Humira, AbbVie’s blockbuster arthritis drug, generated more than $200 billion in cumulative revenue before biosimilar competitors could reach American patients at scale. The original patent on adalimumab, the active molecule, expired in 2016. Biosimilars were approved by the FDA starting in 2016. Yet the first biosimilar did not reach the U.S. market until 2023, seven years after that foundational patent lapsed. AbbVie had, in the interim, assembled a portfolio of more than 250 patents around the drug, covering everything from dosing syringes to manufacturing processes to specific patient populations.
That seven-year gap cost American patients and payers an estimated $19 billion in excess drug spending, according to a 2023 analysis by the RAND Corporation [1]. It is one of the most well-documented examples of a broader pharmaceutical strategy that goes by two names: the “patent thicket” and “evergreening.” Understanding how these strategies work, where they are legally permitted, and how regulators and competitors are pushing back is now essential knowledge for anyone operating in pharmaceutical markets, healthcare policy, or life sciences investment.
This article deconstructs both phenomena from the ground up, covering the mechanics of patent construction, the regulatory frameworks that enable or constrain these strategies, the economic consequences for patients and payers, and the specific legal and legislative challenges that are reshaping how the industry thinks about intellectual property.
Part One: The Architecture of a 20-Year Monopoly
What the Patent Clock Actually Measures
The standard framing of pharmaceutical patents is deceptively simple: a drug company discovers a molecule, files a patent, and receives 20 years of exclusive rights. The clock starts ticking from the filing date. After two decades, generic manufacturers can copy the drug and sell it at a fraction of the price.
This framing is accurate as far as it goes, but it omits the layers of complexity that pharmaceutical companies exploit to extend their effective market exclusivity well beyond that initial 20-year window.
The U.S. patent system, governed primarily by the Patent Act of 1952 and amended substantially by the Drug Price Competition and Patent Term Restoration Act of 1984 (the Hatch-Waxman Act), creates a specific set of interactions between pharmaceutical patents and regulatory drug approval that are different from any other industry. Those interactions are the raw material that makes evergreening possible.
Under Hatch-Waxman, a generic manufacturer seeking approval for a copy of a brand-name drug must file an Abbreviated New Drug Application (ANDA) with the FDA. As part of that filing, the generic company must certify one of four things about each patent the brand-name company has listed in the FDA’s “Orange Book”: that the patent has expired, that the patent will expire before the generic launches, that the patent is invalid, or that the patent is not infringed by the generic product.
That fourth certification, known as a Paragraph IV certification, is a declaration of war. When a generic manufacturer files a Paragraph IV certification, it is telling the world that it believes the listed patent should not stand between it and the market. The brand-name manufacturer then has 45 days to sue the generic company for patent infringement. If it does, an automatic 30-month stay takes effect, preventing the FDA from approving the generic drug while the patent litigation plays out.
This 30-month stay is one of the most powerful tools in the pharmaceutical patent arsenal. It transforms a patent dispute into a three-year delay in generic entry, during which the brand-name manufacturer continues to collect exclusivity pricing. And because brand-name manufacturers choose which patents to list in the Orange Book, they have strong incentives to list as many patents as possible, including patents of questionable validity, to trigger additional litigation and stays.
How Drug Companies Layer Patents
A pharmaceutical patent portfolio is rarely built around a single foundational patent. The typical architecture of a mature drug’s patent estate looks more like a series of concentric rings, each filed at a different time, each covering a slightly different aspect of the product.
The innermost ring is the composition of matter patent, which covers the active ingredient itself. This is the most powerful patent because it prevents anyone from making, using, or selling the molecule for any purpose without a license. Composition of matter patents on novel small molecules are relatively rare in the modern era, since most truly novel compounds were discovered and patented decades ago. But when a company does secure a composition of matter patent, it is the crown jewel of the portfolio.
The second ring covers formulations. A company might hold the original molecule patent and separately patent a tablet formulation, a liquid formulation, an extended-release formulation, a sublingual formulation, and a transdermal formulation. Each of these is a separately patentable invention under U.S. law, provided it meets the requirements of novelty, non-obviousness, and utility. Formulation patents typically expire later than the original compound patent because they are filed later in the drug development process.
The third ring covers methods of treatment. A patent claiming a method of using a compound to treat a specific disease is patentable separately from the compound itself. A drug approved for one indication might accumulate method patents covering each additional indication as it is discovered. These method patents can be filed years after the drug enters clinical trials, and they can be used to block generic competition even after the compound patent expires, if the generic product would necessarily infringe the method claims when used by patients and physicians.
The fourth ring covers manufacturing processes, metabolites, prodrugs, polymorphs, and salt forms. Polymorphs, in particular, are a significant source of secondary patents. A polymorph is a different crystalline form of the same molecule. Identical in therapeutic effect, but different in physical structure. The same compound can exist in dozens of different polymorphic forms, and each one is potentially patentable. When a generic manufacturer synthesizes the original compound using standard chemistry, it may inadvertently produce a patented polymorph and face infringement liability even though the underlying molecule is off-patent.
The fifth ring covers dosing regimens and patient populations. A patent claiming “a method of treating rheumatoid arthritis in elderly patients by administering 40mg of adalimumab every two weeks” is, legally speaking, a distinct invention from the underlying molecule, the formulation, or the general method of treating rheumatoid arthritis. Dosing patents filed late in a drug’s commercial life can extend protection years beyond the original compound.
The result of this layered architecture is what academics and policymakers call a patent thicket: a dense web of overlapping patents that any generic manufacturer must navigate before it can enter the market. Navigating that thicket is expensive. It requires filing Paragraph IV certifications against each relevant patent, triggering separate litigation for each one, and surviving validity challenges and infringement defenses simultaneously.
Part Two: Defining Evergreening and Its Mechanisms
The Core Logic of Evergreening
Evergreening is the practice of making incremental modifications to an existing drug, obtaining new patents on those modifications, and using those patents to extend market exclusivity past the expiration of the original compound patent. The term is pejorative in health economics and academic pharmacology, though the pharmaceutical industry tends to describe the same activity as “continued innovation” or “lifecycle management.”
The distinction between genuine innovation and strategic evergreening is real, but it is difficult to draw in practice. An extended-release formulation of an existing drug genuinely benefits some patients by reducing the dosing frequency and improving adherence. A new polymorph that is physically easier to manufacture might reduce production costs and ultimately benefit consumers. The problem is that these improvements can be real or trivial, and the patent system applies the same protection regardless.
What separates evergreening from legitimate secondary patenting is the strategic intent. When a company files a new formulation patent two years before the original compound patent expires, aggressively markets the new formulation to shift prescription volume from the old product to the new one, and then sues any generic manufacturer that attempts to market the old compound, the sequence of events reveals the underlying logic. The formulation change is not primarily about patient benefit. It is about resetting the exclusivity clock.
Data from the Initiative for Medicines, Access & Knowledge (I-MAK) published in 2018 found that the 12 best-selling drugs in the United States had accumulated an average of 71 patents each, with the average exclusivity period extended to 38 years beyond the original filing date [2]. The study specifically identified drugs where the majority of patent applications were filed after the original drug entered clinical development, precisely the window in which secondary patenting dominates.
DrugPatentWatch, a comprehensive database tracking pharmaceutical patent expirations and litigation, routinely documents cases in which brand-name manufacturers file new patent applications in the final years before a key exclusivity expiry, triggering fresh rounds of Orange Book listing and Paragraph IV litigation. Their data makes the pattern visible in a way that individual case studies cannot.
The Orange Book as a Strategic Instrument
The FDA’s Orange Book, formally titled “Approved Drug Products with Therapeutic Equivalence Evaluations,” is a publicly available list of approved small-molecule drugs and the patents that the brand-name manufacturers claim cover them. It is the central mechanism through which pharmaceutical patents interact with the regulatory approval process for generics.
The Orange Book was created by the Hatch-Waxman Act to facilitate generic entry by giving generic manufacturers a clear map of the patent landscape they needed to challenge. In practice, it has also become a tool for brand-name manufacturers to maximize litigation leverage.
Brand-name manufacturers self-list their patents in the Orange Book. The FDA does not verify whether listed patents actually cover the approved drug or whether the claims are valid. It simply records what the brand-name company submits. For many years, this created an environment in which companies could list patents of marginal relevance, trigger 30-month stays through Paragraph IV litigation, and then settle with the generic company on terms favorable to both parties, but adverse to consumers.
Pay-for-delay settlements, also called reverse payment settlements, are agreements in which a brand-name pharmaceutical company pays a generic manufacturer to delay entering the market. The payment is made in the context of resolving Paragraph IV litigation. The brand-name company, facing a genuine risk that its patent will be invalidated in litigation, pays the generic company more than the generic would earn from entering the market, in exchange for the generic’s agreement to stay out of the market until a specified future date.
The Federal Trade Commission estimated that pay-for-delay settlements cost American consumers approximately $3.5 billion per year in excess drug costs as of 2010 [3]. The Supreme Court addressed the practice in FTC v. Actavis (2013), holding that reverse payment settlements are subject to antitrust scrutiny under the rule of reason. The decision did not ban the practice but created meaningful legal risk for settlements in which the payment is large and inexplicable relative to ordinary litigation risk.
Evergreening Case Studies: Three Drugs, Three Strategies
AstraZeneca and Nexium: The Switch from Prilosec
AstraZeneca’s transition from omeprazole (Prilosec) to esomeprazole (Nexium) is the canonical evergreening case study. Omeprazole is a proton pump inhibitor, a drug that reduces stomach acid. It was one of the best-selling drugs in the world through the 1990s. Its U.S. patent protection was set to expire in 2001.
Esomeprazole is the S-enantiomer of omeprazole. Omeprazole is a racemic mixture, meaning it contains equal amounts of two mirror-image molecular forms (enantiomers). AstraZeneca isolated the S-enantiomer and obtained patents on it as a separate compound, a separate formulation, and as a method of treating gastroesophageal reflux disease. Nexium launched in 2001, just as Prilosec’s generics were entering the market.
