When a generic drug company in New Jersey files an Abbreviated New Drug Application with the FDA, it enters a legal system specifically engineered to use patent law as a gatekeeper to market access. When that same company’s European subsidiary files a marketing authorization application with the European Medicines Agency, patent law is almost entirely irrelevant to the regulatory outcome. The regulator simply does not care.
This is not an accident or an oversight. It is a deliberate architectural choice that has shaped drug pricing, litigation volumes, market competition, and public health outcomes across two of the world’s largest pharmaceutical markets for decades. The divergence between the US patent linkage model and the EU data exclusivity model is one of the most consequential and least-discussed splits in global pharmaceutical policy, and it has real money attached to it: brand-name pharmaceutical companies spent an estimated $6.8 billion defending drug patents in US litigation between 2000 and 2020 [1], much of it made possible by a regulatory structure that the European Union has explicitly and repeatedly refused to adopt.
This article examines why these two systems exist, how each one works mechanically, what the economic consequences are, and what generic manufacturers, brand-name companies, and healthcare payers need to understand to operate in both markets.
Part One: The Architecture of Protection
What Patent Linkage Actually Is
Patent linkage is a regulatory mechanism that ties a generic drug’s approval to the resolution of patent disputes over the reference listed drug. Under a patent linkage regime, the regulatory agency responsible for approving generics either delays its approval or withholds it entirely until the generic manufacturer either proves the relevant patents are invalid or not infringed, waits for them to expire, or obtains a license.
The critical word is “ties.” The patent system and the drug approval system are legally coupled. A generic cannot get its marketing authorization in clean isolation from the patent question. The regulator, even if it has no substantive expertise in patent law, becomes a participant in the intellectual property dispute by virtue of withholding approval pending resolution.
The United States operates the world’s most developed patent linkage system. It was created by the Drug Price Competition and Patent Term Restoration Act of 1984, universally known as the Hatch-Waxman Act, named for its Senate sponsor Orrin Hatch and House sponsor Henry Waxman [2]. The Act was a compromise: it gave innovator pharmaceutical companies the ability to restore patent term lost during regulatory review, and it gave generic manufacturers an expedited pathway to market through the Abbreviated New Drug Application process. The linkage between those two innovations created a system that neither side fully anticipated would become as litigious as it has.
The Orange Book and Its Function
The FDA publishes a document called the “Approved Drug Products with Therapeutic Equivalence Evaluations,” which every pharmaceutical professional knows as the Orange Book. Brand-name manufacturers must list in the Orange Book every patent they claim covers their approved drug product, including patents on the active pharmaceutical ingredient, the drug formulation, and approved methods of use [3].
This listing requirement is central to how patent linkage works. When a generic manufacturer submits an ANDA, it must certify with respect to every Orange Book-listed patent. There are four possible certifications. Paragraph I and II certifications state that either no patent is listed or the patent has expired, in which case the FDA can approve the generic immediately. A Paragraph III certification states that the patent will expire on a specified date, and the generic agrees to wait. The Paragraph IV certification is where the real action happens: it states that the patent is either invalid or will not be infringed by the generic product [4].
Filing a Paragraph IV certification is automatically treated by statute as an act of patent infringement. The brand-name manufacturer can sue the generic applicant within 45 days of receiving notice of the certification. If the brand sues, the FDA is legally required to impose a 30-month stay on ANDA approval, during which the patent case is litigated [5]. The generic stays off the market while the lawyers work.
The first generic manufacturer to file a successful Paragraph IV challenge against any given patent is entitled to 180 days of market exclusivity before other generics can enter. This “first-to-file” exclusivity is a financial incentive worth hundreds of millions of dollars for popular drugs, and it has made Paragraph IV litigation one of the most profitable legal strategies in the pharmaceutical industry [6].
The Mechanics of the 30-Month Stay
The 30-month stay deserves careful attention because it is the mechanism most responsible for delayed generic entry in the United States, and it is the provision that critics most consistently target when arguing for reform.
When a brand-name company lists a patent in the Orange Book and a generic files a Paragraph IV challenge, the brand’s decision to sue within 45 days automatically triggers a 30-month period during which the FDA cannot approve the ANDA. This is true regardless of the merits of the underlying patent claims. The stay is automatic. The court does not evaluate likelihood of success. The patent does not need to be commercially significant. It simply needs to be listed in the Orange Book.
The consequences of this structure are well-documented. Brand-name manufacturers learned quickly that listing additional patents in the Orange Book, particularly patents covering relatively minor innovations like specific polymorphs, enantiomers, or extended-release formulations, could trigger additional 30-month stays and extend market exclusivity far beyond what the primary compound patent would have provided [7]. This practice, often called “evergreening” or more precisely “Orange Book stuffing,” became a central element of brand-name competitive strategy in the 1990s and 2000s.
Congress partially addressed this in the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, which limited brands to a single 30-month stay per drug product regardless of how many patents were listed [8]. But the fundamental architecture remained intact: patent disputes could and would delay generic entry as a direct consequence of regulatory linkage.
Paragraph IV Litigation: The Numbers
The scale of Paragraph IV litigation in the United States is extraordinary by any measure. According to the FDA’s own data, between 2000 and 2019, 73% of all new molecular entities approved by the FDA faced at least one Paragraph IV challenge [9]. For the top-selling drugs, the rate was effectively 100%.
A study by researchers at Harvard Medical School found that brand-name manufacturers sued generic filers in approximately 73% of cases where a Paragraph IV certification was filed, and that the average time from ANDA filing to generic market entry in contested cases was approximately 3.5 years [10]. The same study estimated that delays in generic entry cost the US healthcare system between $3.5 billion and $12 billion annually depending on the drugs involved.
The 180-day first-filer exclusivity has itself generated complex strategic behavior. Brand-name manufacturers discovered that they could enter into agreements with first-filing generic companies to delay market entry in exchange for payments, a practice known as “pay-for-delay” or “reverse payment” settlements. The Federal Trade Commission estimated that pay-for-delay settlements cost consumers and taxpayers approximately $3.5 billion per year in higher drug costs [11]. The Supreme Court’s 2013 ruling in FTC v. Actavis, Inc. held that such settlements could violate antitrust law, subjecting them to a “rule of reason” analysis [12], but the practice did not disappear; it simply became more structurally complex.
What Data Exclusivity Actually Is
Data exclusivity is a protection regime that prevents regulatory authorities from relying on the clinical trial data submitted by the originator drug company to approve a generic version for a defined period after the originator drug’s approval. It is a protection of the regulatory data package, not of any specific patent claim.
The concept recognizes that generating clinical data is expensive. A Phase III clinical trial for a new drug can cost anywhere from $300 million to over $1 billion [13]. If a generic manufacturer could immediately rely on that data to obtain its own approval, the originator would not be able to recover its investment. Data exclusivity gives the originator a window during which no competitor can use the shortcut of referencing that data.
Crucially, data exclusivity says nothing about patents. A drug’s patent might still have fifteen years to run when data exclusivity expires, or data exclusivity might expire years after the compound patent does. The two protections operate on completely separate timelines with no formal legal connection between them.
