives to challenge those listings. The FDA has enforcement authority over what gets listed but has historically exercised it lightly, a gap that regulators and courts have narrowed in recent years.
DrugPatentWatch tracks Orange Book listings, ANDA filings, and Paragraph IV certifications in real time, giving pharmaceutical executives, generic manufacturers, and investors early visibility into which drugs are being targeted for generic entry and when the legal battles are likely to be resolved. For anyone whose business decisions depend on knowing when a drug’s exclusivity will actually end — not just when the compound patent expires on paper — that kind of surveillance is operationally essential.
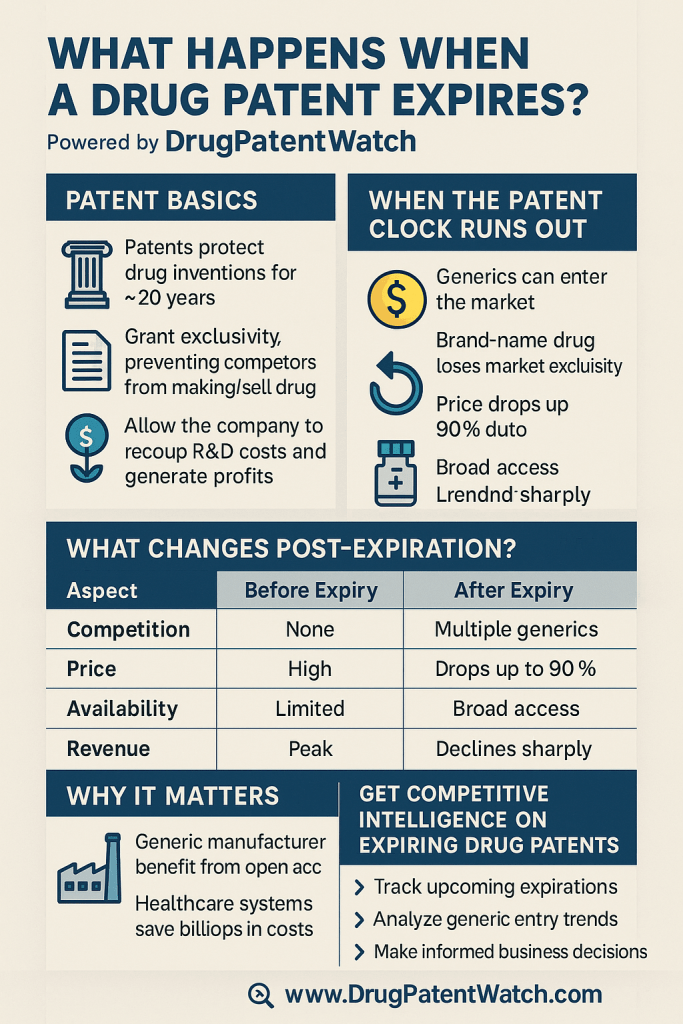
Part Two: The Patent Cliff — Revenue Erosion Mechanics
How Fast Does Revenue Fall?
The speed of revenue erosion after patent expiry depends on four variables: the number of generic entrants, the therapeutic category, the channel mix, and whether the innovator deploys an authorized generic.
The research literature and market data converge on a consistent pattern for small-molecule oral drugs in categories with multiple generic entrants: branded drug prices fall rapidly, and volume follows. Within 12 months of generic entry with six or more competitors, branded market share typically drops to less than 20% of prior volume, with prices falling 70% to 90% from the branded level [5]. <blockquote> “Within one year of patent expiry for major oral solid-dose drugs, generic substitution rates at U.S. retail pharmacies typically exceed 80%, and the branded product retains less than 5% of prescriptions by the second year.” — IQVIA Institute for Human Data Science, Medicine Use and Spending in the U.S., 2024 [6] </blockquote>
This substitution rate is not uniform across channels. In the retail pharmacy setting, generic substitution is nearly automatic — pharmacists are mandated by state laws and incentivized by PBM contracts to dispense generics when therapeutically equivalent options are available. In the institutional (hospital) channel, formulary decisions involve committees and purchasing contracts, which introduces a lag. In specialty pharmacy channels serving complex conditions, branded stickiness can persist longer, particularly when patient support programs, insurance dynamics, or clinical inertia favor the branded product.
The revenue curve also differs significantly between small molecules and biologics. For a standard oral solid-dose drug, the cliff is exactly that: steep, fast, severe. For an injectable biologic, competition from biosimilars produces a slope rather than a cliff — a gradual erosion over years rather than months. The reasons for this difference are structural and are covered in detail in Part Four.
The Lipitor Case: The Cliff as a Historical Fact
Pfizer’s handling of atorvastatin’s patent expiry remains the canonical case study because the cliff hit with unusual severity and because Pfizer’s defensive strategies were more visible than most.
Pfizer held atorvastatin’s compound patent until November 30, 2011 [7]. In preparation for expiry, it deployed multiple approaches simultaneously: it launched its own authorized generic (AG) on the day of expiry, locking up the Watson Pharmaceuticals generic supply chain; it signed deals with multiple PBMs and pharmacy chains to maintain formulary positions for the branded product at reduced prices; and it launched an aggressive patient assistance and pricing program specifically targeted at Medicare Part D patients.
None of it worked as hoped. Within six months, atorvastatin was the most dispensed generic in U.S. history. Pfizer’s branded Lipitor revenues fell from roughly $5.3 billion in the first half of 2011 to less than $1.5 billion in the same period of 2012 [8]. The AG revenues partially offset this loss, but by design they were priced far below the branded level.
The Lipitor case illustrated a basic asymmetry: the tools available to an innovator for post-cliff defense are powerful enough to shift the timing of revenue erosion by months, but not by years. The fundamental dynamic — that a small-molecule drug with multiple generic entrants will lose most of its revenue within 18 months of patent expiry — is remarkably consistent across products and therapeutic categories.
The Plavix Case: A Faster Cliff
Sanofi-Aventis and Bristol-Myers Squibb’s clopidogrel (Plavix) showed that cliffs could arrive even faster under different competitive conditions. When its main U.S. patents expired in May 2012, generic entry was extensive and immediate. Within three months, branded Plavix held less than 15% of prescriptions [9]. The speed of substitution reflected both the widespread use of the drug (it was among the top-selling drugs in the world) and the highly competitive nature of the generic supply chain for a product that manufacturers had been preparing to enter for years.
The Plavix cliff also illustrated the “paragraph IV lottery” dynamic: generic companies that successfully challenge a patent before expiry can be rewarded with 180-day exclusivity — the period during which only they and the innovator (via AG) can sell the drug. In the Plavix case, the generic challengers had lost their court case, meaning full generic entry came only at natural expiry but was immediate and overwhelming when it did.
The Difference Between a Cliff and a Slope
Not all patent expiries produce cliffs. The term “slope” is used by analysts to describe scenarios where revenue erosion is gradual — spread over several years rather than months. Several conditions produce slopes rather than cliffs.