The science behind the switch was thin. Studies comparing esomeprazole to omeprazole showed minimal clinical differences in the primary endpoint of acid suppression. AstraZeneca’s own clinical data submitted to the FDA showed marginal benefit at best. But the patent strategy was effective. AstraZeneca spent more than $500 million marketing Nexium directly to consumers, successfully shifting prescription volume from an off-patent product to a newly patented one. By 2003, Nexium was the second-best-selling drug in the United States.
This strategy is sometimes called a “chiral switch.” It is not inherently fraudulent, and there are cases in which isolating a single enantiomer produces genuine clinical benefits, such as with escitalopram (Lexapro) versus citalopram (Celexa). But in the case of Nexium, the primary driver was IP management, not patient benefit.
Pfizer and Neurontin: Method Patent Enforcement
Gabapentin (Neurontin) presents a different model. The compound patent on gabapentin expired in 2004, opening the market to generic competitors. But Pfizer held method-of-use patents covering gabapentin for the treatment of post-herpetic neuralgia, one of the drug’s FDA-approved uses. These method patents were listed in the Orange Book.
When generic manufacturers entered the market after the compound patent expired, Pfizer sued them for infringement of the method patents. Pfizer argued that even though the generics could legally make and sell gabapentin for other uses, they were liable for “induced infringement” because they knew that physicians would prescribe the generic for post-herpetic neuralgia, infringing the method patent.
The litigation raised a question that remains unsettled today: can a method-of-use patent on a listed indication effectively block generic entry for the entire drug, even after the compound patent has expired? Federal courts have given inconsistent answers over the years, and the legal risk of method patent enforcement continues to cloud the economics of generic entry for drugs with off-label use patterns that overlap with patented indications.
Celgene and Thalomid: REMS as a Barrier to Generic Entry
Thalidomide’s reintroduction as Thalomid for multiple myeloma required the FDA to impose a Risk Evaluation and Mitigation Strategy (REMS) because of the drug’s catastrophic teratogenicity. The REMS required extensive controls on distribution and monitoring to prevent fetal exposure.
Celgene used the REMS system, which was not designed as an IP mechanism, as a barrier to generic entry. To file an ANDA for a generic version of Thalomid, a generic manufacturer needs samples of the brand-name drug to conduct bioequivalence testing. Celgene refused to sell those samples to generic manufacturers, citing the REMS program’s distribution controls.
The Federal Trade Commission sued Celgene in 2019 for using the REMS program anticompetitively [4]. The case settled with Celgene agreeing to changes in its sample-sharing practices. Congress subsequently passed the CREATES Act in 2019, which created a private right of action for generic manufacturers denied access to samples necessary for bioequivalence testing, specifically to address the REMS manipulation problem.
The Celgene case illustrates how evergreening and patent thicket strategies are not limited to the patent system itself. Regulatory mechanisms designed for legitimate safety purposes can be repurposed as competitive barriers, and the legal and regulatory systems have been slow to close these gaps.
Part Three: The Regulatory Framework and Its Gaps
Hatch-Waxman at 40: The Architecture of a Compromise
The Drug Price Competition and Patent Term Restoration Act of 1984 was a genuine bargain between two competing interests. Generic drug manufacturers wanted a faster pathway to market, one that did not require them to repeat all the safety and efficacy testing that the brand-name company had already done. Brand-name manufacturers wanted compensation for the years of patent protection consumed by the regulatory review process, which typically ate 8 to 10 years of a 20-year patent term before a drug could reach the market.
Hatch-Waxman gave the generic industry the abbreviated approval pathway they wanted and gave brand-name manufacturers two things: patent term restoration (adding back up to five years of patent protection to compensate for regulatory review time) and data exclusivity (preventing generic companies from relying on the brand-name company’s clinical data for a fixed period after approval).
Data exclusivity is distinct from patent protection. A company can hold data exclusivity on a drug with no patents, or patents and data exclusivity can run concurrently. For small-molecule drugs, basic data exclusivity runs five years from approval, and can be extended to seven and a half years with certain types of supplemental applications. For new clinical investigations, three years of data exclusivity applies to the specific condition studied in the new trials.
The patent term restoration provision is capped at a maximum of five years of added protection, and the total patent term including the extension cannot exceed 14 years from the date of approval. These provisions were designed to be balanced, but the accumulation of secondary patents that was not contemplated in 1984 has skewed the balance substantially toward brand-name manufacturers.
The Biologics Price Competition and Innovation Act of 2009 (BPCIA), which created the pathway for biosimilar approval, established a parallel but distinct framework with even longer exclusivity periods: 12 years of data exclusivity from the date of first approval of the reference biologic, plus one year of market exclusivity for the first approved interchangeable biosimilar. The BPCIA’s patent dance provisions, which govern how the brand-name biologic manufacturer and the biosimilar applicant exchange patent information and engage in litigation, are notoriously complex and have generated their own body of evergreening concerns distinct from the small-molecule context.
The Orange Book Reform Debate
The Orange Book listing system has been a target for reform for decades. Critics argue that the system allows brand-name manufacturers to list patents of questionable validity and relevance, triggering 30-month stays on generic approval with minimal regulatory scrutiny.
The FDA has taken steps to tighten Orange Book listing standards over time. A 2021 proposed rulemaking would have required brand-name manufacturers to provide more detailed certifications about the relevance of listed patents to the specific approved drug. The FDA finalized rules in 2024 requiring additional specificity in patent declarations and creating an expedited process for challenging improper listings.
The Federal Trade Commission has used its authority under Section 5 of the FTC Act to challenge improper Orange Book listings as unfair methods of competition. In October 2023, the FTC announced challenges to dozens of Orange Book listings that it alleged were improper, targeting listings where the patent did not appear to claim the approved drug product [5]. This marked the most aggressive use of FTC authority against the Orange Book since the agency’s policy statement on the issue in 2003.
The FTC’s renewed activism reflects a broader shift in how regulators and antitrust enforcers view pharmaceutical patent practices. The Biden administration’s July 2021 executive order on competition in the American economy specifically directed the FTC to take action on pharmaceutical patent and pricing practices, signaling political and institutional support for more aggressive enforcement.
The Patent Trial and Appeal Board as a Challenger’s Tool
The America Invents Act of 2011 created the Patent Trial and Appeal Board (PTAB), an administrative tribunal within the USPTO that hears challenges to issued patents through two main proceedings: inter partes review (IPR) and post-grant review (PGR).
PTAB proceedings have become a major tool for generic manufacturers, payers, and patient advocacy organizations to challenge secondary pharmaceutical patents. IPR is limited to prior art challenges based on patents and printed publications. PGR is broader, available in the first nine months after a patent’s grant, and allows challenges on any ground of invalidity.
The economics of PTAB proceedings favor challengers in some respects. The cost of an IPR petition is a fraction of full patent litigation, often $300,000 to $700,000 compared to $3 million or more for a district court case through trial. And the burden of proof at PTAB is lower: a challenger needs to show unpatentability by a preponderance of the evidence, rather than the clear and convincing evidence standard that applies in district court.
Pharmaceutical patent owners have responded to the PTAB threat by challenging the constitutionality of IPR proceedings, seeking to limit the scope of PTAB review through claim construction arguments, and supporting legislative efforts to insulate pharmaceutical patents from PTAB review. The Coalition for Affordable Prescriptions (CAPE), which is funded by generic manufacturers, has filed hundreds of IPR petitions against secondary pharmaceutical patents, with mixed success. Some secondary patents have been canceled, some have been upheld, and many cases have settled on terms that accelerated generic entry.
A 2020 analysis published in the Journal of Law and the Biosciences found that IPR petitions against pharmaceutical patents had a patent cancellation rate of approximately 37%, compared to an overall cancellation rate of about 60% for all IPR petitions [6]. The lower cancellation rate in pharmaceuticals may reflect the higher quality of pharmaceutical patents on average, or it may reflect the sophistication of pharmaceutical patent prosecution teams, or both. The data does not clearly answer the question, but it does suggest that PTAB is a useful tool rather than a silver bullet for patent thicket challenges.
Part Four: The Mechanics of Pediatric Exclusivity and Other Non-Patent Extensions
Pediatric Exclusivity as a Commercial Tool
The Best Pharmaceuticals for Children Act (BPCA), originally enacted in 1997 and made permanent in 2012, grants brand-name pharmaceutical manufacturers an additional six months of market exclusivity in exchange for conducting and submitting pediatric studies of their drug. The exclusivity applies as a “tack-on” to any existing patent or exclusivity period protecting the drug, meaning it extends every listed patent and every applicable exclusivity period by six months.
In theory, pediatric exclusivity rewards companies for generating data in a population that is chronically understudied in pharmaceutical research. Children metabolize drugs differently from adults, and extrapolating adult dosing to pediatric patients can produce both ineffective and dangerous outcomes. The policy intent is legitimate.
In practice, pediatric exclusivity has become one of the more reliable mechanisms for extending the commercial life of high-revenue adult drugs. The FDA issues written requests for pediatric studies based on public health need, but companies can also voluntarily request to be issued a written request for a drug they want to study. The financial calculation is straightforward: six months of additional exclusivity on a drug generating $3 billion annually in U.S. revenues is worth approximately $1.5 billion. The cost of conducting a pediatric study rarely exceeds $20 million to $40 million.
AbbVie received pediatric exclusivity for adalimumab in 2012, adding six months of exclusivity to each relevant patent and exclusivity period. Pfizer received pediatric exclusivity for sildenafil (Viagra) in 2014, extending exclusivity on a drug that generates billions annually in revenue. In both cases, the pediatric studies produced some useful dosing data for children with specific conditions, but the primary commercial value of the exclusivity extension was in the adult markets the drugs primarily served.
The Federal Trade Commission has criticized the pediatric exclusivity program as a mechanism for exclusivity extension that provides outsized economic returns relative to the public health value generated. A 2002 GAO report found that approximately one-quarter of pediatric exclusivity extensions went to drugs for which there was no significant pediatric indication, meaning the six-month extension applied entirely to adult uses [20]. The structure of the law requires the FDA to grant the exclusivity if the sponsor submits acceptable pediatric studies in response to a written request, regardless of whether the pediatric indication turns out to be commercially relevant.