In the United States, data exclusivity for new chemical entities runs five years under the Hatch-Waxman Act, with an additional three years for new clinical studies that support changes to an approved drug, such as new formulations or indications [14]. Biologics receive twelve years of data exclusivity under the Biologics Price Competition and Innovation Act of 2009 [15]. During these periods, the FDA will not approve an ANDA or biosimilar application that relies on the originator’s data. But once the period ends, the FDA’s ability to rely on that data is restored, regardless of what patents remain in force.
How Data Exclusivity Differs from Patent Rights
The distinction between data exclusivity and patent rights matters enormously in practice, and it is where many observers conflate two separate systems into one.
A patent gives its holder the right to exclude others from making, using, selling, or importing the patented invention for the duration of the patent term. Patent infringement is a civil wrong that the patent holder must actively enforce in court. No government agency enforces patents on behalf of patent holders; enforcement is the patent holder’s responsibility.
Data exclusivity, by contrast, is an administrative protection enforced automatically by the regulatory agency. The agency itself will not accept or act on certain types of applications during the exclusivity period. No lawsuit is required. No litigation takes place. The protection operates quietly and automatically within the regulatory system.
This distinction has large practical consequences. Data exclusivity creates a hard floor below which no generic can enter, regardless of patent status. If the compound patent expires two years before data exclusivity does, the generic still cannot enter using the abbreviated pathway until data exclusivity expires, because the regulatory agency will not process the application. Conversely, once data exclusivity expires, a generic can seek approval and the regulatory agency has no reason to delay, even if patents remain. Those patents still exist, still have legal force, and could still be enforced in court, but that is the originator’s problem to manage, not the regulator’s.
Part Two: The European Model
The EU’s Deliberate Rejection of Patent Linkage
The European Union has a patent linkage system for exactly zero of its approved pharmaceutical products. This is not because no one proposed it. When the EU was constructing its modern pharmaceutical regulatory framework in the early 2000s, the question of whether to import a Hatch-Waxman-style linkage regime was explicitly raised and explicitly rejected.
The European Commission’s position, set out in its 2001 Directive 2001/83/EC and clarified in numerous subsequent guidance documents and court rulings, is that the European Medicines Agency and the national competent authorities responsible for drug approval have a mandate limited to evaluating pharmaceutical quality, safety, and efficacy [16]. Intellectual property questions are outside their remit. The EMA does not search patent databases. It does not ask applicants to certify their relationship to listed patents. It does not delay approvals pending patent litigation. It grants marketing authorizations based on the scientific package in front of it, and that is where its legal authority begins and ends.
The Court of Justice of the European Union reinforced this separation in a series of rulings. In Generics (UK) Ltd v. Smith Kline and French Laboratories Ltd and in subsequent cases, the CJEU made clear that national patent law and EU pharmaceutical regulatory law occupy separate legal domains [17]. A national patent authority cannot prevent a national regulatory authority from granting a marketing authorization on patent grounds, and a national regulatory authority cannot delay granting a marketing authorization to give a patent holder time to litigate.
The 8+2+1 Framework: How EU Data Exclusivity Works
The EU’s protection system for originator drugs is built around what practitioners call the “8+2+1” framework, established by Directive 2004/27/EC [18].
The first eight years are a period of “data exclusivity” in the pure sense: generic manufacturers cannot file marketing authorization applications that reference the originator’s data. The generic company can conduct its own research and prepare its dossier, but it cannot formally submit a reliance-based application during this window.
The next two years are “market exclusivity” or “marketing protection.” The generic can now file and the EMA can process and even approve the application, but the approved generic cannot be commercially placed on the EU market. The regulatory queue can start moving, but the product cannot actually be sold.
The final “+1” year is available when the originator obtains a new indication for its product during the first eight years of protection and that new indication brings significant clinical benefit. This extends the market exclusivity period to eleven years total, rewarding genuine therapeutic innovation with an additional competitive buffer.
The combined 10-year period (or 11 years with the bonus) applies to “new active substances.” For substances already approved in the EU before 2005, transitional provisions apply. For products approved after the Directive took effect, the framework is uniform across all EU member states, creating a single regulatory calendar for generic entry timing.
What the EMA Does and Does Not Do in IP Disputes
The EMA’s role in the generic approval process is precisely bounded. It evaluates the quality of the generic’s pharmaceutical dossier: the bioequivalence data, the manufacturing quality documentation, the analytical methods, the stability data. It checks that the generic’s active substance is the same as the reference listed drug and that the bioavailability profile is sufficiently equivalent to permit reliance on the originator’s safety and efficacy data.
The EMA does not check whether the generic manufacturer holds a patent license. It does not ask whether the originator’s compound patent is still in force. It does not invite the originator to submit patent information or flag potential infringement. It issues marketing authorizations to generics that meet the scientific criteria for approval, and when a patent holder believes its rights have been infringed, it must go to a national court to obtain relief.
This is a deliberate allocation of institutional competence. EU regulators argued, and the Commission agreed, that introducing patent questions into the regulatory process would create precisely the delays, costs, and strategic litigation behaviors that have characterized the US system. The regulator’s scientific expertise does not extend to patent validity, claim construction, or infringement analysis. Forcing it to engage with those questions would slow approvals, create legal uncertainty, and shift power decisively toward patent holders who could exploit regulatory delays as a commercial weapon.
The Bolar Exemption in the EU Context
One specific provision illustrates how the EU’s framework operates differently from the US model. Article 10(6) of Directive 2001/83/EC includes a “Bolar exemption” equivalent: conducting the studies, trials, and practical requirements necessary to apply for a marketing authorization during a patent or supplementary protection certificate term is not an act of patent infringement [19].
The EU Bolar exemption is, on paper, more generous than the US original Bolar provision codified in the Hatch-Waxman Act. The EU version covers not just the clinical and pre-clinical studies needed for the regulatory submission, but also the practical administrative steps required to obtain approval. Generic manufacturers can begin conducting bioequivalence studies, preparing manufacturing batches for submission, and engaging in the regulatory process while the originator’s patent is still running, provided they do not actually commercialize the product.
This creates a situation where a generic can have its marketing authorization in hand and its manufacturing line ready to run on the day the originator’s last patent expires, without having infringed any patent rights. The EU system is designed to allow this. The US system nominally allows it too, but the combination of Paragraph IV certification requirements, automatic infringement presumption, and 30-month stays means that the practical experience of generic manufacturers navigating both systems is radically different.
Supplementary Protection Certificates: The EU Equivalent of Patent Term Extension
The EU’s counterpart to Hatch-Waxman patent term extension is the Supplementary Protection Certificate, or SPC, governed by Regulation (EC) No 469/2009 [20]. Like US patent term extension, the SPC compensates originators for the patent term consumed during the regulatory review process.
An SPC can be granted for an active ingredient or combination of active ingredients in a medicinal product that has received its first EU marketing authorization. The certificate takes effect when the underlying patent expires and can provide up to five years of additional protection, plus a further six-month extension if pediatric studies have been conducted under the EU pediatric regulation [21].