Complex formulations that are difficult to replicate — controlled-release systems, transdermal patches, sterile injectables — can delay generic entry by 12 to 24 months beyond compound patent expiry, because generic companies need time to demonstrate bioequivalence through clinical studies. This delay compresses the steep initial drop into a longer, shallower curve.
Therapeutic categories where physicians are reluctant to substitute — certain anti-epileptics, thyroid drugs, and immunosuppressants with narrow therapeutic indices — also see slower branded erosion. Prescribers maintain branded or reference-product prescriptions in these categories even when AB-rated generics are available.
Biologics, as noted, produce slopes almost by definition. The interchangeability standards that apply to biosimilars — the clinical and pharmacokinetic data required to allow pharmacist-level substitution without prescriber authorization — are stringent enough that very few biosimilars have achieved interchangeable status in the U.S. Without interchangeability, substitution requires active physician decision-making, which slows uptake considerably.
Part Three: The Generic Entry Playbook
How Generic Companies Attack a Patent
Generic pharmaceutical companies do not simply wait for patents to expire. They maintain large, professionally staffed patent challenge operations dedicated to finding patents that are vulnerable to invalidity or non-infringement arguments. The legal framework for these challenges is the Hatch-Waxman Act, which created the Paragraph IV certification process specifically to facilitate them.
When a generic company files an ANDA with a Paragraph IV certification against a listed patent, it must notify the patent holder, who then has 45 days to sue [10]. If the innovator sues within that window, FDA approval of the ANDA is automatically stayed for 30 months — a period designed to give courts time to rule on the patent dispute. If the innovator fails to sue within 45 days, the ANDA can proceed without a stay.
The first company to file a Paragraph IV ANDA against a particular patent is entitled to 180 days of generic exclusivity — a period during which no other ANDA filer can receive final approval [11]. This creates a first-mover incentive structure in the generic industry that turns patent challenge into a competitive sport. Generic companies with strong patent litigation capabilities have used this 180-day exclusivity to capture profits sufficient to fund their entire litigation programs.
The economics of this system deserve scrutiny. During the 180-day exclusivity window, there are effectively two players in the generic market: the first filer and, if deployed, the innovator’s authorized generic. With two players rather than ten, prices settle at a level significantly above the eventual commodity price. By the time the exclusivity window expires and multiple additional generics enter, the revenue potential of the drug has already declined substantially.
The ANDA Filing Surge Before Patent Expiry
Companies like DrugPatentWatch track ANDA filings as leading indicators of competitive intent. The number of ANDAs filed against a particular drug — and how far before expiry they arrive — tells you how many generic companies are positioning for entry and how confident they are in their ability to replicate the product.
A drug with 15 or 20 ANDA filers awaiting approval at patent expiry will experience a cliff so steep it looks like a vertical line on a chart. A drug with two or three ANDA filers will experience a more gradual initial erosion, though the long-term trajectory is the same.
For sophisticated pharmaceutical investors and executives, tracking ANDA filing activity against a competitor’s products can provide 12 to 24 months of advance warning about the magnitude of an incoming cliff. This is not speculative analysis — it is documented public information, since ANDAs and Paragraph IV notifications appear in FDA databases and court dockets.
Paragraph IV Litigation Outcomes
Innovators win Paragraph IV cases more than they lose, but the margin is not as large as patent holders typically claim in investor presentations. Studies of Hatch-Waxman litigation outcomes from 1984 through the early 2020s show that innovators prevailed in roughly 54% to 59% of cases that went to final judgment [12]. The remaining cases either settled (often with pay-for-delay provisions, discussed in Part Six) or resolved in favor of generic entry.
The practical consequence of this win rate is that patent protection is probabilistic rather than certain. An innovator with a compound patent running to 2032 faces genuine risk that a successful Paragraph IV challenge could bring generic entry in 2027. Risk-adjusted patent expiry dates — which incorporate litigation probability — differ meaningfully from nominal expiry dates, and sophisticated investors and competitors work with risk-adjusted numbers when modeling competitive scenarios.
The 180-Day Exclusivity Window in Practice
The 180-day exclusivity period is simultaneously one of the most profitable events in the pharmaceutical generic industry and one of the most complex legal constructs in regulatory law.
The FDA’s Office of Generic Drugs monitors whether a first-filer has “forfeited” its exclusivity rights — through failure to market, failure to obtain tentative approval, or other triggering events — before it begins. Forfeiture provisions exist because Congress recognized that a first-filer could use its exclusivity as a blocking mechanism, preventing all other generic entry without actually marketing its own product.
The commercial dynamics during 180-day exclusivity are favorable to the first-filer but not as favorable as the innovator might fear. Unless the innovator deploys an authorized generic in the same window — a strategy it often pursues specifically to erode the first-filer’s margins — the first-filer and innovator compete in a two-player market at prices well above the eventual commodity level. For major branded drugs, the 180-day exclusivity window can generate hundreds of millions of dollars in profits for the first-filer.
Part Four: Biosimilar Competition — A Different Animal
Why Biologics Don’t Have Cliffs
The difference between a small-molecule drug and a biologic runs deeper than chemistry. A small-molecule drug is a precisely defined chemical structure that can, in principle, be synthesized exactly. A biologic is a complex protein or other macromolecule produced by living cells, whose three-dimensional folding, post-translational modifications, and higher-order structure are inherently difficult to replicate with perfect fidelity.
This distinction is why generic biologics are called “biosimilars” rather than “generics.” They are not chemically identical copies; they are highly similar products that must demonstrate they have no clinically meaningful differences in safety and efficacy from the reference product. The Biologics Price Competition and Innovation Act (BPCIA) of 2009 created the legal pathway for biosimilar approval in the U.S., analogous to what Hatch-Waxman did for small-molecule generics [13].
The regulatory requirements for biosimilar approval are considerably more demanding than for small-molecule ANDAs. Biosimilar applicants must conduct extensive analytical comparisons, and often clinical pharmacology and clinical studies, to establish biosimilarity. The result is that biosimilar development costs are far higher than generic drug development — typically in the range of $100 million to $250 million per biosimilar program — and the lead time required is longer [14].
The BPCIA also created a 12-year reference product exclusivity period for biologics, running from the date of approval, during which no biosimilar can be approved on the basis of the reference product’s safety and efficacy data [15]. This exclusivity period is independent of patent protection and cannot be extended.
The Humira Example: Biosimilar Entry Finally Arrives
AbbVie’s adalimumab (Humira) spent years as the world’s best-selling drug, generating over $20 billion annually in global revenues [16]. Its compound patents had expired in other markets years before U.S. biosimilar entry, and several biosimilar developers had been preparing for years. The U.S. BPCIA exclusivity ran out in January 2023.