Orphan Drug Exclusivity and Its Commercial Exploitation
The Orphan Drug Act of 1983 was designed to incentivize development of drugs for rare diseases, defined as conditions affecting fewer than 200,000 Americans. Drugs that receive orphan drug designation from the FDA qualify for seven years of market exclusivity from approval, separate from and potentially longer than patent protection. They also qualify for a 50% tax credit on qualifying clinical trial costs and expedited FDA review.
The economic logic was that drugs for rare diseases served small markets that would not attract private investment without additional incentives. A seven-year exclusivity period, even without patent protection, could provide enough of a returns window to justify the investment in orphan drug development.
The orphan drug program has become one of the pharmaceutical industry’s favorite tools for generating high-value exclusivity on drugs that serve patient populations much larger than the rare disease definition implies. Companies have pursued orphan designation for specific subpopulations of common diseases, obtained seven-year exclusivity for treating that narrow population, and then prescribed the drug broadly for the wider disease, charging orphan drug prices for a product that serves millions of patients.
Gleevec (imatinib) is the paradigmatic example. Novartis obtained orphan drug designation for imatinib in chronic myelogenous leukemia, which qualified as a rare disease at the time of designation. The drug subsequently proved effective in multiple malignancies and has been prescribed to hundreds of thousands of patients worldwide. The initial orphan exclusivity gave Novartis pricing power that it maintained through subsequent compound and formulation patents, charging prices that made the drug unaffordable in many countries without price controls.
The FDA has tightened orphan designation standards since 2017, requiring companies to provide more detailed evidence that the disease meets the prevalence threshold and that the indication is genuinely for the rare disease rather than a subset carved out of a common condition. But the fundamental structure of the orphan drug program, which creates seven-year exclusivity without any requirement that the drug serve an unmet medical need or that pricing be related to the cost of development, has not been reformed.
New Dosage Form and New Population Exclusivities
Beyond pediatric exclusivity and orphan exclusivity, the Hatch-Waxman Act creates several other forms of regulatory exclusivity that can extend market protection. Three years of data exclusivity applies to new clinical investigations that support a supplemental application, including applications for new formulations, new dosing regimens, new routes of administration, and new patient populations.
This three-year “clinical investigation” exclusivity is distinct from the five-year new chemical entity exclusivity. It applies to the specific new use or condition of use supported by the new clinical data, not to the drug as a whole. A generic manufacturer can still enter the market for the original approved indication while the new indication is protected by clinical investigation exclusivity.
But the three-year exclusivity, combined with Orange Book patent listings for the new formulation or indication, can create a combination of regulatory exclusivity and patent protection that effectively extends the brand-name manufacturer’s commercial advantage. If a brand-name company successfully shifts prescriptions from the original formulation to a new extended-release formulation protected by both a new patent and three-year clinical investigation exclusivity, the net effect is an exclusivity extension that generic manufacturers cannot easily work around, even after the original compound patent expires.
The compound effect of stacking pediatric exclusivity, orphan drug exclusivity, clinical investigation exclusivity, patent term restoration, and secondary patents can produce total effective exclusivity periods that far exceed any single exclusivity mechanism. The individual pieces each have legitimate policy justifications. The problem is that companies can combine them in ways that the legislative drafters did not contemplate and that produce total exclusivity periods far beyond what any single mechanism was designed to provide.
Part Five: Pay-for-Delay in Depth: The Economics of Patent Settlements
The Mathematics of Anticompetitive Settlements
Pay-for-delay settlements, formally called reverse payment settlements, represent a case where the incentive structures of the pharmaceutical patent system produce outcomes that are economically rational for both parties but harmful to consumers and competition.
The logic is cleanest when seen through the lens of a basic asymmetry. A brand-name pharmaceutical manufacturer with a drug generating $3 billion per year faces the prospect of a generic manufacturer entering the market and reducing the brand-name’s revenues by 80% to 90% as generic prices erode the market. If the patent covering the drug is challenged in Paragraph IV litigation and the generic manufacturer wins, the brand-name company loses most of those revenues. If the brand-name company wins, it preserves them.
Suppose the brand-name company estimates a 50% probability that the generic manufacturer will prevail in the patent litigation. The expected value of fighting the litigation is the 50% chance of winning, preserving $3 billion per year, multiplied by however many years of exclusivity remain. The expected value of settling is whatever terms the parties can negotiate.
If the brand-name company pays the generic manufacturer $500 million to delay entry for five years, both parties are better off than they would be in expected value terms if the litigation proceeded. The brand-name company pays $500 million but preserves five additional years of exclusivity worth, say, $7 billion in present value. The generic manufacturer receives $500 million without having to invest in manufacturing, marketing, or litigation risk. The loser in this transaction is the consumer, who pays brand-name prices for five years longer than they would in competitive equilibrium.
This is the transaction that the FTC argued should be presumptively anticompetitive, and that the Supreme Court addressed in FTC v. Actavis in 2013. The Court’s rule of reason standard requires courts to balance the anticompetitive harm against any procompetitive justification, without a presumption either way. The practical effect of Actavis has been to subject large settlements to antitrust scrutiny, but not to eliminate them, since many settlements can be justified under rule of reason analysis.
Post-Actavis Trends in Settlement Structures
After the Actavis decision, pharmaceutical companies adapted their settlement structures to reduce antitrust exposure. Pure cash payments from brand-name to generic manufacturers became less common. More sophisticated deal structures emerged, including: “no-authorized generic” commitments, in which the brand-name company agrees not to market its own authorized generic when the first generic enters the market, which is valuable to the first generic manufacturer because it gives the generic a temporary monopoly; supply agreements under which the brand-name company buys product from the generic manufacturer at profitable terms; and licensing agreements that give the generic manufacturer early entry into some markets but not others.
The “no-authorized generic” commitment is particularly effective as an indirect payment. When the first generic enters the market after a patent litigation settlement, it typically faces competition from an authorized generic that the brand-name company markets itself or licenses to another party. This authorized generic competition reduces the first generic’s profits. A commitment not to launch an authorized generic for a period is therefore worth substantial money to the settling generic manufacturer, and it achieves the same economic effect as a direct cash payment without the same antitrust visibility.
The FTC has argued that no-authorized generic commitments should be treated as reverse payments under Actavis because they transfer economic value from brand-name to generic in exchange for delayed market entry. Several circuit courts have agreed, treating non-cash transfers as analytically equivalent to cash payments when valuing reverse payments for antitrust analysis. The precise legal standards for non-cash reverse payments remain contested across different federal circuits.
A 2023 FTC report on branded pharmaceutical manufacturers’ settlements with potential generic entrants found that the total number of settlements involving potential pay-for-delay remained elevated compared to pre-Actavis levels, though the proportion involving large cash payments had declined [21]. The data suggests that Actavis changed the form of settlements more than it eliminated their anticompetitive potential.
Part Six: The Polymorph Problem in Detail
Why Crystal Structure Is Patentable
Polymorphism, the ability of a solid material to exist in more than one crystal form, is a ubiquitous property of pharmaceutical compounds. Different polymorphs of the same compound have the same molecular formula and the same chemical bonds, but their atoms arrange themselves differently in the crystal lattice. These structural differences can affect physical properties including solubility, melting point, stability, and ease of manufacture.
The legal framework for patenting polymorphs follows from the basic patent law principle that a new form of an existing compound, if it meets the novelty, non-obviousness, and utility requirements, is patentable. U.S. patent law does not require that a new form be therapeutically superior to an existing form. The question is only whether the polymorph is structurally distinct (novel) and whether discovering it required inventive effort rather than routine experimentation (non-obvious).
The practical problem with pharmaceutical polymorph patents is that many pharmaceutical compounds have dozens of known polymorphs, and systematic screening of polymorphic forms has become a standard part of pharmaceutical development. When generic manufacturers attempt to synthesize an off-patent compound using standard chemistry, they may inadvertently produce a polymorph that is covered by a later-filed secondary patent, even though the underlying compound itself is clearly in the public domain.
This was precisely the situation with ranitidine, the active ingredient in Zantac. The compound patent on ranitidine expired in 1995. But a polymorph patent on a specific crystalline form of ranitidine, “Form 2,” had been filed years after the original compound patent and was set to expire later. When generic manufacturers attempted to produce ranitidine, some found that their synthesis processes produced Form 2, potentially infringing the polymorph patent. The Form 2 patent was eventually found to be invalid in litigation, but not before creating years of additional uncertainty about generic entry.
The Ritonavir Crisis: When Polymorphism Disrupts Supply
The ritonavir case of 1998 illustrates the reverse problem: when polymorphism creates genuine technical challenges rather than strategic ones. Ritonavir (Abbott’s Norvir) was an early protease inhibitor used in HIV treatment. In 1998, Abbott discovered that a new polymorph of ritonavir, Form II, was crystallizing in the manufacturing process and in product already in commerce. Form II had much lower bioavailability than Form I, the original polymorph.
Abbott had to withdraw Norvir from the market for approximately a year while it reformulated the product in a manner stable against Form II crystallization. This was a genuine pharmaceutical crisis caused by unexpected polymorphism, the opposite of strategic polymorph patenting.
The ritonavir episode prompted the FDA to increase its attention to polymorph-related stability and bioavailability issues in drug approval and post-approval monitoring. It also made the pharmaceutical industry more systematic about polymorph screening during development. The unintended consequence is that the industry’s comprehensive polymorph screening programs, developed for legitimate technical reasons, also generate the data necessary to file broad polymorph patent portfolios.
Regulatory Guidance on Polymorph Patents
The USPTO has issued guidance on examining polymorph patent applications that is stricter than its pre-2000 standards. Under current guidelines, a polymorph patent application faces heightened obviousness scrutiny if the new polymorph was discovered by routine screening methods that are standard practice in the field. Discovering a new polymorph through comprehensive polymorph screening is generally not considered inventive if the screening method itself is routine.
Despite this guidance, many polymorph patents continue to issue through the USPTO, partly because “routine” is a contested characterization, partly because patents issued before the tightened guidelines remain in force, and partly because examiners face resource constraints that limit the depth of their prior art searches for secondary pharmaceutical applications.