The SPC system has generated its own considerable body of litigation, much of it focused on what constitutes a “product” protected by the SPC and whether combination products or products approved in some EU member states before others create eligibility complications. The CJEU has delivered dozens of rulings interpreting SPC eligibility, some of which contradicted earlier decisions, creating genuine commercial uncertainty for companies trying to plan generic entry timelines.
What the SPC does not do is link itself to the generic approval process. An SPC extends the period during which commercial sale of the generic would infringe the originator’s rights, but the regulatory approval for the generic can proceed in parallel. The generic has its marketing authorization ready. It simply cannot sell until the SPC expires, and then it launches immediately.
Part Three: Why the Systems Diverge
Different Legal Philosophies on IP-Regulatory Coupling
The US and EU systems reflect genuinely different philosophical positions on the appropriate relationship between intellectual property law and pharmaceutical regulation, and tracing those philosophies helps explain why neither system is simply going to adopt the other’s approach.
The US approach reflects a legislative bargain struck in 1984 in which both sides got something. Generic manufacturers got the ANDA pathway, which eliminated the requirement to run independent clinical trials and dramatically reduced the cost of bringing generics to market. Originators got patent term restoration and the Orange Book listing mechanism that effectively gave them regulatory enforcement of their patent rights. The linkage was the price the generic industry paid for the ANDA system. Neither side negotiated away what they had won, and subsequent reform efforts have struggled because any change to the linkage system reopens the underlying bargain.
The EU approach reflects a different institutional design. The EMA was created as a scientific body with a specific and limited mandate. Asking it to adjudicate patent disputes would require it to develop expertise it was never designed to have, would expose it to legal challenge from parties on both sides of patent disputes, and would politicize what was intended to be a technocratic approval process. The Commission’s 2008 Pharmaceutical Sector Inquiry found that brand-name companies had filed over 700 applications for interim injunctions in EU member states to delay generic market entry between 2000 and 2007 [22]. The Commission’s explicit conclusion was that this litigation was a competitive tool used by originators to maintain market exclusivity beyond the legitimate scope of their IP rights. Introducing a formal patent linkage mechanism at the regulatory level would have amplified this problem significantly.
Trade Pressure and the TRIPS Agreement
The United States has consistently used international trade agreements to push other countries toward adopting patent linkage, despite the EU’s resistance. The Trade-Related Aspects of Intellectual Property Rights Agreement, or TRIPS, negotiated under the World Trade Organization in 1994, establishes minimum standards for IP protection globally but does not require patent linkage [23].
However, US bilateral and regional free trade agreements go further. The US-Korea Free Trade Agreement, the Central America-Dominican Republic Free Trade Agreement, and several others contain provisions requiring trading partners to implement patent linkage regimes [24]. The Trans-Pacific Partnership, before the US withdrew in 2017, contained similar provisions that were substantially weakened in the successor Comprehensive and Progressive Agreement for Trans-Pacific Partnership.
The European Union has not signed any free trade agreement that requires patent linkage. When the EU negotiated the Comprehensive Economic and Trade Agreement with Canada, which entered into force provisionally in 2017, the intellectual property chapter included enhanced data exclusivity provisions but explicitly excluded any patent linkage requirement [25]. The EU’s position is that patent linkage is not a TRIPS obligation, is not required by sound public health policy, and would violate the institutional mandate of its regulatory agencies.
This creates a genuine divergence in the global regulatory landscape. Countries negotiating FTAs with the United States face pressure to adopt patent linkage. Countries negotiating with the EU do not. Middle-income countries that have signed agreements with both face the challenge of implementing legally consistent regimes across potentially conflicting obligations.
The 2008 EU Pharmaceutical Sector Inquiry: A Defining Document
The European Commission’s 2008 Pharmaceutical Sector Inquiry is worth examining in some detail because it shaped EU policy toward patent linkage for the decade that followed and remains the most comprehensive official analysis of strategic IP behavior in the European pharmaceutical market [26].
The Commission examined 219 molecules that lost patent protection between 2000 and 2007 and found a range of practices by originator companies designed to extend commercial exclusivity beyond the scope of legitimate patent rights. These included filing additional patent applications to build “patent clusters” around established products, using SPCs in ways that the Commission considered legally questionable, filing divisional patent applications to extend the effective protection period, and initiating patent litigation purely for its commercial delay effect rather than as genuine enforcement of valid IP rights.
The inquiry found that generic entry delays cost the EU healthcare system approximately 3 billion euros per year [27]. The Commission did not recommend the introduction of patent linkage to address this; it recommended enhanced antitrust enforcement, greater transparency in SPC applications, and cleaner separation between patent protection and regulatory approval.
The inquiry’s finding that originator companies filed approximately 698 patent-related cases against generics during the study period, of which originators won fewer than 60%, illustrates the extent to which patent litigation was being used as a delay tactic rather than a genuine enforcement tool [28]. The Commission’s conclusion was that more litigation, embedded in the regulatory process through linkage, would make this problem worse rather than better.
Part Four: Strategic Implications for Generic Manufacturers
Filing Strategy in the United States
A generic manufacturer operating in the US market needs to make a series of strategic decisions before filing an ANDA for any branded product still protected by Orange Book-listed patents. Those decisions have significant financial consequences and require deep knowledge of both the regulatory and patent landscapes.
The first question is timing. Filing a Paragraph IV certification opens the door to the 30-month stay, meaning the brand can trigger automatic delay simply by suing within 45 days of receiving notice. A generic manufacturer must assess the probability of being sued, the likely duration and outcome of the litigation, and the value of being first to file against the potential costs of a multi-year legal battle before product launch.
Tools like DrugPatentWatch are essential for this analysis. DrugPatentWatch compiles and analyzes Orange Book patent listings, ANDA filing histories, patent expiration data, and litigation outcomes to give generic manufacturers and their advisors a structured picture of the competitive landscape around any branded drug. For a generic company evaluating whether to challenge the patent protection around a blockbuster medication, knowing exactly which patents are listed, when they expire, which have been successfully challenged before, and what litigation has already been filed can be the difference between a commercially viable strategy and a costly dead end.
Once the decision to file is made, the generic manufacturer must decide whether to file with a Paragraph III certification and wait for patent expiration, or to take the more aggressive Paragraph IV route and invite litigation. Paragraph IV makes sense when the generic company has strong invalidity arguments, when the relevant patents are weak or narrowly drafted, when the 180-day exclusivity bounty is large enough to justify the litigation cost, or when competition from other generics would erode the value of waiting anyway.
The 180-day exclusivity calculation is not simple. The value of being the first generic is highest when the brand drug has large sales volume, when the brand sets a high list price, and when there are few other generics expected to enter quickly. For a drug with $2 billion in annual US sales, 180 days of exclusivity as the only generic can generate hundreds of millions of dollars in profit. For a drug with $50 million in sales, the math looks very different.
Filing Strategy in the European Union
Generic manufacturers entering the EU market face a structurally simpler regulatory pathway, though not necessarily a commercially simpler one.