When biosimilar entry finally came, it was not a cliff. Seven adalimumab biosimilars launched in the U.S. in 2023, with more following [17]. But unlike Lipitor’s experience, branded Humira did not collapse. AbbVie had spent years negotiating exclusive or preferred formulary positions with major PBMs at substantially reduced net prices (through rebates). The result was a pricing structure that retained large portions of the patient base under managed care while dramatically reducing net revenue per unit.
By late 2023 and into 2024, the biosimilar market had captured a modest but growing share. AbbVie reported that Humira’s U.S. revenues declined roughly 35% to 40% from peak levels — significant, but nowhere near the 80% collapse seen for major small-molecule cliffs [18]. The pattern aligns with the slope rather than the cliff model.
Several structural factors explain the slower biosimilar uptake: only a fraction of adalimumab biosimilars had achieved “interchangeable” status with the FDA, meaning pharmacist substitution without prescriber authorization was not uniformly available; payer formulary decisions are made annually and take time to cycle in new products; physicians treating complex immune conditions — rheumatoid arthritis, psoriasis, Crohn’s disease — are often reluctant to switch stable patients; and AbbVie’s rebate strategy made the branded product competitively priced on a net basis for large payers.
The Interchangeability Hurdle
The interchangeability designation is critical to understanding biosimilar market dynamics. A biosimilar that achieves interchangeable status can be substituted for the reference product by a pharmacist in most states without contacting the prescriber — the same automatic substitution that applies to small-molecule AB-rated generics.
Achieving interchangeability requires demonstrating, through switching studies, that alternating between the reference product and biosimilar does not produce greater safety or efficacy differences than using the reference product alone. This additional clinical work adds cost and time to biosimilar development, and not all developers pursue it.
Without interchangeability, biosimilar adoption is driven by physician prescribing decisions, health plan formulary management, and patient education — all slower and more friction-laden processes than automatic pharmacist substitution. This is why biosimilar market penetration in the U.S. lags that of European markets, where interchangeability standards are different and payer incentives for biosimilar adoption are often stronger.
Part Five: Lifecycle Management — What Innovators Actually Do
The Strategic Toolkit
When pharmaceutical executives talk about “lifecycle management” (LCM), they mean the set of strategies deployed to extend a drug’s commercial life beyond its initial patent exclusivity. Some of these strategies are legitimate innovations that provide genuine patient benefit. Others are regulatory and legal maneuvers that extend exclusivity without material therapeutic value. Most pharmaceutical portfolios contain both.
Understanding LCM is essential for generic companies, investors, and payers, because LCM strategies directly determine when meaningful competition can actually begin — regardless of what the nominal compound patent expiry date suggests.
Strategy 1: Authorized Generics
An authorized generic (AG) is a generic version of a branded drug marketed by the innovator itself, typically through a subsidiary or licensing arrangement. AGs are often deployed on the day of patent expiry, specifically to erode the first-filer generic’s 180-day exclusivity advantage.
The logic is straightforward: by competing in the generic market from day one, the innovator splits the two-player exclusivity market with the first-filer rather than leaving all the upside to the challenger. The AG competes on price, which drives down margins for the first-filer, reducing the profitability that makes Paragraph IV challenges attractive in the first place.
Critics of AGs argue they reduce generic competition by eroding the financial incentive for Paragraph IV challenges, effectively discouraging future patent challenges across the industry. The FTC has studied this question multiple times and reached mixed conclusions. The empirical evidence suggests AGs modestly reduce the profitability of 180-day exclusivity periods without materially changing long-run competitive outcomes [19].
Strategy 2: Reformulation and New Delivery Systems
Developing a new formulation of an existing compound — extended-release versus immediate-release, a once-daily version versus a twice-daily version, a transdermal patch versus an oral tablet — can generate new patent protection and potentially new regulatory exclusivities.
AstraZeneca’s extended-release metoprolol (Toprol-XL) ran for years after the base metoprolol patents expired, protected by formulation patents and backed by clinical data showing improved tolerability. Forest Laboratories’ escitalopram (Lexapro) was positioned as a purer, better-tolerated version of citalopram, with its own patent protection.
The commercial success of formulation-based LCM depends heavily on whether physicians and payers accept the clinical differentiation argument. For a drug with a genuine pharmacokinetic rationale for extended release — one where steady plasma levels matter clinically — the reformulation can represent real improvement. For drugs where the extended-release version is developed primarily to extend exclusivity, payers often refuse preferred formulary placement and physicians are skeptical, limiting the commercial upside.
Strategy 3: New Indications and Method-of-Use Extensions
FDA approval of a new indication for an existing drug generates three years of additional market exclusivity for that indication [20]. This exclusivity does not prevent generics from being dispensed for the drug’s original uses, but it does restrict them from being approved for the new indication.
Pfizer pursued this aggressively with sildenafil — the compound that became Viagra. When the pulmonary arterial hypertension indication was pursued and approved (as Revatio), it generated additional exclusivity, though the commercial significance was modest relative to the primary erectile dysfunction market.
New indication strategies are most powerful when they expand a drug into a much larger patient population or when the new indication is clinically significant enough to justify premium pricing that erodes more slowly than the original indication’s market.
Strategy 4: Pediatric Exclusivity
Under the Best Pharmaceuticals for Children Act, companies that conduct FDA-requested pediatric studies of a drug can receive six months of additional marketing exclusivity bolted onto all existing patents and exclusivities [21]. This six-month extension applies to all formulations and indications, making it one of the most cost-effective LCM strategies available.
For a drug generating $3 billion annually, six months of additional exclusivity is worth roughly $1.5 billion in additional protected revenue. The cost of conducting the required pediatric studies is typically $30 million to $100 million — a return-on-investment that makes pediatric exclusivity one of the clearest value-creation opportunities in pharmaceutical development.
Strategy 5: Patent Thickets
A patent thicket is a dense collection of overlapping patents that a generic or biosimilar company must navigate before entering the market. Unlike individual patents, which can be challenged through the Paragraph IV process, a thicket requires either challenging every patent individually (expensive and time-consuming) or negotiating around them.
Humira is the most cited example: AbbVie built a portfolio of more than 130 patents around adalimumab, many of which were filed in the years after the drug’s initial approval [22]. Even if a biosimilar company was confident it could invalidate any individual patent, the collective burden of challenging the entire portfolio was daunting enough to delay serious U.S. biosimilar competition for years after patents in other markets had expired.
Critics, including Members of Congress and the FTC, have characterized certain pharmaceutical patent thickets as anticompetitive. The legal standard for patent misuse or antitrust violation is high, however, and companies have generally been able to maintain thickets without successful challenge.
Strategy 6: Citizen Petitions and REMS Programs
Risk Evaluation and Mitigation Strategies (REMS) are FDA-required safety programs for drugs with significant safety concerns. When a REMS requires special distribution controls, restricted access programs, or mandatory safety testing, it can create practical barriers to generic entry — because generic companies need samples of the reference product for bioequivalence testing, and if those samples are only available through controlled distribution channels, the innovator controls access.