The EPO has been more aggressive in rejecting polymorph patent applications. The EPO’s problem-and-solution approach to inventive step requires that the claimed polymorph solve a technical problem, such as improved stability or processability, in a non-obvious way. Simply discovering a new polymorph that has the same therapeutic properties as a known form does not satisfy the EPO’s inventive step requirement. This difference in examination standards means that polymorph patents that are valid in the United States are often rejected or invalid in Europe, contributing to the jurisdictional variation in effective exclusivity periods.
Part Seven (Extended): International Patent Landscapes in Comparative Detail
Canada’s Linkage Regulation and Its Limits
Canada’s pharmaceutical patent system includes a patent linkage mechanism analogous to Hatch-Waxman, known as the Patented Medicines (Notice of Compliance) Regulations. Under this system, brand-name manufacturers can list patents on Health Canada’s Patent Register, and generic manufacturers must address those patents before receiving regulatory approval.
Canada reformed its linkage regulations substantially in 2017 in connection with the Canada-European Union Comprehensive Economic and Trade Agreement (CETA). The reforms introduced an eight-year data protection period for new pharmaceutical products (extended to 10 years for pediatric studies), more stringent rules for patent listing, and new IP protections that Canadian public health advocates argued tilted the system further toward brand-name manufacturers.
The Canadian reforms illustrate a broader dynamic in which bilateral and multilateral trade agreements push countries toward higher pharmaceutical IP standards than they would choose independently. CETA, the Trans-Pacific Partnership (TPP, now the Comprehensive and Progressive Agreement for Trans-Pacific Partnership), and the United States-Mexico-Canada Agreement (USMCA) all include pharmaceutical IP provisions negotiated partly under pressure from U.S. trade representatives acting on behalf of U.S. pharmaceutical industry interests.
Japan’s Patent System and Incremental Innovation
Japan has traditionally been more receptive to secondary pharmaceutical patents than Europe. The Japan Patent Office (JPO) applies a lower non-obviousness standard for pharmaceutical compounds than the EPO, and Japan grants patents for new uses, dosing regimens, and patient populations relatively readily. Japan also provides data exclusivity of eight years for new drugs and six years for new dosage forms, supplemented by re-examination periods that provide additional protection against generic entry.
Japanese pharmaceutical companies have historically relied heavily on secondary patents and incremental product modifications. The practice of creating “me-too” drugs, slightly modified versions of existing compounds, has been more prevalent in Japan than in the United States or Europe. Critics have argued that the JPO’s permissive standards for secondary pharmaceutical patents discourage development of truly novel drugs, since the returns to incremental patenting can be captured with lower investment.
Japan has been reforming its pharmaceutical IP system to encourage more genuine innovation, partly in response to international pressure and partly in response to domestic concerns about the competitiveness of Japanese pharmaceutical companies in global markets. But the transition from a culture of incremental patenting to one focused on breakthrough innovation is slow and reflects deep-seated industry structures.
The TRIPS Flexibilities That Developing Countries Rarely Use
TRIPS Article 31 permits compulsory licensing of patented inventions without the patent holder’s consent under specified conditions, including national emergencies, other circumstances of extreme urgency, or cases of anti-competitive practices. In the pharmaceutical context, compulsory licensing is theoretically available as a tool to ensure access to needed medicines when patent holders refuse to license on reasonable terms.
In practice, compulsory licensing has been used by developing countries far less often than the legal framework would permit, for reasons that include diplomatic pressure from developed countries, bilateral trade agreement provisions that constrain or penalize compulsory licensing, limited technical and administrative capacity to operationalize compulsory licenses, and the difficulty of manufacturing complex pharmaceutical products domestically.
Thailand issued compulsory licenses for three HIV/AIDS drugs in 2006 and 2007, triggering significant pressure from the United States and the European Union. India has issued compulsory licenses sparingly, including a notable 2012 license for sorafenib (Nexavar), a cancer drug that Bayer was selling at prices inaccessible to most Indian patients. Brazil, Ecuador, and a handful of other middle-income countries have issued compulsory licenses in specific cases.
The Doha Declaration of 2001 affirmed the right to issue compulsory licenses, and the 2003 TRIPS decision and subsequent 2005 amendment created a mechanism allowing countries without manufacturing capacity to import compulsorily licensed drugs. But administrative complexity has limited the use of this mechanism. Between 2003 and 2020, only one country, Rwanda, successfully imported a drug through the mechanism, and the transaction took several years to complete.
The gap between the legal framework for flexibilities and the actual use of those flexibilities means that the practical access problem in developing countries remains severe, despite nominal legal solutions.
Part Eight (Extended): Antitrust Law and Patent Thickets
Walker Process Claims and Fraudulent Patent Procurement
Beyond the Actavis framework for pay-for-delay settlements, antitrust law provides another avenue for challenging patent thickets through “Walker Process” claims. Walker Process Equipment, Inc. v. Food Machinery & Chemical Corp. (1965) established that a company can be liable under Section 2 of the Sherman Act for attempting to enforce a patent that was obtained through inequitable conduct, i.e., fraud on the patent office.
Inequitable conduct occurs when a patent applicant fails to disclose material information to the USPTO during patent prosecution, with intent to deceive. In the pharmaceutical context, a company that obtains a secondary patent by failing to disclose prior art that would have led the examiner to reject the application, and then enforces that patent to block generic entry, may face both Walker Process antitrust liability and unenforceability of the patent.
Walker Process claims have been raised in several pharmaceutical patent cases but have rarely succeeded, because the intent to deceive requirement is difficult to prove. Courts require clear and convincing evidence of a specific intent to deceive the patent office, rather than mere negligent failure to disclose. Pharmaceutical companies employ experienced patent prosecution counsel who are aware of this standard and who structure prosecution to minimize the risk of inequitable conduct findings.
The Federal Circuit significantly constrained inequitable conduct doctrine in Therasense, Inc. v. Becton, Dickinson & Co. (2011), holding that inequitable conduct requires proof of both materiality and specific intent to deceive, and tightening the materiality standard. After Therasense, Walker Process claims became even more difficult to sustain, and some commentators have argued that the heightened standard has made it practically impossible to challenge even egregious prosecution conduct through this mechanism.
Sham Litigation and the Noerr-Pennington Doctrine
Pharmaceutical patent holders who file Paragraph IV infringement suits are generally protected from antitrust liability under the Noerr-Pennington doctrine, which holds that petitioning the government, including through litigation, is constitutionally protected activity exempt from antitrust scrutiny. A brand-name company that files suit against a generic manufacturer under the 45-day window cannot ordinarily be sued for antitrust violations simply because it filed the suit.
The exception to Noerr-Pennington protection is “sham litigation”: a lawsuit that is objectively baseless (no reasonable litigant could realistically expect to prevail) and subjectively motivated by desire to use the litigation process as an anticompetitive tool. In Professional Real Estate Investors v. Columbia Pictures (1993), the Supreme Court established a two-part test for sham litigation that courts have applied in the pharmaceutical context.
Sham litigation claims are rarely successful in the Paragraph IV context for two reasons. First, a brand-name company that wins a Paragraph IV case (i.e., the patent is found valid and infringed) defeats the claim retrospectively, since a prevailing litigant’s lawsuit cannot have been objectively baseless. Second, even if the brand-name company loses, demonstrating that no reasonable litigant could have expected to prevail requires a high level of legal clarity about the invalidity of the asserted patent, a clarity that is rarely present before litigation concludes.
The combination of Noerr-Pennington immunity, the Walker Process intent requirement, and the Actavis rule of reason standard creates a legal environment in which pharmaceutical patent thicket strategies face antitrust risk primarily for clear reverse payment settlements, not for the underlying patent prosecution and litigation tactics.
State Attorneys General as an Emerging Force
State attorneys general have become an increasingly active force in pharmaceutical patent antitrust enforcement. State AGs have authority to bring antitrust claims under both federal antitrust law (in their role as parens patriae for their citizens) and state antitrust law. They are not subject to the same resource and institutional constraints that have limited federal enforcement in some periods.
A multistate coalition of 46 state attorneys general filed suit in 2019 against several generic pharmaceutical manufacturers, alleging price-fixing and bid-rigging in the generic drug market. While this case targets the generic side of the market rather than brand-name patent thicket strategies, it illustrates the capacity and willingness of state AGs to take on pharmaceutical competition cases.
California’s AG has brought independent antitrust actions against pharmaceutical pay-for-delay settlements under California’s Cartwright Act, which applies a different and in some respects broader standard for anticompetitive conduct than federal antitrust law. Several other states have filed or are considering similar actions. The decentralized nature of state AG enforcement creates the possibility that pay-for-delay settlements challenged under federal law in a particular circuit can face state law challenges that apply standards more favorable to plaintiffs.
Part Nine (Extended): The Investment and Payer Perspective
How Patent Cliffs Drive Pharmaceutical M&A
For pharmaceutical investors and financial analysts, the patent cliff, the period when a major drug’s key exclusivity protections expire and generic entry dramatically reduces revenues, is one of the most important events in a company’s commercial lifecycle. Understanding the true shape of a drug’s patent portfolio, not just the nominal expiry of the compound patent, is essential for accurate financial modeling.
The gap between nominal and effective patent expiry is substantial for many major drugs. A company whose compound patent expires in three years but whose secondary patent portfolio runs for eight years faces a different financial trajectory than a company whose entire patent portfolio expires in three years. The secondary patents create legal risk and litigation costs for generic manufacturers that translate into delayed entry and preserved revenues.
Patent cliff analysis, as practiced by pharmaceutical equity analysts, now routinely incorporates secondary patent assessment. Analysts track Orange Book listings, PTAB petition activity, district court litigation timelines, and settlement patterns to build probabilistic models of generic entry timing. DrugPatentWatch’s patent expiry tracking data feeds directly into these models, providing the raw data on patent portfolios and litigation status that supports more accurate financial forecasting.