Because the EMA does not engage with patent questions, the generic manufacturer’s regulatory task is to demonstrate bioequivalence and pharmaceutical quality. There is no equivalent of the Orange Book certification requirement, no automatic infringement presumption, and no 30-month stay. The generic submits its dossier, the EMA or national authority evaluates it, and if the scientific package is adequate, approval is granted.
The strategic complexity in the EU comes from a different direction: SPCs, national patent injunctions, and the fragmented nature of patent enforcement across 27 member states. While the marketing authorization process is centralized for many products through the EMA’s centralized procedure, patent rights are still national rights enforced in national courts. A patent holder can obtain an interim injunction against a generic manufacturer in Germany, France, or the Netherlands independently, potentially fragmenting the generic’s ability to launch even after regulatory approval is secured.
The practice of “torpedo litigation” in European patent law further complicates this landscape. An originator can file a declaratory judgment action in a jurisdiction with a slow court system, which, under Brussels Regulation rules on jurisdiction, can delay the generic from filing in a faster jurisdiction. This tactic has been substantially curtailed by EU procedural reforms but has not disappeared entirely [29].
Generic manufacturers with EU ambitions therefore need to track SPC expiry dates with precision, monitor injunction applications across key markets, and plan their manufacturing and commercial launch timelines around the most conservative national legal scenarios. The data tools that help with this analysis, including DrugPatentWatch’s international patent expiry tracking capabilities, are as important for EU strategy as they are for US strategy, even though the regulatory linkage mechanism that defines US timing does not exist in the EU.
Cross-Jurisdictional Planning: Running Both Playbooks
For large generic manufacturers operating in both markets, which includes virtually all of the major players, the challenge is running two completely different strategic playbooks simultaneously for the same product.
A generic pharmaceutical company evaluating whether to develop a generic version of a hypothetical blockbuster drug faces a US timeline built around Paragraph IV litigation risk and 30-month stays, and a separate EU timeline built around data exclusivity windows, SPC expiries, and national patent enforcement. Those timelines may not align. A drug whose data exclusivity expires in the EU years before its Orange Book patents expire in the US creates a situation where the generic can launch commercially in Europe while still mired in US litigation.
This asymmetry creates genuine strategic opportunities. EU generic revenue can fund US litigation. A company that launches successfully in Germany or France generates cash flow that makes the US legal battle more affordable. Conversely, a company that wins a high-profile US Paragraph IV challenge gains experience, credibility with investors, and sometimes directly transferable technical knowledge about the drug’s manufacturing that strengthens its EU competitive position.
The regulatory data generated for one market can often be leveraged for the other, though this is less straightforward than it might appear. US bioequivalence studies conducted under FDA standards are generally acceptable to the EMA, and vice versa, but the specific study designs, reference products used, and analytical requirements can differ in ways that require additional work. Global generic manufacturers develop internal regulatory teams that understand both systems and can identify where data packages can be shared and where parallel work is needed.
Part Five: Brand-Name Strategy in Both Systems
Evergreening: The Same Goal, Different Mechanics
The term “evergreening” describes a family of strategies by which originator pharmaceutical companies attempt to extend their effective commercial exclusivity beyond the expiry of the primary compound patent. The strategies differ significantly between the US and EU systems because the systems provide different tools.
In the United States, the most common evergreening strategies involve Orange Book manipulation. This includes filing patents on formulations, dosage forms, delivery systems, and methods of use and listing them in the Orange Book; obtaining approval for new formulations and switching commercial focus to them before compound patent expiry so that any substitutable generic must also address the formulation patents; and using 30-month stays to delay generic entry on each new Orange Book listing.
The FDA has increasingly scrutinized Orange Book listings and has the authority to require removal of patents it determines do not meet the listing criteria. The FTC has also taken an active enforcement posture, suing pharmaceutical companies for improper Orange Book listings under Section 5 of the FTC Act. In 2023, the FDA finalized rules tightening the criteria for Orange Book patent listings for combination products and drug-device combinations [30]. These reforms have had some effect, but the fundamental linkage architecture remains available for genuine patent holders to exploit.
In the European Union, where the Orange Book mechanism does not exist, evergreening takes different forms. The primary tools are the SPC system and national patent filings. Originator companies file divisional patent applications, which are new applications derived from a parent application, to obtain additional patent rights covering variants of their products. They file continuation patents on manufacturing processes, specific crystalline forms, and delivery systems. They seek SPCs on new formulations or new combination products.
The EU’s patent linkage absence means that none of these additional patents can trigger automatic regulatory delays. A generic manufacturer facing a thicket of secondary EU patents must still navigate them, but through the courts rather than through the regulator. This changes both the cost structure and the power dynamic: courts apply uniform legal standards, allow invalidity challenges, and can assess damages, while regulatory delay mechanisms do not.
Data Package Strategy and the US-EU Comparison
Originator companies in both markets use data exclusivity strategically, but the ways they leverage it differ.
In the United States, the five-year data exclusivity for new chemical entities provides a hard floor, as noted. But the additional three-year exclusivity for new clinical studies creates incentives to conduct additional studies on approved drugs. An originator that obtains FDA approval for a new indication, formulation, or patient population for an already-approved drug gets three additional years of data exclusivity for that new use. This incentivizes genuine investment in label expansion but also creates the possibility of strategically timed label additions designed primarily to reset exclusivity timelines.
In the European Union, the 8+2+1 system is similarly subject to strategic use. The “+1” year extension for new therapeutic indications requires a genuine showing of significant clinical benefit, and the EMA’s Committee for Medicinal Products for Human Use evaluates this scientifically. The bar is meaningfully higher than simply obtaining FDA approval for an additional indication, and the EMA has denied “+1” extension requests that it found did not meet the clinical benefit threshold [31].
The EU system also contains a global marketing authorization rule: data exclusivity is calculated from the date of first approval in any EU member state, not from the date of approval in each country. This prevents originator companies from creating artificial exclusivity-renewal cycles by obtaining new national approvals in sequence. A drug approved in Germany in 2015 that is later approved in Bulgaria in 2020 does not get a fresh eight-year clock in Bulgaria; the Bulgarian generic exclusivity period began running in 2015 [32].
The Patent Thicket Problem
A “patent thicket” is a situation in which a large number of overlapping patents, often filed by the same originator company, must all be navigated before a generic or biosimilar manufacturer can bring a competing product to market. The term is most associated with the US biologics market, where reference product sponsors like AbbVie filed over 160 US patents on adalimumab (Humira), creating a portfolio that kept biosimilar competitors off the US market for years after biosimilars were already competing in Europe [33].
Patent thickets are a legal phenomenon that exists independently of the patent linkage mechanism, but the US linkage system significantly amplifies their commercial effect. If a generic or biosimilar manufacturer must address every patent in a thicket through the Paragraph IV or biosimilar patent dance process, the cumulative litigation burden can be prohibitive. The originator does not need to win every case; it simply needs to make the litigation expensive enough and slow enough that potential entrants recalculate the economics of market entry.