Several companies used REMS-related distribution restrictions specifically to delay generic entry. Congress and the FDA responded with provisions in the FDA Safety and Innovation Act requiring innovators to provide REMS-compliant access to reference product samples for bioequivalence testing [23]. The problem has been largely addressed, though enforcement actions against companies that created artificial distribution barriers have continued.
Part Six: Pay-for-Delay — The Legal and Economic Framework
Reverse Payment Settlements
Among the most controversial LCM strategies is the “reverse payment” or “pay-for-delay” settlement. When a generic company files a Paragraph IV certification and the innovator sues, the parties often settle rather than proceed to trial. In many settlements, the innovator pays the generic company — in cash, exclusive licensing rights, or other value — in exchange for the generic company agreeing not to enter the market until a specified date.
The transaction has an obvious economic logic: if the patent is genuinely likely to be upheld (and the generic’s entry blocked), both parties are no better off litigating. If the patent is weak, both parties may prefer a settlement that guarantees a future entry date over the uncertainty of trial. The economic problem is that settlements involving large payments from innovator to generic leave the payer — the patient or insurance company — paying branded prices for an unnecessarily long period, with the settlement value effectively extracted from that payer through artificially maintained branded pricing.
FTC v. Actavis: The Supreme Court Draws a Line
The Federal Trade Commission spent years arguing that reverse payment settlements were per se antitrust violations. The Supreme Court’s 2013 decision in FTC v. Actavis rejected the per se approach but held that these agreements could violate antitrust law and should be evaluated under the “rule of reason” [24]. The decision opened the door for antitrust litigation of individual settlements, and several subsequent cases resulted in significant settlements.
The practical effect of Actavis has been to moderate but not eliminate reverse payment agreements. Companies have become more careful about the structure and size of payments, often converting cash payments into supply agreements, co-promotion arrangements, or other forms of value transfer that are less obviously problematic. The FTC’s Annual Report on Pharmaceutical Patent Settlements tracks these arrangements and has noted that the percentage of settlements involving “no-authorized-generic” commitments and other indirect forms of value transfer has remained significant [25].
For generic companies, the post-Actavis environment means that settlements that once appeared straightforward now carry antitrust risk. For innovators, the risk of an FTC enforcement action or private antitrust suit has constrained the range of settlement terms they can reasonably offer.
Part Seven: Regulatory Exclusivities Beyond Patents
The Non-Patent Exclusivity System
Patent protection and regulatory exclusivity are legally distinct mechanisms that can overlap, interact, or operate independently. Understanding both is necessary to accurately predict when generic or biosimilar entry is legally possible.
The FDA grants several forms of non-patent market exclusivity:
New Chemical Entity (NCE) Exclusivity provides five years of data exclusivity from the date of approval for a drug containing a previously unapproved active moiety [26]. During this period, no ANDA referencing the NDA can be submitted — not just approved, but submitted. The practical effect is to delay the entire Paragraph IV challenge process by five years, since generic companies cannot even file their applications until the exclusivity expires.
New Clinical Investigation Exclusivity (three-year exclusivity) applies when an NDA or supplemental NDA includes new clinical investigations essential to the approval [27]. This applies to new formulations, new combinations, new routes of administration, and new indications. Unlike NCE exclusivity, it does not prevent ANDA submission — it only prevents FDA approval of an ANDA for the applicable change.
Orphan Drug Exclusivity provides seven years of marketing exclusivity for drugs approved for rare diseases (defined as affecting fewer than 200,000 Americans) [28]. This exclusivity is condition-specific — it applies only to the particular orphan indication, not to the drug as a whole. However, for drugs that are primarily or exclusively used for orphan indications, it can be the dominant form of exclusivity.
Biologic Exclusivity under the BPCIA, as noted, provides 12 years from the date of approval for reference products, plus a four-year data exclusivity period during which no biosimilar application can be submitted [29].
How Exclusivities Interact with Patents
The interaction between exclusivities and patents creates a legal landscape that requires careful analysis for each product individually. A drug might have a compound patent expiring in Year 10, an NCE exclusivity running to Year 5, formulation patents running to Year 14, and pediatric exclusivity adding six months to everything — producing an effective exclusivity window that extends to Year 14.5, despite a nominal compound patent expiry in Year 10.
This is why databases like DrugPatentWatch are operationally valuable to competitive intelligence analysts: they aggregate Orange Book patent listings, FDA exclusivity records, ANDA filing activity, and litigation outcomes into a unified view that shows not just when patents expire on paper, but when meaningful generic competition can realistically begin.
For payer organizations trying to model drug expenditures, knowing that a drug’s effective exclusivity extends four years beyond its compound patent expiry is the difference between accurate budget forecasting and significantly understating drug spend.
Part Eight: Financial Impact on Innovator Companies
Modeling Revenue Post-Expiry
Pharmaceutical company investors and equity analysts spend considerable effort modeling post-cliff revenue for drugs facing near-term patent expiration. The key variables in these models are:
The number of competitors entering at expiry is the most powerful determinant of pricing dynamics. Two generic entrants in a 180-day exclusivity window price very differently from twelve entrants in a fully open market. ANDA filing counts, available from FDA databases, provide the most direct leading indicator.
The channel mix of the branded product matters significantly. A drug with high managed care penetration and tight formulary management will see faster branded substitution than one with a significant cash-pay or Medicare Part D patient base, where formulary levers are weaker.
Manufacturing complexity determines how quickly generic supply can scale to meet demand. For a simple oral solid-dose drug, generic supply can ramp in months. For a sterile injectable with specialized manufacturing requirements, supply constraints can delay price erosion for a year or more after the legal barriers to entry are removed.
Therapeutic category dynamics influence physician behavior. Statins, proton pump inhibitors, and ACE inhibitors tend to see rapid generic adoption. Anti-epileptics, thyroid hormones, and certain psychiatric medications tend to see slower substitution due to physician concerns about bioequivalence in narrow therapeutic index drugs.
The AbbVie Model: Building Through the Biosimilar Transition
AbbVie’s approach to managing the Humira biosimilar transition illustrates both the limits of LCM and the financial engineering that companies use to soften the blow.
In the years before biosimilar entry, AbbVie invested aggressively in building out its immunology pipeline — acquiring Allergan for $63 billion in 2020, a transaction that added Botox, Juvederm, and a range of non-Humira products to the portfolio [30]. The Allergan acquisition was, in part, a hedge against the Humira cliff: by the time biosimilars arrived, AbbVie had substantial revenue from non-adalimumab sources.
AbbVie also positioned Skyrizi (risankizumab) and Rinvoq (upadacitinib) as successor immunology products in the years before Humira’s U.S. biosimilar exposure. By the time the cliff arrived, the combined Skyrizi/Rinvoq revenue run rate was growing rapidly enough to partially offset Humira’s decline.
This pre-cliff portfolio construction — identifying successor products, acquiring diversifying revenue streams, and managing investor expectations through the transition — is the financial equivalent of building a second cliff upstream that produces new revenue as the first cliff collapses underneath you.