The strategic logic of pharmaceutical M&A is also deeply connected to patent portfolio management. Acquisitions of companies with drugs approaching patent expiry can be rationalized partly by the acquirer’s ability to apply its patent prosecution resources to building a denser secondary patent portfolio around the acquired drug. Conversely, companies whose patent portfolios are genuinely thin, lacking robust secondary protection, trade at valuation discounts relative to peers with more durable exclusivity profiles.
How Pharmacy Benefit Managers and Payers Respond
Pharmacy benefit managers (PBMs) and health plans have developed formulary management strategies specifically designed to account for the timing of generic entry. These strategies involve monitoring patent expiry timelines, preparing formulary tier changes in advance of generic entry, and in some cases negotiating with brand-name manufacturers for rebates that are partly conditioned on the payer’s agreement not to shift formulary coverage upon generic entry.
The last category, rebate agreements that implicitly compensate brand-name manufacturers for the payer’s restraint in formulary management, functions economically as a form of pay-for-delay in the third-party payer context. A payer that accepts a large rebate on a brand-name drug in exchange for maintaining the brand-name’s preferred formulary tier may be deferring the cost reductions associated with formulary-driven generic substitution.
PBMs have been under increasing scrutiny from regulators and legislators for their own pricing and contracting practices, and the complexity of pharmaceutical distribution economics makes it difficult to assess how much payer contracting behavior reflects genuine cost management versus tacit collusion with manufacturers. The point is that patent thicket effects are amplified or mitigated by formulary management decisions, and the full economic picture of delayed generic entry cannot be assessed without considering how payers and PBMs manage the transition from brand to generic.
The Institutional Investor Perspective
From the perspective of institutional pharmaceutical investors, secondary patenting and lifecycle management strategies are generally value-accretive in the short and medium term. A more durable exclusivity profile supports higher revenue forecasts, lower uncertainty about the timing of revenue decline, and better financial modeling. ESG considerations are beginning to change this calculus at the margin, with some institutional investors incorporating access-to-medicines frameworks into their pharmaceutical company assessments.
The Access to Medicine Foundation publishes biannual rankings of major pharmaceutical companies on metrics including their IP strategies in developing countries, their licensing practices, and their approach to patent enforcement. Companies with aggressive secondary patenting strategies in low- and middle-income countries rank lower on access metrics, and some institutional investors have incorporated these rankings into their ESG scoring frameworks.
The financial materiality of ESG risk in pharmaceutical investing is still debated. But as Medicare drug price negotiation, state pricing boards, and international reference pricing create downward pressure on pharmaceutical revenues in markets where the patent thicket strategy has historically generated the most value, the financial case for aggressive secondary patenting strategies is becoming more nuanced.
Who Pays and How Much
The economic consequences of patent thickets fall disproportionately on patients without pharmaceutical insurance, government payers, and private insurers who bear the cost of brand-name drug prices in markets where generics would otherwise be available.
The standard economic model of generic drug entry predicts that the entry of a single generic competitor reduces prices by approximately 20% to 30%, and that the entry of multiple generic competitors can drive prices down to 10% to 20% of the brand-name price within 18 months [7]. These price reductions do not occur during the years of delayed entry that patent thickets create.
A 2020 study in JAMA Network Open estimated that U.S. consumers and payers spent an additional $4.7 billion on branded versions of drugs in 2018 due to delayed generic entry attributable to patent thicket litigation [8]. This estimate is conservative in methodology and almost certainly understates the total impact, because it considers only drugs that were actively involved in Paragraph IV litigation during the study period.
The social welfare consequences are not uniformly distributed. Low-income patients who lack comprehensive drug coverage, and who cannot afford brand-name prices, often go without medication during periods of delayed generic entry. This is a direct health consequence, not merely an economic one. Studies of statin access, for instance, have found measurable differences in cardiovascular outcomes in patient populations that have lower rates of medication adherence attributable to cost barriers [9].
The Biosimilar Market: Evergreening at Scale
The economics of delayed biologic entry are more severe than in the small-molecule space, for two reasons. First, biologics are substantially more expensive than small-molecule drugs, meaning the per-unit excess cost during exclusivity is larger. Second, the patent thickets around biologics tend to be denser, because biologics are more complex molecules that generate more opportunities for secondary patenting around manufacturing processes, formulations, and cell culture methods.
The Humira biosimilar situation is the most visible example, but it is not unique. Data from the Association for Accessible Medicines shows that by 2022, 10 biologics with aggregate annual sales of more than $80 billion had biosimilars approved by the FDA but not yet on the market, largely due to ongoing patent disputes [10]. The gap between FDA approval and market entry in the biosimilar context averages approximately three years, compared to less than one year for small-molecule generics.
AbbVie’s Humira patent portfolio illustrates the density that is achievable with sustained effort. Between 1991 and 2021, AbbVie and its predecessors filed more than 250 patents related to adalimumab. Of these, fewer than 20 covered the core antibody molecule and its basic formulation. The remaining 230-plus patents covered dosing syringes, pre-filled pens, manufacturing cell lines, purification methods, specific patient populations, specific co-administration protocols, and other secondary aspects of the product. <blockquote> “AbbVie’s patent strategy around Humira is not exceptional in its type, only in its scale. The company applied to every technology that touches the drug and filed in every jurisdiction that would grant a patent. What is unusual is how transparent the strategy became once biosimilar developers started litigating.” – Robin Feldman, Professor of Law, UC College of the Law San Francisco, speaking to a Senate Judiciary subcommittee in 2021 [11] </blockquote>
The settlement agreements that AbbVie reached with biosimilar developers in the United States allowed biosimilar entry in 2023 but prohibited entry in Europe significantly earlier, in 2018. European patients have had access to biosimilar adalimumab for five years that U.S. patients did not. This discrepancy reflects differences in patent law across jurisdictions, but it also reflects the specific terms of settlement agreements that AbbVie negotiated with individual biosimilar developers, terms that included commitments not to challenge certain patents in exchange for licenses to enter specified markets at specified times.
The Market Exclusivity Arithmetic
The actual effective market exclusivity period for a drug can be calculated by combining all the sources of exclusivity that apply to it: the compound patent, patent term restoration, data exclusivity, and any additional protection created by secondary patents.
For a typical small-molecule drug developed in the 2000s and 2010s, the arithmetic looks roughly like this: the original compound patent has a nominal 20-year term from filing. Clinical development and regulatory review consume roughly 10 to 12 years from the initial filing date. Patent term restoration adds back up to five years. Basic data exclusivity adds five years from approval (running concurrently with the restored patent term in most cases). The total statutory protection period from approval is therefore approximately 10 to 14 years in the absence of secondary patents.
Secondary patents extend this period by creating new litigation risk for any generic manufacturer that attempts to enter the market. The practical effect of a secondary patent is not necessarily to give the brand-name manufacturer legal rights that would be enforceable to completion, since many secondary patents are of questionable validity. The practical effect is to make entry economically risky for a generic manufacturer, since a wrong bet on a court ruling can mean infringement liability, injunctions, and treble damages.
This risk economics explains why some Paragraph IV certifications against clearly weak patents result in no litigation at all from the brand-name manufacturer. The brand-name company may not believe its secondary patent is valid enough to withstand challenge, and filing suit when the patent is likely to be invalidated creates the risk of adverse precedent. The more common outcome in this scenario is that the generic company enters the market “at risk” while litigation proceeds, and the brand-name company tolerates the entry rather than seek an injunction it might lose.
Part Five: Global Perspectives and International Variation
Why the Same Drug Has Different Patent Expiries in Different Countries
The U.S. pharmaceutical patent system does not exist in isolation. A drug’s effective exclusivity period varies dramatically by country, depending on each country’s patent laws, data exclusivity rules, patent examination standards, and the specific portfolio of patents the brand-name manufacturer has sought and obtained in each jurisdiction.
The European Patent Convention creates a single examination process through the European Patent Office (EPO), but national courts enforce patents separately. The EPO has historically been more skeptical of secondary pharmaceutical patents than the USPTO, applying a stricter inventive step standard that makes it harder to patent polymorphs, enantiomers, and minor formulation changes as separate inventions. The result is that patent thickets around major drugs are generally less dense in Europe than in the United States.
The Unitary Patent system, which took effect in June 2023, creates a single patent with unitary effect across all participating EU member states. Combined with the Unified Patent Court, which hears infringement and validity disputes for unitary patents, this creates a new forum for pharmaceutical patent disputes in Europe. Early cases before the UPC have involved several pharmaceutical patents, and the court is still developing its jurisprudence. The extent to which the UPC will align with EPO’s stricter secondary patent standards or move in a different direction is an open question.
India presents the most aggressive statutory restriction on pharmaceutical secondary patenting of any major economy. Section 3(d) of India’s Patents Act, amended in 2005, prohibits the grant of patents on new forms of known substances unless those new forms show enhanced efficacy compared to the known substance. This provision was specifically designed to prevent evergreening by requiring that secondary patents demonstrate genuine therapeutic improvement, not just structural novelty.
Novartis challenged Section 3(d) at the Indian Supreme Court in 2013, arguing that imatinib mesylate (the salt form of imatinib, sold as Gleevec) was patentable as a new form of the parent compound because it showed improved bioavailability. The Court rejected the argument, holding that bioavailability is a property of the substance itself, not a measure of therapeutic efficacy, and therefore did not meet the enhanced efficacy standard of Section 3(d) [12].
The Novartis v. Union of India decision attracted intense criticism from the pharmaceutical industry and the U.S. government, which characterized it as a violation of the TRIPS Agreement. It attracted equally intense praise from public health advocates, who saw it as the most important judicial decision on access to medicines in the twenty-first century. India’s approach has not been widely replicated among other developing countries, in part because the United States includes pharmaceutical IP protections in bilateral and regional trade agreements that effectively preclude Section 3(d)-style provisions.
The TRIPS Agreement and the Doha Declaration
The Agreement on Trade-Related Aspects of Intellectual Property Rights (TRIPS), negotiated as part of the Uruguay Round that established the WTO in 1994, requires all WTO members to provide at least 20-year patent protection for pharmaceutical products. Before TRIPS, many developing countries did not recognize pharmaceutical product patents at all, allowing their domestic industries to manufacture and sell generic versions of patented drugs developed in wealthy countries.