In the EU, a patent thicket creates legal exposure but not regulatory delay. A generic manufacturer that has received its EMA marketing authorization can launch its product, accepting the risk that doing so might infringe one or more patents in the thicket, with the calculation being that it will win enough patent challenges to make commercial entry viable. This is not an approach available in the United States, where the mandatory 30-month stay prevents launch during active litigation regardless of the manufacturer’s confidence in its legal position.
Part Six: Economic Outcomes
Generic Entry Timing: The Evidence
The most direct measure of whether a regulatory framework succeeds in enabling generic competition is how quickly generics enter the market after patent expiry and how deeply prices fall once they do.
On entry timing, the evidence across both systems is mixed and context-dependent. A landmark study published in the Journal of the American Medical Association found that in the United States, the median time from patent expiry to generic entry for the top-selling drugs was approximately six months, but that the presence of patent litigation significantly extended this to a median of 35 months when litigation was active [34]. The study identified Paragraph IV litigation as the primary driver of delay, not patent strength or regulatory complexity.
European data tells a more varied story. A study by the European Commission’s Joint Research Centre found that generics entered EU markets within one year of patent/SPC expiry for approximately 75% of molecules analyzed, but that the speed of entry varied substantially by country, with Germany and the United Kingdom showing the fastest entry and southern European markets showing slower timelines due to national regulatory and pricing approval processes that are separate from marketing authorization [35].
The more important comparison is the counterfactual: what would US generic entry timing look like without the 30-month stay mechanism? A 2019 study by economists at the Congressional Budget Office estimated that eliminating the 30-month stay and requiring generic manufacturers to post bond before launching would accelerate generic entry by an average of 18 months for contested drugs, saving the healthcare system approximately $2.5 billion annually [36].
Price Effects After Generic Entry
Both systems ultimately produce substantial price competition once generics enter the market, but the speed and magnitude of price decline differ.
In the United States, the 180-day first-filer exclusivity creates a period of duopoly pricing during which the single generic competitor typically prices at 15-30% below brand price. This represents a meaningful discount but not the sharp price reduction that occurs when multiple generics enter simultaneously. Once exclusivity expires and additional generics launch, prices typically fall to 20-30% of brand price within 18 months [37].
In European markets, where no 180-day exclusivity mechanism exists, multiple generics typically enter simultaneously on the first day of market availability. Price competition among multiple entrants from day one is more aggressive, and prices can fall to 30-40% of brand price within the first year of generic competition in well-functioning markets [38]. However, EU member states also apply price controls and reference pricing systems that affect the absolute price levels at which generics compete, making direct comparison with US prices complicated. <blockquote> “Generic drugs account for 90% of all prescriptions dispensed in the United States, yet represent only 20% of total drug spending. The savings generated by generic competition exceed $300 billion annually.” – Association for Accessible Medicines, 2023 Generic Drug & Biosimilar Access & Savings in the U.S. Report [39] </blockquote>
The EU data is comparable: generics represent approximately 70% of prescriptions in major EU markets and approximately 25% of drug spending by value, with savings to national health systems estimated at 35-50 billion euros annually [40]. The slightly lower US penetration rate by prescription count reflects in part the longer delays in generic entry caused by patent linkage litigation.
Healthcare System Costs: A Direct Comparison
When public health economists compare the direct costs of the US and EU pharmaceutical frameworks, they reach conclusions that are largely unfavorable to the US patent linkage model from a payer’s perspective.
A 2020 study published in JAMA Internal Medicine compared US and European net prices for 79 brand-name drugs marketed in both regions and found that US net prices were, on average, 256% of prices in comparable European countries [41]. The study identified patent protection period length as one contributing factor, though not the only one: US payer fragmentation, absence of central price negotiation, and pharmacy benefit manager practices also contribute significantly to the price differential.
The portion of this differential attributable specifically to patent linkage litigation delays is difficult to isolate. But the FTC’s consistent finding that pay-for-delay settlements cost US consumers approximately $3.5 billion annually, combined with the documented generic entry delays from 30-month stays, points to linkage as a meaningful driver of higher US pharmaceutical spending [42].
For the biosimilar market, where the stakes are highest given the prices of biologics, the patent linkage comparison between the US and EU is even more stark. The biosimilar regulatory pathway in the US, the Biologics Price Competition and Innovation Act’s “patent dance” procedure, creates a multi-step compulsory pre-litigation information exchange between reference product sponsors and biosimilar applicants before any litigation can proceed [43]. The process is complex, has generated significant case law on whether participation is mandatory, and has contributed to the much slower development of US biosimilar competition compared to the EU.
Part Seven: Case Studies
Clopidogrel (Plavix): The US Litigation Machine in Action
Clopidogrel, marketed as Plavix by Sanofi and Bristol-Myers Squibb, is one of the most instructive cases in the history of US pharmaceutical patent litigation. It illustrates how the patent linkage system can protect a blockbuster drug for years beyond what a straightforward patent analysis would suggest.
Clopidogrel’s compound patent was set to expire in November 2011. However, Sanofi also held US Patent No. 4,847,265, covering the bisulfate salt form of clopidogrel. This salt form patent was listed in the Orange Book. Generic manufacturers filed Paragraph IV certifications challenging the salt form patent, arguing it was obvious in light of the prior art on the compound itself.
In litigation that wound through the courts for years, the US Court of Appeals for the Federal Circuit ultimately upheld the salt form patent in 2007, ruling that the enhanced bioavailability and stability of the bisulfate salt were not obvious to a person of ordinary skill in the art at the time of the patent filing [44]. This ruling extended effective Plavix exclusivity to 2012, contributing to cumulative US sales exceeding $70 billion over the drug’s commercial life.
The European experience was different. The corresponding SPC for Plavix in the EU expired in July 2013, and national courts in several member states reached different conclusions about the validity of secondary patents. In France, generic entry was delayed. In Germany and the United Kingdom, generic manufacturers launched earlier based on different judicial assessments of patent validity. The absence of patent linkage meant that each national market played out according to its own courts’ analysis of local patent law, with no EMA delay imposed across the board.
Imatinib (Gleevec/Glivec): Data Exclusivity in Practice
Imatinib, Novartis’s blockbuster cancer drug, illustrates the data exclusivity system and the limits of secondary patent protection in a different way. The drug’s compound patent expired in multiple markets between 2013 and 2016. Novartis famously sought and was denied patent protection for the beta crystalline form of imatinib mesylate in India, where the Supreme Court ruled that the crystalline form did not meet the enhanced efficacy standard required for patentability under Section 3(d) of India’s Patents Act [45].
In the United States, imatinib faced Paragraph IV challenges beginning in 2013, and the resulting litigation was resolved through settlements that permitted generic entry in early 2016. In Europe, the original Glivec patent expired in 2016, and generic imatinib entered multiple European markets within months under national tenders. The SPC for the original compound had expired, there were no valid secondary patents capable of being enforced to delay generic marketing authorization, and the data exclusivity period under the 8+2+1 framework had long since run.