Pfizer’s Post-Lipitor Challenge
Pfizer’s situation after Lipitor’s cliff was less well-managed. The company did not have a comparable replacement revenue stream in the same commercial magnitude. It used the period following the Lipitor cliff to aggressively cut costs, return capital to shareholders, and pursue pipeline acquisitions that would eventually produce new blockbusters like Xeljanz, Eliquis (co-commercialized with BMS), and, ultimately, the COVID-19 vaccine and antiviral.
The lesson from Pfizer’s experience is that patent cliffs are not primarily financial events — they are strategic transitions. Companies that have built pipeline succession years in advance are prepared for them. Companies that have relied on a single franchise for a disproportionate share of revenue face a longer and more painful recovery.
Investor Implications
For equity investors, patent cliff risk is material, frequently disclosed, and sometimes still mispriced. Companies with large cliffs approaching in the next three to five years will typically acknowledge the exposure in SEC filings but may present lifecycle management and pipeline depth more optimistically than the data supports.
The pattern of investor response to cliff events has been consistent: stocks tend to price in the cliff gradually as expiry approaches, with the sharpest re-ratings occurring when the first Paragraph IV filing appears (signaling that generic entry is imminent) and when initial generic market share data appears post-launch. Companies that have credibly demonstrated pipeline succession tend to weather cliff events with less permanent valuation damage.
For short sellers, patent cliff timing combined with pipeline analysis can identify companies where the market is pricing in more post-cliff resilience than the fundamentals support. For long investors, cliffs that have been overestimated — where branded durability or biosimilar uptake challenges are stronger than the consensus expects — can create entry points.
Part Nine: Generic Industry Economics Post-Expiry
The Generic Manufacturer’s Business Model
The economics of generic pharmaceutical manufacturing are unlike those of innovator drug development in almost every dimension. Innovators compete on clinical differentiation and market access; generic companies compete on manufacturing efficiency, regulatory speed, and supply chain reliability.
The profitability of the generic business model is highly concentrated in the period immediately following patent expiry for major drugs — particularly during the 180-day exclusivity window. Outside of that window, the generic pharmaceutical market is characterized by intense price competition, thin margins, and commodity economics.
Over the past decade, generic drug prices in the U.S. have fallen dramatically. Studies tracking generic drug prices since the late 2000s show that many commodity generics are priced at a fraction of their levels a decade ago — the result of aggressive purchasing by pharmacy chains, PBMs, and group purchasing organizations (GPOs), combined with increasing consolidation among buyers and global supply chain expansion [31].
For generic manufacturers, the strategic implication is that the first-to-file, first-to-market advantage on major drugs is disproportionately important. A generic company that captures the 180-day exclusivity on a billion-dollar brand can generate profits sufficient to fund dozens of lower-value commodity products. Companies that build their strategic planning around this model — investing heavily in patent challenge capabilities and ANDA filing pipelines — tend to generate substantially better returns than those focused purely on commodity product manufacturing.
Consolidation and the Indian and Chinese API Supply Chains
The generic drug industry has undergone significant consolidation since the 2000s. Major acquisitions have created global generic companies with integrated manufacturing, formulation, and regulatory capabilities across multiple markets simultaneously.
The underlying API for most generic drugs is manufactured in India or China. This global supply chain creates operational resilience challenges that came into sharp focus during the COVID-19 pandemic, when disruptions to Indian manufacturing facilities exposed the fragility of U.S. generic drug supply. Several major generic drug shortages in 2020 and 2021 were directly linked to API supply disruptions in these markets [32].
For innovator companies tracking competitive entry timelines, the state of the generic supply chain matters. A drug whose API manufacturing is concentrated in a single Indian facility may experience delayed or limited generic entry even after patent expiry if that facility faces FDA import alerts, capacity constraints, or supply chain disruptions.
The Pricing Spiral
The dynamics of generic drug pricing after full market opening follow a predictable spiral: prices fall rapidly as competitors enter, margins compress until manufacturers with insufficient scale exit the market, and the remaining manufacturers stabilize at a price level that covers their marginal cost of production plus a minimal margin. In some commodity generics, this terminal price is below the cost of manufacturing for U.S.-based producers, effectively making the U.S. dependent on Indian or Chinese manufacturers for permanent supply.
This pricing dynamic has created concerns about the long-run sustainability of the U.S. generic drug supply chain. Congressional interest in domestic API manufacturing, combined with FDA incentive programs for domestic production, reflects recognition that the competitive dynamics of the generic market have produced a structurally fragile supply chain even as they have delivered lower prices for patients.
Part Ten: PBM and Payer Dynamics in the Post-Expiry Market
Formulary Mechanics
Pharmacy benefit managers (PBMs) are the most powerful actors in determining how quickly branded drugs lose market share after patent expiry. PBMs design the formularies that determine what patients pay for specific drugs — and those formularies are explicitly designed to steer patients toward lower-cost alternatives.
When a drug loses patent protection, the three major PBMs (CVS Caremark, Express Scripts, and OptumRx) typically move the branded version to a non-preferred or excluded tier while placing the generic on the preferred or lowest-cost tier within one to two formulary cycles [33]. The speed of this transition depends on whether the PBM has pre-negotiated arrangements — including the deployment of the branded drug on a “when generic available” exclusion clause — and on the availability of AB-rated generics at the time of the formulary update.
For patients enrolled in commercial health plans that use these formularies, the out-of-pocket differential between a non-preferred branded drug and a preferred generic can be substantial — often the difference between a $5 to $15 co-pay for the generic and a $50 to $150 co-pay for the branded version. This cost differential, not physician prescribing patterns, is the primary driver of generic substitution in the commercial market.
Rebate Erosion and Net Pricing
The net price that innovators receive for branded drugs — after paying rebates and discounts to PBMs, health plans, and government programs — is significantly lower than the list price. For major branded drugs in competitive categories, net prices may be 40% to 60% below list, with rebates paid in exchange for preferred formulary positioning [34].
When generic competition enters, the economics of the rebate system shift dramatically. Branded drugs facing multiple generic competitors have very limited leverage in formulary negotiations — the alternative (the generic) is simply less expensive, and no rebate can change the fundamental economics. The branded drug may attempt to compete on net price by offering very large rebates, but this strategy erodes margins and rarely succeeds in maintaining meaningful formulary position against established generic competition.
For biosimilars, the rebate dynamics are different. AbbVie successfully used large upfront rebate commitments on Humira to maintain preferred formulary positioning even after biosimilar entry. PBMs that had signed multi-year contracts with significant Humira rebate commitments had limited incentive to immediately preference biosimilars whose rebate offers were smaller in absolute dollar terms, even if biosimilars were priced lower per unit.
This perverse outcome — where a more expensive branded drug maintains formulary position over cheaper biosimilars through rebate structures — has drawn significant criticism from patient advocates, biosimilar manufacturers, and academic health economists. It illustrates a structural misalignment in the PBM rebate model that regulators have not fully resolved.