TRIPS created a global floor for pharmaceutical patent protection, but it also included flexibilities that allow countries to issue compulsory licenses for patented drugs in certain circumstances, including national health emergencies. The Doha Declaration on TRIPS and Public Health, agreed in 2001 following the HIV/AIDS crisis in sub-Saharan Africa, affirmed that the TRIPS Agreement should be interpreted in a manner supportive of WTO members’ rights to protect public health.
Compulsory licensing has been used relatively rarely, partly because the process is complex and diplomatically costly, and partly because TRIPS-plus provisions in bilateral trade agreements negotiated by the United States and European Union impose additional IP requirements beyond the TRIPS floor, including data exclusivity requirements and patent linkage provisions that constrain the ability of developing countries to issue compulsory licenses without triggering trade sanctions.
The practical consequence is that the global regulatory framework for pharmaceutical patents tilts strongly toward brand-name manufacturer interests in high-income countries and creates complex tradeoffs between IP protection and access in low- and middle-income countries.
Part Six: The Biologic Frontier
How Biosimilar Patent Thickets Differ from Small-Molecule Thickets
The patent dynamics around biologics differ from those around small molecules in ways that make biosimilar entry more difficult and expensive than generic drug entry.
Small-molecule drugs are synthesized through defined chemical reactions that produce identical copies of the active compound. A generic version of aspirin is molecularly identical to the brand-name version. Bioequivalence testing, which shows that the generic delivers the same amount of active drug to the bloodstream at the same rate, is sufficient to establish that the generic is therapeutically equivalent.
Biologics are produced by living cells. They are proteins, antibodies, or other complex molecules whose precise three-dimensional structure, glycosylation patterns, and other characteristics depend on the specific cell line, culture conditions, and manufacturing processes used. A biosimilar is not molecularly identical to the reference biologic. It is highly similar, which is why the regulatory standard for approval is “no clinically meaningful differences” rather than pharmaceutical equivalence.
This manufacturing complexity creates a different category of patentable subject matter. Where a small-molecule drug’s patent portfolio centers on molecular structure, formulation, and method of use, a biologic’s patent portfolio can also cover the specific cell line used to produce the protein, the culture media formulation, the purification steps, the fill-finish process, the container closure system, and every other aspect of the manufacturing chain. Each of these elements is independently patentable if it meets the novelty and non-obviousness requirements.
The BPCIA patent dance, set out in 42 U.S.C. § 262(l), requires biosimilar applicants to engage in a sequential exchange of confidential patent information with the reference product sponsor before litigation can be filed. The process begins when the biosimilar applicant provides the reference product sponsor with its complete regulatory submission, an act of disclosure that has no analog in the small-molecule Hatch-Waxman system. The reference product sponsor then discloses its patent portfolio and identifies which patents it believes are infringed. The parties negotiate about which patents to litigate first. If they cannot agree, the BPCIA provides default rules.
The patent dance has generated significant litigation over procedural questions, including whether participation in the dance is mandatory (it is not, the Supreme Court held in Sandoz Inc. v. Amgen Inc. in 2017) and what notices are required before a biosimilar can launch commercially. These procedural questions have delayed biosimilar launches in ways that compound the delays caused by substantive patent disputes.
The 12-Year Exclusivity Question
The BPCIA’s 12-year data exclusivity period for reference biologics was the subject of intense lobbying during the statute’s drafting and passage. The pharmaceutical industry sought 14 years. The Obama administration initially proposed 7 years. Congress settled on 12 years.
The 12-year exclusivity period is separate from patent protection. It prevents biosimilar applicants from relying on the reference biologic’s clinical data until 12 years have elapsed from the reference biologic’s first approval. This means that even if a biosimilar developer were able to invalidate all relevant patents, it still could not obtain FDA approval for a biosimilar product within the first 12 years of the reference biologic’s commercial life. Data exclusivity operates as an absolute bar, not subject to litigation or PTAB challenge.
President Biden’s proposed American Families Plan included a reduction of the 12-year exclusivity period to 7 years, arguing that the existing period goes beyond what is necessary to incentivize biologic innovation. The proposal did not advance. The Inflation Reduction Act of 2022, while it included significant pharmaceutical pricing provisions including Medicare drug price negotiation, did not modify the BPCIA exclusivity period.
The interaction between data exclusivity and patent protection creates a layered exclusivity structure for biologics that is more complex and more durable than the analogous structure for small molecules. Data exclusivity provides a hard floor below which biosimilar entry cannot occur regardless of patent outcomes. Secondary patents then extend protection above that floor. The combination produces effective exclusivity periods that regularly exceed 20 years from drug approval for major biologics.
Part Seven: Legislative and Policy Responses
The Inflation Reduction Act’s Drug Pricing Provisions
The Inflation Reduction Act of 2022 included the most significant changes to pharmaceutical pricing policy in the United States since Medicare Part D in 2003. While the IRA’s primary mechanism, Medicare drug price negotiation, is not directly an IP policy, it interacts with pharmaceutical patent strategy in ways that will affect how companies think about their patent portfolios going forward.
Under the IRA, Medicare can negotiate prices for a defined set of high-cost drugs. Small-molecule drugs become eligible for negotiation nine years after their FDA approval. Biologics become eligible 13 years after their approval. Drugs that are selected for negotiation face a negotiated maximum fair price that is lower than the existing Medicare price.
The differential treatment of small molecules versus biologics in the IRA replicates the differential data exclusivity periods and creates a perverse incentive: pharmaceutical companies developing new drugs may prefer biologic drug platforms over small-molecule platforms partly because the longer negotiation delay gives them more years of unencumbered pricing. This incentive effect was noted by health economists before the IRA’s passage and has not been resolved by any subsequent legislation.
The IRA also imposed inflation rebate requirements, requiring pharmaceutical companies to pay rebates to Medicare if they raise drug prices faster than inflation. This provision creates independent pressure on the pricing strategies that often accompany patent thicket maintenance, since companies that use exclusivity to hold prices high face financial penalties if they raise those prices further once a drug is subject to the inflation rebate requirement.
The Patent Eligibility Restoration Act and Its Implications
Patent eligibility for pharmaceutical inventions has been shaped by a series of Supreme Court decisions that restricted the types of subject matter that qualify for patent protection. Mayo Collaborative Services v. Prometheus Laboratories (2012) and Association for Molecular Pathology v. Myriad Genetics (2013) held that laws of nature, natural phenomena, and abstract ideas are not patentable, and that claims drawn too closely to these categories do not satisfy the subject matter eligibility requirement of 35 U.S.C. § 101.
These decisions created uncertainty about the patent eligibility of claims covering methods of treatment, diagnostic methods, and certain biotechnology inventions. The pharmaceutical industry has argued that this uncertainty discourages investment in diagnostic and personalized medicine innovation.
Bipartisan legislation to reform § 101, known at various stages as the Patent Eligibility Restoration Act, has been proposed but not enacted as of mid-2025. The legislation would substantially expand the scope of patentable subject matter, including by removing the categorical exclusions for laws of nature and natural phenomena. Critics, including some academic researchers and public health advocates, have argued that the legislation would expand the scope of pharmaceutical patenting beyond what is needed to incentivize innovation and would make it easier to patent incremental improvements that currently struggle to meet the eligibility standard.
State-Level Policy Responses
In the absence of comprehensive federal reform, some states have enacted their own policies targeting pharmaceutical patent practices. California enacted SB 1364 in 2022, requiring pharmaceutical manufacturers to give the state’s Medicaid program advance notice of new drug price increases and to disclose information about patent status and upcoming patent expirations. While this is primarily a price transparency measure, it creates public accountability for the relationship between patent strategy and pricing.
Maryland, Maine, and several other states have enacted or are considering prescription drug affordability boards with authority to set upper payment limits for certain high-cost drugs. These boards operate independently of patent status, which means they can set price limits even during a drug’s patent-protected period. The constitutionality of state drug pricing limits is contested, and several states have faced legal challenges from pharmaceutical industry groups.
The most direct state-level challenge to pharmaceutical patent practices came in a 2021 decision from the California Court of Appeal, which held that a pay-for-delay settlement involving a California health plan’s drug coverage could be challenged under California’s antitrust law even after the parties had argued that the FTC’s federal oversight was exclusive. The decision opened the door for state-level antitrust actions against patent settlements, supplementing the federal enforcement framework established by FTC v. Actavis.
Part Eight: The Innovation Counterargument
What the Industry Actually Spends
Any honest assessment of pharmaceutical patent thickets must grapple with the industry’s central counterargument: that extensive patent protection is necessary to fund the research and development that produces new medicines in the first place. The argument is not wrong, but it is often overstated and selectively applied.
The average cost to bring a new drug from discovery through FDA approval is a subject of genuine empirical dispute. The most widely cited estimate, produced by Tufts University’s Center for the Study of Drug Development, put the figure at $2.6 billion in 2014 [13]. Critics of this estimate, including Public Citizen and Doctors Without Borders, have argued that it substantially overstates actual R&D spending by using a capital cost methodology that counts the opportunity cost of invested capital rather than just cash expenditure, and by focusing on the most expensive class of drug development projects.
DiMasi, Hansen, and Grabowski, the researchers behind the Tufts estimate, have consistently defended their methodology, and versions of the estimate have been published in peer-reviewed journals. Alternative estimates from I-MAK and others put the out-of-pocket cash cost of drug development at approximately $648 million per approved drug, a figure that is more than double the industry-reported R&D spending per approved drug if you exclude failed compounds but substantially less than the Tufts figure [14].
The specific claim that secondary patents are necessary to fund innovation is the weakest part of the industry’s argument. Secondary patents on polymorphs, formulations, and dosing devices do not fund the discovery of new therapeutic compounds. They fund the legal and regulatory work necessary to obtain and enforce the secondary patents. The capital that would otherwise flow to generic competitors instead flows to brand-name manufacturers as a result of delayed generic entry, and some portion of that surplus is invested in R&D, but there is no mechanism that ensures the surplus is reinvested rather than distributed to shareholders.