The imatinib case is notable because it shows how different systems treat the same scientific innovation differently. The US system generated years of litigation over secondary patents. The EU system had clear expiry dates for both data exclusivity and SPC protection, and generics entered on those dates with minimal regulatory friction.
Atorvastatin (Lipitor): The 180-Day Exclusivity Race
Atorvastatin, Pfizer’s Lipitor, was at the time of its patent expiry the best-selling drug in pharmaceutical history, with annual US sales exceeding $13 billion. The story of its patent expiry and generic entry is a study in the strategic behavior that the 180-day first-filer exclusivity creates.
Ranbaxy Laboratories, an Indian generic manufacturer, filed the first Paragraph IV certification against the Lipitor patents in 2003, claiming the patents were invalid. The resulting litigation lasted nearly a decade, resolving through a settlement that allowed Ranbaxy to launch a generic in November 2011, the date of the basic compound patent expiry [46]. As the first filer, Ranbaxy was entitled to 180 days of exclusive generic marketing.
However, Ranbaxy’s regulatory status was complicated by FDA enforcement actions related to manufacturing quality violations at its Indian plants. The FDA issued import alerts on Ranbaxy products, creating uncertainty about whether Ranbaxy could actually launch on the agreed date. Watson Pharmaceuticals, which had also filed an ANDA and was waiting for Ranbaxy’s exclusivity to expire, faced the prospect that Ranbaxy’s exclusivity period would run without Ranbaxy actually selling product, using up the exclusivity window without providing price competition.
This scenario, where a first-filer holds exclusivity but cannot or does not launch, effectively delays all subsequent generics. The 2012 “park and pay” reforms to the Hatch-Waxman Act created mechanisms to forfeit first-filer exclusivity in certain circumstances [47], but the Lipitor situation illustrated how the 180-day mechanism, designed to incentivize Paragraph IV challenges, can paradoxically delay generic competition when first-filer circumstances are complicated.
In Europe, multiple generic atorvastatin products entered the market beginning in 2012 across different member states. There was no 180-day exclusivity for any single generic, no first-filer race, and price competition among multiple entrants drove prices down rapidly. The EU experience with atorvastatin has been cited in academic literature as a case study in how the absence of first-filer exclusivity can produce faster, deeper price competition than the US 180-day mechanism [48].
Part Eight: Biosimilars and the Patent Dance
The US BPCIA and the Biosimilar Patent Dance
The Biologics Price Competition and Innovation Act of 2009 created the US regulatory pathway for biosimilars, analogous to the ANDA pathway for small molecule generics. But unlike the Hatch-Waxman pathway, which matured over decades of practice and litigation, the BPCIA introduced a novel and complex patent resolution procedure from the outset [49].
The “patent dance” is a multi-step exchange of information between the reference product sponsor (the originator biologic company) and the biosimilar applicant. The biosimilar applicant discloses its FDA application and manufacturing information. The reference product sponsor identifies patents it believes would be infringed by the biosimilar. The parties exchange lists of patents and positions on each. They negotiate which patents will be litigated in an initial wave and which will be held back for a second round of litigation that cannot begin until commercial launch.
The procedure was designed to front-load patent disputes and resolve them before commercial launch rather than at launch. In practice, it has generated enormous litigation over whether each step of the patent dance is mandatory, what the consequences of non-participation are, and how the 12-year data exclusivity period interacts with the patent dance timeline [50].
The Supreme Court addressed several of these questions in Sandoz Inc. v. Amgen Inc. in 2017, ruling that participation in the patent dance information exchange is optional but that certain notice requirements to the reference product sponsor are mandatory [51]. The ruling clarified parts of the framework but did not eliminate the fundamental complexity.
EU Biosimilar Framework: An Instructive Contrast
The EU approved the world’s first biosimilar in 2006, roughly a decade before the US had a functional biosimilar pathway. European regulators at the EMA developed scientific guidelines for biosimilar approval through a process of international collaboration and iterative refinement that is now recognized as global standard-setting [52].
The EU biosimilar approval process involves no equivalent of the patent dance. The biosimilar applicant submits a comprehensive comparability dossier to the EMA demonstrating that the biosimilar has no clinically meaningful differences from the reference biologic in terms of quality, safety, and efficacy. The EMA evaluates the dossier on scientific merits. If approved, the biosimilar receives a marketing authorization. Patent questions are the reference product sponsor’s problem to handle in national courts.
As of 2023, over 90 biosimilars had been approved in the EU compared to approximately 40 in the United States [53]. The price differential is substantial: EU biosimilar prices are typically 20-35% below reference product prices, and in some therapeutic categories where multiple biosimilars compete, discounts of 60-70% from reference product list prices have been observed through national tendering systems [54]. US biosimilar discounts, while growing, averaged approximately 10-20% off list price in 2022, partly reflecting the slower pace of market entry and the complexity of the patent landscape.
Part Nine: Tools for Intelligence and Compliance
Tracking the Patent Landscape: What Practitioners Actually Need
Patent and regulatory intelligence is not an academic exercise. It is a commercial necessity for anyone making capital allocation decisions in the pharmaceutical industry. A generic manufacturer deciding whether to invest $50 million in developing a generic formulation needs to know not just when the compound patent expires but whether there are secondary patents capable of triggering 30-month stays, what the SPC situation looks like across key EU markets, what litigation has already been filed by or against other ANDA applicants for the same drug, and how the data exclusivity timeline interacts with all of these patent dates.
DrugPatentWatch is one of the most widely used tools for this analysis in the US context. The platform aggregates Orange Book data, ANDA filing information, Paragraph IV certifications, litigation records, and patent term extension information to provide a consolidated view of the competitive patent landscape for any given drug. For a generic company evaluating the clopidogrel or atorvastatin situation in retrospect, or evaluating today’s equivalent situation for a drug approaching patent expiry, DrugPatentWatch provides the structured data that makes systematic analysis possible rather than requiring researchers to manually compile information from multiple FDA and court databases.
For EU analysis, the EMA’s public assessment reports and the European Patent Office’s patent information system provide the core data sources, but integrating them with SPC expiry information, national court decisions, and marketing authorization timelines requires either substantial internal expertise or access to commercial intelligence platforms. The complexity of the EU’s national fragmentation, where the same drug might face different SPC situations in Germany, France, and Italy based on the specific claims of national SPCs granted under the pre-2009 national SPC systems, makes this analysis genuinely challenging.
Regulatory Intelligence as Competitive Advantage
The companies that consistently execute well in the generic pharmaceutical market treat patent and regulatory intelligence as a core competency rather than a due diligence function triggered by specific filing decisions. They maintain ongoing surveillance of the patent landscape for drugs in their therapeutic focus areas, track litigation outcomes for competitors’ ANDA filings, monitor FDA and EMA approval rates and timelines for comparable products, and update their commercial timeline models regularly.