Part Eleven: Patent Intelligence in Practice
Tracking the Landscape
The competitive intelligence advantage in pharmaceutical markets belongs to those who can accurately translate patent data into commercial timelines. This is harder than it sounds, because:
Orange Book listings may include patents that are legally vulnerable and unlikely to survive challenge. Nominal expiry dates on those patents overstate effective exclusivity.
Paragraph IV filings, conversely, may target patents that courts ultimately uphold — meaning that nominal expiry dates for those patents accurately reflect the earliest realistic generic entry date.
Regulatory exclusivities run on timelines that are separate from and sometimes longer than patent protection.
Lifecycle management activities — new formulations, new indications, pediatric studies — can add exclusivity on top of existing protection in ways that aren’t immediately visible from the Orange Book alone.
DrugPatentWatch aggregates these sources into structured data that enables systematic monitoring. For a pharmaceutical executive tracking competitive entry risk against a key product, or a generic company prioritizing its ANDA pipeline, or an investor building financial models around post-cliff revenue assumptions, the ability to cross-reference patent listings, exclusivity records, ANDA filing histories, and litigation outcomes in a single platform is operationally valuable. The alternative is maintaining a team of patent attorneys and regulatory specialists to track each product manually — an approach that is expensive, slower, and more prone to missing the early signals that precede competitive entry.
The most useful applications of patent intelligence in practice include:
Identifying drugs where multiple generic companies have filed Paragraph IV certifications against the same patent — a signal that the patent is viewed as vulnerable and that early generic entry is being actively pursued.
Tracking when ANDA approvals move from “tentatively approved” to “final approved” status — a status change that often coincides with patent expiry or litigation resolution and immediately precedes commercial launch.
Monitoring biosimilar applications filed with the FDA for reference products whose 12-year exclusivities are approaching expiration — useful for both potential biosimilar developers assessing the competitive landscape and for investors modeling the biosimilar entry curves for specific biologics.
Watching citizen petition filings, which are public documents that sometimes reveal LCM strategies being deployed to delay generic or biosimilar approval — occasionally providing the clearest signal that an innovator is uncertain about the durability of its patent position.
Part Twelve: The 2025–2030 Patent Cliff Wave
The Drugs at Risk
The pharmaceutical industry is approaching what analysts have called the largest patent cliff in its history. The pipeline of major drugs losing exclusivity between 2025 and 2030 represents an estimated $300 billion or more in branded pharmaceutical revenues that will be exposed to generic or biosimilar competition [35].
The drugs in this cohort include some of the most commercially significant products of the past decade:
Stelara (ustekinumab), Janssen’s IL-12/23 inhibitor for psoriasis and Crohn’s disease, with U.S. revenues exceeding $6 billion annually, faced biosimilar entry beginning in 2023 and has seen escalating biosimilar competition into 2025 [36].
Eliquis (apixaban), Bristol-Myers Squibb and Pfizer’s Factor Xa inhibitor and among the highest-revenue drugs in the U.S., has compound patents that began expiring in 2026, with complex patent litigation determining the precise entry timeline for generics [37].
Keytruda (pembrolizumab), Merck’s PD-1 inhibitor and the world’s best-selling drug, faces a more complex biosimilar landscape given both the biological complexity of the molecule and the strength of its label — spanning more than 30 tumor types [38]. The core biologic patent expires in 2028, with the 12-year biologic exclusivity running to similar or slightly later dates.
Ozempic/Wegovy (semaglutide), Novo Nordisk’s GLP-1 receptor agonist, has patent protection running through the early 2030s in most markets, though the precise timeline is subject to ongoing litigation from generic and biosimilar challengers who are already preparing entry strategies [39].
Dupixent (dupilumab), Sanofi and Regeneron’s IL-4/13 antagonist for atopic dermatitis and other Type 2 inflammatory conditions, represents a significant future cliff event with core biologics exclusivity running into the early 2030s.
Revenue at Risk Calculations
Calculating “revenue at risk” from patent cliffs requires distinguishing between nominal listed revenues and the net revenues after rebates and discounts. For many biologics with significant rebate obligations, the list price overstates commercial value, and the net revenue decline associated with biosimilar entry may be smaller than headline numbers suggest — while the rebate savings to payers may be less dramatic than advertised.
The useful analytical framework is to ask: what is the net present value of revenues the innovator can defend after patent expiry, given realistic lifecycle management success rates, biosimilar/generic competition timelines, and payer formulary dynamics? For most products, this analysis produces a significantly lower number than the projections that investor relations presentations tend to highlight.
Structural Differences from Prior Cliff Waves
The 2025–2030 cliff wave differs from previous cycles in several respects that affect how revenue erosion will unfold.
The proportion of at-risk revenues attributable to biologics rather than small molecules is significantly higher than in previous cliff waves. This means a larger share of the revenue at risk will erode as slopes rather than cliffs — somewhat reducing the severity of individual quarter impacts but extending the period of competitive pressure over multiple years.
The biosimilar industry is more mature than at any prior cliff cycle. Companies that developed biosimilars for adalimumab, bevacizumab (Avastin), and trastuzumab (Herceptin) have built manufacturing and regulatory capabilities that they can deploy more rapidly for subsequent biologics. This suggests that biosimilar competition for the 2025–2030 cohort will be more vigorous and faster to ramp than earlier entrants experienced.
PBM and payer sophistication in managing biosimilar formularies has also improved. Major health plans and PBMs have built institutional knowledge of how to structure formulary decisions, step therapy requirements, and prior authorization criteria to accelerate biosimilar adoption — and have financial incentives to deploy that knowledge more aggressively than in the adalimumab cycle.
Part Thirteen: International Dimensions
Why Patent Expiry Dates Differ by Country
A drug’s patent expiry date in the United States is often not the same as its expiry date in Europe, Japan, or emerging markets. This divergence has significant competitive and commercial implications.
Patent terms, supplementary protection certificates (the European equivalent of patent term extensions), regulatory exclusivity periods, and the timing of initial product approvals all vary by country. A compound patent filed simultaneously in the U.S. and Europe will have the same 20-year term from filing, but if the drug was approved two years earlier in the U.S. than in Europe, its effective period of commercial exclusivity post-approval differs between the two markets.
European supplementary protection certificates (SPCs) can extend patent protection by up to five years, similar in concept to U.S. patent term extensions but with different calculation methodologies [40]. Countries in the EU calculate SPC durations individually based on their own approval dates, so SPCs differ in length across EU member states for the same product.
For generic and biosimilar companies, this patchwork of expiry dates creates a global market entry sequencing problem. A biosimilar that has already launched in Europe for two or three years — building manufacturing scale, regulatory experience, and market access data — is better positioned for U.S. entry than one entering a market simultaneously with no prior commercialization experience. European biosimilar markets have therefore served as a proving ground that accelerates U.S. entry readiness for the highest-value biologics.