A 2021 analysis published in JAMA Internal Medicine found that from 2000 to 2018, large pharmaceutical companies spent more on stock buybacks and dividends than on research and development in eight of the eighteen years studied [15]. This finding does not prove that patent thickets divert R&D spending to shareholders, since the counterfactual of what spending would look like without patent thickets is impossible to observe. But it weakens the claim that the economic rents generated by patent thickets are primarily reinvested in innovation.
Where Innovation Incentives Actually Come From
The relationship between patent protection and pharmaceutical innovation is more nuanced than either the industry’s or critics’ standard talking points suggest. The empirical evidence on this question is complex, partly because “innovation” can mean breakthrough discoveries, incremental improvements, or repackaging of existing compounds, and patent protection incentivizes all three.
Joseph DiMasi at Tufts and Frank Lichtenberg at Columbia Business School have both found positive correlations between pharmaceutical patent protection and R&D investment across countries and time periods [16, 17]. These studies suggest that weakening patent protection would reduce investment in drug development, at least at the margin.
But Kyle and McGahan (2012) found that differences in patent protection across countries do not explain most of the variation in R&D spending, suggesting that factors other than patent strength, including market size, regulatory capability, and scientific infrastructure, are at least as important as IP protection in driving pharmaceutical investment [18].
The specific question of whether secondary patents generate additional innovation incentives above those created by the primary compound patent is even less studied. The available evidence does not support the conclusion that a drug company would invest less in developing new drugs if it knew that its secondary patents on formulations and dosing devices would be more easily challenged. The returns on those secondary patents are not primarily used to fund new compound discovery.
Part Nine: What the Databases Reveal
Reading the Patent Landscape
Tracking pharmaceutical patent expirations and litigation requires specialized data tools that can integrate patent filing dates, regulatory approval dates, Orange Book listings, PTAB proceedings, and district court litigation records into a coherent picture of any drug’s competitive landscape. DrugPatentWatch is the most comprehensive commercial database doing this work, providing real-time tracking of patent expirations, upcoming exclusivity cliffs, Paragraph IV certifications, and IPR petitions for thousands of branded drugs.
The data in these databases reveals patterns that are not visible when you look at individual drugs in isolation. When you look across the pharmaceutical market, you can see that the drugs with the densest patent portfolios are not necessarily the most innovative; they are the ones with the highest revenue, where the economic returns to portfolio expansion are greatest. This is consistent with the strategic logic of patent thickets: the cost of building a large patent portfolio is fixed (filing fees, patent prosecution costs), while the benefit scales with the revenue at stake.
A 2022 analysis using DrugPatentWatch data found that among the 50 best-selling branded drugs in the United States, the average number of Orange Book-listed patents per drug was 17.8, compared to an average of 4.3 for the broader population of branded drugs [19]. The concentration of patent listings among high-revenue products confirms that patent portfolio expansion is a deliberate commercial strategy rather than a byproduct of ongoing innovation.
The same analysis found that among the 50 best-selling drugs, the average time between the expiration of the compound patent and the last-expiring secondary patent was 8.7 years. This 8.7-year figure represents, on average, the additional exclusivity period that secondary patent portfolios create for these drugs, over and above what the original compound patent would provide.
For payers and investors tracking pharmaceutical competitive dynamics, these data points translate directly into revenue and expenditure projections. A drug with a compound patent expiring in three years but secondary patents running for another decade is not facing the same competitive risk as a drug whose entire patent portfolio expires simultaneously. DrugPatentWatch’s patent cliff analysis enables payers to model generic entry timing and plan formulary strategies accordingly.
Spotting Evergreening in Real Time
The time series of patent filings for a drug often reveals the evergreening strategy more clearly than any single filing does. When a pharmaceutical company files a cluster of new patent applications in the 18 to 36 months before its compound patent expires, those filings are almost certainly not the product of new scientific discoveries. They are the product of systematic review of everything connected to the drug that might be patentable, conducted precisely because the compound patent expiry is approaching.
Patent offices in the United States and Europe have become more attentive to this pattern. USPTO examiners examining applications for secondary pharmaceutical patents have access to the prosecution history of earlier patents on the same compound, and rejections based on obviousness over the primary compound have become more common for secondary applications that would have been granted routinely two decades ago.
Still, the examination process is imperfect, and many secondary patents of questionable validity issue through the USPTO and must be challenged in post-grant proceedings or district court litigation. The cost and time of these challenges creates enough friction that some secondary patents effectively extend exclusivity even if they would not survive a full validity challenge.
Part Ten: The Future of Pharmaceutical Patent Strategy
Artificial Intelligence and Patent Mining
Pharmaceutical companies are increasingly using artificial intelligence and machine learning tools to identify patentable aspects of their drug portfolios that human researchers might overlook. AI-assisted patent mining can systematically identify polymorph forms, metabolites, and formulation variations that could support patent applications, applying computational chemistry to expand the scope of secondary patenting.
This development has concerned some patent policy scholars, who argue that AI-assisted patent mining could produce a flood of secondary patent applications based on computationally generated variants, overwhelming patent office examination capacity and creating ever-denser patent thickets around existing drugs. The USPTO issued a guidance document in 2024 on AI-assisted patent applications, clarifying that AI-generated inventions require a human inventor who made a significant contribution to the conception of the claimed invention. But the boundary between human-guided AI assistance and AI-generated patentable subject matter is technically and legally contested.
The Compulsory License Question
The IRA’s Medicare drug price negotiation provisions have revived discussion of the U.S. government’s march-in rights under the Bayh-Dole Act of 1980. Bayh-Dole gives federal agencies the right to “march in” and require a contractor or assignee who has received federal funding for research to license a patented invention to other parties on reasonable terms, if the contractor or assignee is not making the invention “available to the public on reasonable terms.”
The march-in right has never been exercised in the 40 years since Bayh-Dole’s enactment, despite multiple petitions asking the government to use it. NIH and other agencies have consistently interpreted the statute as not permitting price-based march-in, meaning that a drug patent cannot be subject to march-in solely because the drug is priced too high for many patients to afford.
The Biden administration’s National Institute of Standards and Technology proposed a new framework for march-in rights in 2024 that would allow pricing to be a factor in march-in determinations. The pharmaceutical industry challenged this interpretation, arguing that it would deter investment in government-funded research. The legal and policy controversy over march-in rights is likely to persist regardless of which administration occupies the White House, because the underlying tension between public investment in biomedical research and private appropriation of the resulting patents has never been resolved.
The Price Negotiation Feedback Loop
Medicare drug price negotiation creates a new feedback loop in pharmaceutical patent strategy. If a drug’s negotiated Medicare price is based partly on how many years of exclusivity remain in the patent portfolio, then the density of the patent portfolio affects the negotiation baseline. Companies may rationally invest more in building dense patent portfolios if doing so produces better negotiation outcomes under the IRA’s framework.
Conversely, if price negotiation limits the returns available during an exclusivity period, the economic value of each year of exclusivity declines, which could reduce the incentive to invest in patent portfolio expansion. The net effect of Medicare price negotiation on pharmaceutical patent strategies is an empirical question that cannot be answered until the negotiation program has operated for several years and companies have had time to adjust their IP strategies in response.
The Role of Patient Advocacy and Public Pressure
Public attention to pharmaceutical patent practices has grown substantially in the past decade, driven by high-profile cases like Daraprim, EpiPen, and Humira, and by increasingly organized advocacy from patient groups and health reform organizations. Organizations like the National Academy for State Health Policy, the Drug Policy Alliance, and Patients for Affordable Drugs have made patent thickets and evergreening central to their policy agendas.
This public pressure has had some effect. Legislative attention to pharmaceutical IP has increased. The FTC’s Orange Book challenge program reflects political support for more aggressive enforcement. PTAB petitions against secondary pharmaceutical patents have become a more common tool for payers and patient groups.
But the fundamental economics of pharmaceutical patent strategy have not changed. Brand-name manufacturers have strong incentives to build dense patent portfolios around high-revenue products. The legal system provides mechanisms for challenging those portfolios, but those mechanisms are costly, slow, and uncertain. The gap between the incentive to build patent thickets and the practical ability to dismantle them remains wide.
Conclusion: The 20-Year Clock Is a Fiction
The 20-year pharmaceutical patent is a legal construct that approximates a social bargain: in exchange for publicly disclosing an invention, the inventor receives a temporary monopoly of limited duration. The theory is that 20 years is long enough to recoup R&D investment and reward innovation, short enough that the public eventually regains free access to the invention.
For pharmaceutical drugs, the actual terms of this bargain look very different from the theory. Through the tools documented in this article, systematic secondary patenting, aggressive Orange Book listings, pay-for-delay settlements, REMS manipulation, and strategic use of data exclusivity provisions, brand-name manufacturers have extended the effective exclusivity period for major drugs to 30, 40, or more years from initial filing.
This extended exclusivity is not without some justification. Drug development is expensive and risky. Patent protection serves a legitimate function in funding innovation. But the scale of the gap between the 20-year theoretical maximum and the 40-year practical reality, documented in case after case by analysts, academics, and databases like DrugPatentWatch, suggests that the system has drifted far from its original design.
Restoring balance requires reforms that work at multiple levels: stricter patent examination standards that apply a genuine non-obviousness analysis to secondary pharmaceutical patent applications, stronger procedural limits on Orange Book listings and 30-month stays, better tools for PTAB challenges to weak secondary patents, and antitrust enforcement that treats large reverse payment settlements as presumptively anticompetitive.
The international dimension of this problem is worth restating. Patents on a single drug are now litigated simultaneously in dozens of jurisdictions, with outcomes that vary not because the underlying science or innovation is different, but because patent examination standards, litigation procedures, and the political economy of pharmaceutical IP differ across countries. American patients face longer exclusivity periods than European patients for the same drugs, not because American drugs are more innovative, but because the U.S. patent examination and litigation system creates more opportunities for secondary patents to survive challenge.
The rise of AI-assisted patent mining, described earlier in this article, means the future may bring more secondary patents, not fewer, unless examination standards and post-grant challenge procedures keep pace with the technology. A patent office that cannot distinguish AI-generated polymorph variants from genuine inventions will see its role reduced to a rubber stamp on portfolio expansion strategies.