This intelligence function is particularly valuable at the intersection of the US and EU systems, where a generic manufacturer needs to make resource allocation decisions across both markets simultaneously. A drug approaching SPC expiry in Germany in 2026 may still be in active Paragraph IV litigation in the United States. Understanding both timelines, knowing which US patents are most likely to succeed or fail in litigation based on claim analysis and judicial precedent, and modeling the EU competitive entry scenario with multiple simultaneous generic entrants requires integrated cross-jurisdictional intelligence.
Part Ten: Reform Debates
EU Pharmaceutical Legislation Reform: The 2023 Proposal
The European Commission published a comprehensive proposal to reform EU pharmaceutical legislation in April 2023, the first major overhaul of the framework since 2004 [55]. The reform package addressed data exclusivity, market protection, and the SPC system in ways that generated significant controversy.
The proposal would reduce the baseline data exclusivity period from eight years to six years for products that do not meet certain criteria related to unmet medical needs, pediatric data, and comparative effectiveness. Products that launch in all EU member states within two years would retain the full eight years. Products addressing high unmet medical needs could receive extended protection of up to 12 years [56].
The proposal explicitly maintained the separation between regulatory data exclusivity and patent rights. Nothing in the reform proposal created a patent linkage mechanism. The Commission’s position on the inappropriate coupling of regulatory approval and patent disputes did not change.
The European Federation of Pharmaceutical Industries and Associations opposed the reduction in data exclusivity baseline, arguing it would reduce incentive for pharmaceutical R&D investment in Europe and encourage companies to prioritize US launches over EU launches [57]. Generic and biosimilar industry groups supported the reform as a measure to accelerate patient access to more affordable medicines.
As of this writing, the proposal is progressing through the EU’s co-legislative process between the European Parliament and the Council of the EU, with a final text expected in 2025 or 2026. The outcome will have significant implications for generic entry timelines across the EU, but will not alter the fundamental absence of patent linkage in the European regulatory system.
US Reform Proposals
Reform of the Hatch-Waxman patent linkage system has been proposed repeatedly in the US Congress without producing major structural change. The CREATES Act, which became law in 2019, addressed a specific abuse where brand manufacturers refused to provide generic manufacturers with samples of their products needed for bioequivalence testing, effectively blocking ANDA filings [58]. This was a targeted fix rather than structural reform of the linkage mechanism.
More sweeping proposals have included limiting the 30-month stay to a single stay per drug product without the ability to restart stays through new patent litigation, requiring cash bonds from patent holders who impose stays to compensate generic manufacturers if the patents are found invalid, creating an expedited Patent Trial and Appeal Board review procedure for Orange Book patents, and modifying the 180-day first-filer exclusivity to prevent parking.
The Affordable Prescriptions for Patients Act, which passed the Senate Judiciary Committee in 2019, would have limited the number of patents a brand manufacturer could assert in litigation against a generic ANDA filer and placed restrictions on certain types of product hopping [59]. The bill did not pass the full Senate.
Any structural reform of Hatch-Waxman faces the same challenge that has blocked reform for four decades: the Act was a legislative compromise, and reopening it invites all parties to re-litigate the underlying bargain. Generic manufacturers, who benefit from the ANDA pathway and the 180-day exclusivity bounty, are not uniformly supportive of reforms that would reduce the value of being a first filer. Brand manufacturers fight any reduction in their ability to use the Orange Book to protect market position. Payers and patient advocates push for faster generic entry. The political coalition needed to pass comprehensive reform has not materialized.
TRIPS Flexibilities and Developing Country Policy
Beyond the US-EU comparison, the question of patent linkage has significant implications for developing countries navigating their obligations under the TRIPS Agreement and the Doha Declaration on TRIPS and Public Health.
The Doha Declaration, adopted in 2001, confirmed that TRIPS must be interpreted and implemented in a way that supports WTO members’ rights to protect public health and promote access to medicines [60]. The Declaration explicitly confirmed that members have the right to grant compulsory licenses, define the grounds for such licenses, and determine what constitutes a national emergency. It also confirmed that TRIPS does not and should not prevent countries from taking measures to protect public health.
Nothing in TRIPS or the Doha Declaration requires patent linkage. When developing countries are pressed by US trade negotiators to adopt patent linkage in bilateral FTA negotiations, they are being asked to go beyond TRIPS minimum standards in ways that the Doha Declaration suggests they are not obligated to accept. Several countries, including Thailand and India, have explicitly declined to adopt patent linkage on public health grounds [61]. The EU’s own rejection of linkage lends credibility to the position that linkage is not a necessary feature of a functioning pharmaceutical market.
Part Eleven: The Future of the Two Systems
Convergence Pressures and Resistance Points
Despite four decades of divergence, there are genuine pressures toward convergence in some respects, even as the fundamental architectural differences persist.
The global harmonization of pharmaceutical regulatory standards through the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has brought FDA and EMA technical requirements for drug approval increasingly close together [62]. Bioequivalence standards, pharmaceutical quality requirements, and clinical trial design standards are now sufficiently aligned that data generated for one regulatory system can generally support applications in the other. This does not create patent linkage in the EU or eliminate it in the US, but it does reduce the regulatory burden of operating in both markets simultaneously.
The Unified Patent Court, which became operational in June 2023 and covers 17 EU member states (with more expected to join), creates a pan-European patent enforcement forum that may accelerate IP dispute resolution in ways that affect generic entry timelines [63]. Before the UPC, a patent holder seeking to block generic entry across Europe needed to file national actions in each country. The UPC allows a single action to produce a binding decision across all participating member states. This could work in either direction: an originator that successfully obtains a pan-European injunction through the UPC could block generic entry across a much larger market simultaneously, but a generic manufacturer that successfully invalidates a patent at the UPC clears the market for all participating member states at once.
The Unitary SPC: A Developing Question
The creation of the Unitary Patent under EU Regulation 1257/2012 raises questions about whether a corresponding Unitary SPC will be created that covers the same 17-plus member states in a single certificate [64]. Currently, SPCs are national rights that must be applied for separately in each member state, creating a patchwork of different expiry dates and potentially different validity determinations.
A Unitary SPC would rationalize this system, providing clear single-market expiry dates for originator protection and simplifying generic entry planning. The Commission has proposed Unitary SPC legislation as part of the 2023 pharmaceutical reform package, though the specific design and eligibility criteria remain under negotiation.
From the perspective of generic manufacturers, a Unitary SPC is a double-edged development. On one hand, it creates predictability: a single expiry date, a single invalidity proceeding, and a single entry date for the entire unitary patent area. On the other hand, a successfully defended Unitary SPC would block a much larger market than any individual national SPC, raising the stakes of SPC invalidity litigation considerably.
AI and Patent Intelligence: The Next Frontier
The volume and complexity of pharmaceutical patent data continues to grow. The USPTO granted over 350,000 patents in fiscal year 2023, and the pharmaceutical sector accounts for a significant and growing share [65]. Orange Book listings are updated monthly. ANDA filings generate new Paragraph IV certifications continuously. EU national patent databases publish new applications and grants in multiple languages across 27 member states.
Machine learning tools applied to patent analysis are increasingly capable of identifying invalidating prior art, predicting litigation outcomes based on historical data, and flagging potential patent thicket situations around drugs approaching commercial development. Generic manufacturers that invest in these capabilities gain a genuine analytical advantage over competitors relying on manual patent review processes.