Compulsory Licensing and Access Challenges
In markets outside the U.S. and Europe, patent protection for pharmaceutical products is more fragile. The TRIPS Agreement (Trade-Related Aspects of Intellectual Property Rights) allows countries to issue compulsory licenses to manufacture or import generic versions of patented drugs in certain public health emergencies or other circumstances [41].
Compulsory licensing has been most prominently used for HIV/AIDS antiretrovirals and, more recently, was the subject of significant debate during the COVID-19 pandemic. Several developing countries issued or threatened compulsory licenses on vaccines and antivirals, though the outcome of those negotiations was complex and largely settled through voluntary licensing agreements and the WHO’s COVAX mechanism.
For pharmaceutical companies, compulsory licensing risk is a consideration in pricing and access strategy in emerging markets. Strategies that keep prices in lower-income markets at levels that reduce the political pressure for compulsory licensing — including tiered pricing, voluntary licensing programs, and partnerships with local manufacturers — have become standard elements of global commercial planning for major pharmaceutical products.
Part Fourteen: The Future of Patent Cliff Dynamics
How Drug Price Negotiation Changes the Calculus
The Inflation Reduction Act of 2022 introduced direct Medicare drug price negotiation for a defined set of high-expenditure drugs, beginning with small molecules that have been on the market for at least nine years and biologics that have been on the market for at least 13 years [42].
This negotiation mechanism fundamentally changes the financial model for drugs approaching but not yet past patent expiry. A drug that would previously have maintained high net pricing through its remaining exclusivity period now faces a Medicare-set negotiated price that may significantly reduce the revenue available in those final exclusivity years. The mechanism effectively front-loads some of the post-expiry pricing pressure onto the pre-expiry period for the highest-expenditure products.
The implications for LCM strategy are significant. Lifecycle management investments that previously made financial sense based on the revenue available during extended exclusivity years must now be re-evaluated against a lower base price in the Medicare channel. Pediatric exclusivity, worth six months at $3 billion in annual revenues, is worth considerably less if Medicare has negotiated a 30% to 40% price reduction for the final years of the drug’s exclusivity.
AI and Accelerated Generic Development
Artificial intelligence applications in pharmaceutical formulation development, bioequivalence prediction, and regulatory documentation are beginning to accelerate the generic drug development process. AI-assisted formulation development can identify bioequivalent generic formulations more rapidly than traditional trial-and-error approaches, potentially reducing the lead time between compound patent expiry and generic launch.
For innovator companies, this means that the window between patent expiry and first generic entry — which has historically provided some runway for patient transition programs and revenue collection — may compress. The practical implications are still emerging, but the directional trend is toward faster generic entry relative to patent expiry dates.
Similarly, machine learning approaches to biosimilar characterization are improving the efficiency of analytical comparability exercises, the most scientifically demanding component of biosimilar development. Faster analytical comparability reduces the time and cost of biosimilar programs, potentially bringing more competitors to market on shorter timelines.
Patent Reform Pressure
Legislative and regulatory pressure on pharmaceutical patent practices has been building for years and is likely to intensify. Congressional proposals to limit the listing of certain formulation and dosage regimen patents in the Orange Book, restrict the availability of SPCs and patent term extensions, and streamline Paragraph IV litigation have been introduced repeatedly, though comprehensive reform has been difficult to enact.
The most durable regulatory change has been at the FDA level: the agency has strengthened its enforcement of Orange Book listing requirements, leading to the voluntary delisting of patents that some companies had listed without adequate legal basis. This enforcement activity has accelerated Paragraph IV challenge timelines for a small number of products where improper listings had been used to trigger automatic 30-month stays.
Key Takeaways
1. Patent expiry is not a date — it is a process. The compound patent expiry date is the starting point of analysis, not the answer. Formulation patents, method-of-use patents, regulatory exclusivities, and pediatric extensions create a layered protection structure that routinely extends effective market exclusivity years beyond compound patent expiry.
2. Cliffs hit small molecules; slopes hit biologics. The structural differences between small-molecule generic entry and biosimilar competition produce fundamentally different revenue erosion patterns. Small molecules face rapid, steep price declines driven by automatic pharmacist substitution. Biologics face slower erosion because interchangeability standards, formulary dynamics, and physician reluctance create friction that delays adoption.
3. The 180-day exclusivity is the most profitable event in the generic industry. First-to-file Paragraph IV challengers capture disproportionate value in the immediate post-expiry period. The financial logic explains why generic companies maintain substantial patent challenge operations, and why innovators deploy authorized generics as a countermeasure.
4. Lifecycle management extends exclusivity but does not prevent cliffs. The most sophisticated LCM programs — formulation innovations, new indications, patent thickets — can add years to a drug’s commercial life, but they do not change the fundamental trajectory. The cliff comes; the question is when.
5. Payers and PBMs are the most powerful agents of post-expiry competition. Physician prescribing patterns matter less than formulary design in determining how fast branded drugs lose volume. PBMs that preference generics through co-pay differentials and formulary exclusions can accelerate substitution faster than almost any other mechanism.
6. The 2025–2030 patent cliff represents a structural transition for the industry. The cohort of biologics facing exclusivity loss in this period is larger than any prior cycle. Companies entering it with credible successor pipelines and diversified revenue bases will absorb it; companies relying on a small number of at-risk products without succession will face significant financial pressure.
7. Patent intelligence is a competitive weapon. ANDA filing activity, Paragraph IV certifications, Orange Book changes, and biosimilar application submissions are all publicly available signals that provide advance warning of competitive entry intentions. Organizations that track them systematically — using tools like DrugPatentWatch — have a meaningful informational advantage over those that don’t.
FAQ
Q1: When multiple patents cover the same drug, which expiry date actually determines when generics can enter?
The effective exclusivity end date is determined by the last-expiring patent (or regulatory exclusivity) that a generic company cannot design around or successfully challenge. This is rarely the compound patent and often a formulation, method-of-use, or pediatric exclusivity extension. A generic company can enter as soon as it can demonstrate that it does not infringe any remaining, valid listed patent — which is why Paragraph IV challenges targeting specific patents can allow entry years before the last listed patent expires. The Orange Book shows all listed patents and their expiry dates; the practical question is which ones are valid and infringed by the proposed generic product.
Q2: Why don’t biosimilars capture more market share faster, given that they are substantially cheaper than reference biologics?
Three structural barriers slow biosimilar adoption in the U.S. First, the interchangeability designation required for automatic pharmacist substitution is difficult and expensive to achieve, limiting most biosimilars to physician-directed prescribing. Second, reference product manufacturers have used large rebate offers to maintain formulary preference with major PBMs, creating a situation where the lower-priced biosimilar is not always the lower-cost option from a formulary management standpoint. Third, physician inertia in complex therapeutic areas — oncology, rheumatology, inflammatory bowel disease — produces genuine reluctance to switch stable patients whose disease is controlled on a reference product. European markets, with different interchangeability standards and stronger regulatory support for biosimilar adoption, show faster uptake rates.