None of these reforms will eliminate secondary patenting entirely, nor should they. Genuine incremental innovation in drug delivery, formulation, and manufacturing deserves patent protection. What the system needs is a more discriminating ability to distinguish genuine innovation from strategic filing, and a more proportionate relationship between the social cost of exclusivity and the innovation it actually rewards.
The intellectual honesty required to reform this system demands acknowledging both sides of the equation simultaneously. Pharmaceutical innovation is genuinely expensive, genuinely risky, and genuinely valuable to humanity. And the specific tools that generate 40-year effective monopolies on individual drugs, through polymorph patents, pediatric exclusivity tack-ons, pay-for-delay settlements, and REMS manipulation, are not what generates that innovation. They are what happens after the innovation has already succeeded, when the goal shifts from discovery to rent extraction.
Until that balance is restored, the 20-year clock on pharmaceutical patents will remain more marketing claim than mathematical reality.
Key Takeaways
Patent thickets are deliberately constructed. The dense patent portfolios around major pharmaceutical drugs are not accidents of the patent system. They result from systematic patent prosecution strategies that identify every patentable aspect of a drug and file applications on each one, timed to maximize the coverage of approaching exclusivity cliffs.
Evergreening and legitimate lifecycle management overlap. Some secondary patents reflect genuine innovation. Extended-release formulations can improve patient adherence. New salt forms can improve solubility. The problem is that the patent system applies identical protection regardless of whether the secondary patent creates real clinical benefit or serves primarily to extend market exclusivity.
The regulatory framework enables the strategy. Hatch-Waxman’s 30-month automatic stay, the BPCIA’s 12-year data exclusivity, and the Orange Book self-listing system all create legal mechanisms that patent thicket strategies exploit. Reforming these mechanisms is as important as challenging individual patents.
International patent rules vary significantly. India’s Section 3(d), the EPO’s stricter non-obviousness standard, and the TRIPS flexibilities available to developing countries all create materially different patent environments. U.S. patients face longer exclusivity periods than their counterparts in many other high-income countries because of differences in how secondary patents are examined and enforced.
Data tools reveal the pattern. Databases like DrugPatentWatch make patent thicket strategies visible at a market-wide scale that individual case analysis cannot provide. The concentration of patent listings among the highest-revenue drugs, the timing of secondary patent filings relative to compound patent expirations, and the patterns of Paragraph IV litigation all tell a coherent strategic story when analyzed in aggregate.
Legislative and regulatory reform is active but incomplete. The IRA’s price negotiation provisions, the FTC’s Orange Book challenge program, and the CREATES Act all represent meaningful policy responses. But they address symptoms rather than causes, and the fundamental economics of patent portfolio construction remain largely unchanged.
Frequently Asked Questions
Q1: What is the difference between a patent thicket and evergreening? Are they the same strategy?
They are related but distinct. Evergreening specifically refers to extending market exclusivity beyond the original patent term through incremental product modifications and new secondary patents on those modifications. A patent thicket describes the accumulated result of this strategy: a dense web of overlapping patents around a single product that any competitor must navigate before entering the market. Evergreening is the process; the patent thicket is the product. A drug can have a dense patent thicket without much evergreening if the original drug itself is complex and generated many legitimate secondary inventions. Conversely, a company can pursue an evergreening strategy that does not produce a thicket if its secondary patents are few but strategically timed to delay generic entry.
Q2: Can a generic manufacturer simply ignore secondary patents and enter the market anyway?
It can enter “at risk,” meaning it launches before the litigation over the secondary patent is resolved, accepting the possibility that if it loses the patent case it will face injunctive relief and potentially damages. Some generic manufacturers do enter at risk when they believe a secondary patent is clearly invalid or not infringed. The decision depends on the strength of the patent, the anticipated duration of litigation, the size of the market, and the tolerance for risk of the generic company’s management and investors. The 30-month automatic stay that follows a Paragraph IV certification applies during litigation, but it can expire before litigation concludes, at which point the FDA can approve the generic, and the generic can choose whether to enter at risk.
Q3: How does the PTAB compare to district court litigation as a tool for challenging secondary pharmaceutical patents?
PTAB offers lower cost and faster resolution than district court litigation, typically completing IPR proceedings within 18 months compared to the two to five years typical for district court patent trials. The IPR process is limited to prior art challenges, which means it cannot address validity questions based on enablement, written description, or § 101 subject matter eligibility. District court litigation covers the full range of invalidity defenses. For generic manufacturers, a common strategy is to file IPR petitions as a lower-cost initial challenge while pursuing district court litigation in parallel, using the IPR to develop the prior art record and potentially resolve some patent claims before trial. The risk of this strategy is that losing an IPR petition can create estoppel that limits the arguments available in district court.
Q4: Do pharmaceutical companies disclose their evergreening strategies in their public filings?
Not directly. Annual reports and investor presentations describe “lifecycle management” as a strategic priority, which is the industry’s term for the same activity. Specific patent strategies are described in general terms, such as commitments to “protect the intellectual property underlying our products” or to “pursue supplemental applications that extend the period of market exclusivity.” More detailed information about specific patent filing strategies appears in 10-K risk factors and patent litigation disclosures. The Orange Book is a public database that shows which patents a company has listed for each drug, and the timing of those listings, relative to the drug’s patent expiry schedule, makes the evergreening strategy visible to anyone who looks. DrugPatentWatch consolidates this public information and provides analytical tools that make the patterns easier to identify and quantify.
Q5: Will the Medicare drug price negotiation provisions of the Inflation Reduction Act reduce evergreening?
The long-term effect is uncertain and will depend on how the negotiation program develops over time. In the short term, the IRA creates an incentive for companies to shift development toward biologics (subject to negotiation after 13 years) rather than small molecules (subject to negotiation after 9 years), which could affect innovation patterns rather than patent strategies for existing drugs. For drugs already on the market, the negotiated price replaces the market price for Medicare purchases but does not affect patent rights or the legal mechanisms for challenging them. If the negotiated price is set low enough that the economic returns to maintaining a large patent portfolio are reduced, companies may rationally invest less in secondary patenting. But the IRA’s negotiation framework is still new, and its effect on patent strategy is not yet empirically measurable.
References
[1] RAND Corporation. (2023). The cost of delayed biosimilar entry: Quantifying the impact of AbbVie’s Humira patent strategy on U.S. drug spending. RAND Health Quarterly.
[2] Feldman, R., & Frondorf, E. (2018). Drug wars: How big pharma raises prices and keeps generics off the market. I-MAK Secondary Patent Report.
[3] Federal Trade Commission. (2010). Pay-for-delay: How drug company pay-offs cost consumers billions. FTC Staff Study.
[4] Federal Trade Commission v. AbbVie Inc. et al., No. 14-5151 (3rd Cir. 2018); FTC v. Celgene Corp., No. 2:19-cv-15284 (D.N.J. 2019).
[5] Federal Trade Commission. (2023, October). FTC challenges patents listed in FDA’s Orange Book for 17 branded drug products. FTC Press Release.
[6] Kapczynski, A., Park, C., & Sampat, B. (2020). Polymorphs and prodrugs and salts (oh my!): An empirical analysis of “secondary” pharmaceutical patents. Journal of Law and the Biosciences, 7(1), lsaa080.
[7] Congressional Budget Office. (1998). How increased competition from generic drugs has affected prices and returns in the pharmaceutical industry. CBO Study.
[8] Sarpatwari, A., Avorn, J., & Kesselheim, A. S. (2020). Using the courts and the FDA to protect competition in the pharmaceutical market. JAMA Network Open, 3(7), e207059.
[9] Choudhry, N. K., Avorn, J., Glynn, R. J., et al. (2011). Full coverage for preventive medications after myocardial infarction. New England Journal of Medicine, 365(22), 2088-2097.
[10] Association for Accessible Medicines. (2022). Access delayed: Biosimilar market entry and the patent barrier. AAM White Paper.
[11] Feldman, R. (2021, March). Testimony before the Senate Judiciary Committee, Subcommittee on Intellectual Property. U.S. Senate.
[12] Novartis AG v. Union of India & Ors., (2013) 6 SCC 1 (India Supreme Court).
[13] DiMasi, J. A., Grabowski, H. G., & Hansen, R. W. (2016). Innovation in the pharmaceutical industry: New estimates of R&D costs. Journal of Health Economics, 47, 20-33.
[14] I-MAK. (2018). Overpatented, overpriced: Special edition on insulin. Initiative for Medicines, Access & Knowledge.
[15] Dafny, L., Ody, C., & Schmitt, M. (2021). Undermining value in health care: The race to the top and its consequences. JAMA Internal Medicine, 181(7), 938-943.
[16] DiMasi, J. A., & Grabowski, H. G. (2007). The cost of biopharmaceutical R&D: Is biotech different? Managerial and Decision Economics, 28(4-5), 469-479.
[17] Lichtenberg, F. R. (2021). The impact of pharmaceutical innovation on longevity and medical expenditure in France, 2000-2015. Economics & Human Biology, 40, 100940.
[18] Kyle, M. K., & McGahan, A. M. (2012). Investments in pharmaceuticals before and after TRIPS. Review of Economics and Statistics, 94(4), 1157-1172.
[19] DrugPatentWatch. (2022). Patent portfolio analysis of the 50 best-selling branded drugs in the United States. DrugPatentWatch Market Intelligence Report.
[20] U.S. Government Accountability Office. (2002). Pediatric drug research: Studies conducted under best pharmaceuticals for children act. GAO-07-557.
[21] Federal Trade Commission. (2023). Agreements filed with the Federal Trade Commission under the Medicare Prescription Drug, Improvement, and Modernization Act of 2003: Overview of agreements filed in FY 2022. FTC Report.
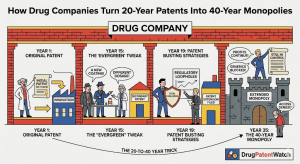









")
