Q3: What is the most common reason a Paragraph IV patent challenge fails?
The most common failure mode is not legal but strategic: generic companies challenge patents that they ultimately cannot prove are invalid or non-infringed. The inventive step for compound patents is often well-documented in prosecution history, making invalidity arguments difficult. Non-infringement arguments fail when courts find that the generic product, even if formulated differently, meets the patent claim elements. Companies that challenge without thorough prior art analysis, or that underestimate the innovation in a formulation patent, tend to lose at trial. The 54% to 59% innovator win rate in litigated cases reflects this reality.
Q4: How do companies decide which drugs are worth pursuing Paragraph IV challenges against, given the cost and uncertainty?
The financial analysis centers on the value of the 180-day exclusivity period if the challenge succeeds, discounted by the probability of success in litigation and the time required to reach a resolution. For a drug generating $1 billion annually in the U.S., a successful challenge might yield $150 million to $300 million in exclusivity-period profits. A Paragraph IV litigation campaign costs $5 million to $20 million. The expected value calculation is favorable even at moderate probability of success, which explains why major drugs attract multiple simultaneous challengers. Companies with strong track records of patent litigation success can demand higher implied probabilities in their pipeline valuations, making their litigation programs a genuine competitive differentiator.
Q5: How should investors think about companies with large patent cliffs approaching in the next five years?
The key analytical question is not the magnitude of the cliff in absolute revenue terms but the ratio of at-risk revenue to projected successor pipeline revenue, adjusted for probability of clinical and commercial success. A company with $5 billion of annual revenue at cliff risk but $8 billion in probability-adjusted pipeline value entering commercialization in the same window is a different investment than one with the same cliff and $2 billion of pipeline value. Beyond the pipeline, investors should examine whether the company has demonstrated disciplined capital allocation — reinvesting cliff-period cash flows into acquisitions or internal development rather than returning all capital to shareholders in a manner that leaves the company underinvested in its post-cliff portfolio. AbbVie’s Allergan acquisition, whatever its critics said at announcement, was the kind of pre-cliff diversification that ultimately proved its value.
Sources
[1] Hirschler, B. (2012). Lipitor’s U.S. generic competition erodes Pfizer revenue. Reuters. https://www.reuters.com/article/pfizer-results
[2] 35 U.S.C. § 154(a)(2). Patent term. United States Code.
[3] Drug Price Competition and Patent Term Restoration Act of 1984, Pub. L. No. 98-417, 98 Stat. 1585 (1984).
[4] 21 U.S.C. § 355(j)(5)(B)(iii). Federal Food, Drug, and Cosmetic Act, ANDA provisions.
[5] Berndt, E. R., & Newhouse, J. P. (2012). Pricing and reimbursement in U.S. pharmaceutical markets. National Bureau of Economic Research Working Paper No. 16297.
[6] IQVIA Institute for Human Data Science. (2024). Medicine use and spending in the U.S.: A review of 2023 and outlook to 2028. IQVIA.
[7] United States District Court, District of Delaware. (2011). Ranbaxy Laboratories Ltd. v. Pfizer Inc. Lipitor patent litigation dockets.
[8] Pfizer Inc. (2012). Annual report on Form 10-K for the fiscal year ended December 31, 2012. U.S. Securities and Exchange Commission.
[10] 21 C.F.R. § 314.95. Paragraph IV certification notification requirements.
[11] 21 U.S.C. § 355(j)(5)(B)(iv). 180-day exclusivity for first applicant.
[12] Hemphill, C. S., & Sampat, B. N. (2012). Evergreening, patent challenges, and effective market life in pharmaceuticals. Journal of Health Economics, 31(2), 327–339.
[13] Biologics Price Competition and Innovation Act of 2009, Pub. L. No. 111-148, Title VII, Subtitle A (2009).
[14] Doshi, P., & Herder, M. (2018). The biosimilar approval pathway: Complexity and uncertainty. JAMA Internal Medicine, 178(10), 1416–1418.
[15] 42 U.S.C. § 262(k)(7). Reference product exclusivity under BPCIA.
[16] AbbVie Inc. (2022). Annual report on Form 10-K for the fiscal year ended December 31, 2022. U.S. Securities and Exchange Commission.
[17] Food and Drug Administration. (2023). Biosimilar product information: Adalimumab. FDA Center for Drug Evaluation and Research.
[18] AbbVie Inc. (2024). Full-year 2023 earnings results and 2024 financial guidance. AbbVie press release.
[19] Federal Trade Commission. (2011). Authorized generic drugs: Short-term effects and long-term impact. FTC Report.
[20] 21 C.F.R. § 314.108. New drug product exclusivity.
[21] Best Pharmaceuticals for Children Act of 2002, Pub. L. No. 107-109, 115 Stat. 1408 (2002).
[22] Initiative for Medicines, Access & Knowledge (I-MAK). (2021). Overpatented, overpriced: How excessive pharmaceutical patenting is extending monopolies and driving up drug prices. I-MAK Report.
[23] FDA Safety and Innovation Act of 2012, Pub. L. No. 112-144, § 1133 (2012). REMS shared system requirements.
[24] Federal Trade Commission v. Actavis, Inc., 570 U.S. 136 (2013).
[25] Federal Trade Commission. (2023). Agreements filed with the Federal Trade Commission under the Medicare Prescription Drug, Improvement, and Modernization Act of 2003. FTC Annual Report.
[26] 21 C.F.R. § 314.108(b)(2). Five-year new chemical entity exclusivity.
[27] 21 U.S.C. § 355(c)(3)(E). Three-year exclusivity for new clinical investigations.
[28] 21 U.S.C. § 360cc. Orphan drug designation and marketing exclusivity.
[29] 42 U.S.C. § 262(k)(7)(A)-(B). Biologic data and reference product exclusivities.
[30] AbbVie Inc. (2020). AbbVie completes Allergan acquisition. Press release, May 8, 2020.
[31] Government Accountability Office. (2021). Drug pricing: Research on savings from generic drug use. GAO-21-282.
[32] Association for Accessible Medicines. (2021). The U.S. generic and biosimilar medicines savings report. AAM.
[33] Dusetzina, S. B., Winn, A. N., Abel, G. A., Huskamp, H. A., & Keating, N. L. (2014). Cost sharing and adherence to thienopyridines for acute coronary syndrome. Journal of the American Medical Association, 312(19), 1998–2009.
[34] Congressional Budget Office. (2022). Prices for and spending on specialty drugs in Medicare Part D and Medicaid. CBO Report, March 2022.
[35] Evaluate Pharma. (2024). World preview 2024, outlook to 2030. Evaluate Ltd.
[36] Janssen Biotech, Inc. (2022). Ustekinumab U.S. patent litigation and biosimilar settlement timeline. SEC filings.