The integration of regulatory data, patent data, litigation records, and commercial market data into unified intelligence platforms is already underway through services like DrugPatentWatch and its competitors. The next generation of these tools will incorporate predictive modeling about regulatory approval timelines, litigation probability distributions, and competitive entry scenarios, giving generic manufacturers the kind of decision-support infrastructure that until recently was only available to the largest companies with substantial internal regulatory intelligence teams.
Conclusion: What Each System Reveals About the Other
The US patent linkage system and the EU data exclusivity-only system are not simply technical regulatory choices. They reflect different answers to a genuinely hard question: how much of the protection that the law gives to pharmaceutical innovators should be embedded in the regulatory process itself, and how much should be left to private enforcement through the courts?
The US answer has been: significant coupling. Orange Book listings, Paragraph IV certifications, 30-month stays, and 180-day exclusivities all create a system where the regulatory agency is structurally enrolled in enforcing intellectual property rights that it has no expertise to evaluate. The benefits of this system for innovators are real: automatic delay of generic entry without any judicial assessment of patent validity or merit. The costs are also real: approximately $3.5 to $12 billion annually in delayed generic savings, a litigation system that consumes substantial legal resources on both sides, and patent strategies that use regulatory mechanics rather than genuine innovation as their primary tool.
The EU answer has been: no coupling at all. Regulatory approval is a scientific and administrative matter. Patent enforcement is a legal matter. Each operates in its own domain. The benefits of this design are clear in the data: faster generic entry in most categories, lower prices within a year of expiration, and a regulatory agency that can focus on what it knows how to do. The costs are less obvious but real: a fragmented national enforcement landscape, SPC complexity, torpedo litigation tactics, and a patent thicket problem that national courts address inconsistently.
Neither system is obviously superior in all respects. The US system, despite its litigation costs, has successfully incentivized Paragraph IV challenges that have invalidated many weak secondary patents that might otherwise have gone unchallenged. The EU system, despite its cleaner architecture, still produces significant variation in generic entry timing across member states due to national regulatory and pricing approval differences that sit entirely outside the patent linkage question.
What the comparison makes clear is that the choice between these systems has consequences that extend well beyond the pharmaceutical sector. When the United States pushes patent linkage in trade negotiations as a condition for market access, it is not simply exporting a technical regulatory mechanism. It is exporting the entire commercial and litigation ecosystem that Hatch-Waxman has generated over forty years. Developing countries evaluating that choice benefit from understanding what both the US and EU experiences have actually shown.
Key Takeaways
The US patent linkage system, established by Hatch-Waxman in 1984, embeds intellectual property enforcement directly into the drug approval process through Orange Book listings, mandatory 30-month stays, and 180-day first-filer exclusivity.
The European Union explicitly rejected patent linkage as incompatible with the EMA’s scientific mandate and the institutional separation of regulatory and intellectual property law. EU regulators approve generics on pharmaceutical grounds; patent holders must enforce their rights through national courts.
EU data exclusivity under the 8+2+1 framework provides ten to eleven years of protection for originator drugs, running entirely independently of patent status. US data exclusivity runs five years for new chemical entities, also independently, but in parallel with Orange Book patent linkage protections.
30-month stays in the United States delay generic entry by an average of 35 months in contested cases, costing the US healthcare system an estimated $3.5 to $12 billion annually in foregone generic savings.
Evergreening strategies differ mechanically between the two systems: US companies use Orange Book manipulation; EU companies rely on divisional patent filings, SPC applications, and national court injunctions.
The US biosimilar patent dance is more complex and has produced slower biosimilar market development than the EU framework, where biosimilar approvals began in 2006 and have consistently outpaced US approvals by volume and price competition depth.
The Unified Patent Court, operational since June 2023, has the potential to both accelerate and concentrate patent enforcement in the EU in ways that will affect generic entry strategies without introducing any patent linkage into the regulatory process.
TRIPS does not require patent linkage. The EU’s consistent refusal to adopt it provides developing countries with a significant counterargument to US trade pressure for linkage in bilateral FTA negotiations.
Patent and regulatory intelligence tools, including platforms like DrugPatentWatch, are essential for manufacturers operating in both systems who need to model generic entry timelines, litigation risk, and competitive landscape across jurisdictions simultaneously.
Frequently Asked Questions
Q1: Can a generic manufacturer in the EU launch its product while the originator’s patent is still in force, given that the EMA does not check patent status?
A: Yes, technically. The EMA grants marketing authorizations on the basis of pharmaceutical quality, safety, and efficacy, and a marketing authorization does not confer the right to sell; it confers the right to seek the right to sell. If an originator holds a valid patent and the generic launches without a license, the originator can obtain a national court injunction to halt sales. The generic manufacturer would be infringing the patent. However, the EU Bolar exemption allows all preparatory steps needed to obtain regulatory approval, including bioequivalence studies and regulatory submissions, to proceed during the patent term without infringement. The line between preparation (permitted) and commercial sale (potentially infringing) is legally clear in principle, though complex in practice for activities like supply chain preparation and commercial manufacturing scale-up.
Q2: Why do some developing countries face US pressure to adopt patent linkage even when the TRIPS Agreement does not require it?
A: US bilateral and regional free trade agreements routinely contain “TRIPS-plus” intellectual property provisions that go beyond WTO minimum standards. Patent linkage is one such provision: the US negotiated it into agreements with Korea, Jordan, Chile, Singapore, Australia, Morocco, and several Central American countries, among others. Countries that want preferential access to the US market through FTA tariff reductions must accept these provisions as part of the deal. The leverage is straightforward: the US market is sufficiently large and valuable that many trading partners accept IP provisions they would prefer not to accept in order to secure access. The EU, which negotiates from a different power base and has different domestic pharmaceutical policy priorities, does not include patent linkage requirements in its FTAs, giving developing countries that prefer not to adopt linkage a template for what a modern, TRIPS-compliant regulatory framework can look like without patent linkage.
Q3: If the EU’s system is arguably better for generic competition, why hasn’t the US moved toward adopting it?
A: Several structural factors preserve the US system. First, the Hatch-Waxman Act created a political bargain that gave both brand manufacturers and generic manufacturers substantial benefits; neither side wants to reopen the deal. Second, US pharmaceutical companies, which collectively spend the most on lobbying of any industry sector in Washington, have a strong financial interest in preserving the Orange Book system. Third, trial lawyers on both sides of patent litigation have built substantial businesses around Paragraph IV cases and have no interest in eliminating the work. Fourth, the first-filer exclusivity mechanism creates an incentive for some generic manufacturers to support the litigation-intensive system, because the 180-day bounty only exists because the litigation pathway exists. The coalition in favor of structural reform has not outweighed the coalition in favor of the status quo.
Q4: How does the Supplementary Protection Certificate system in the EU actually compare to patent term restoration under Hatch-Waxman in terms of the effective protection they provide?
A: Both mechanisms compensate innovators for patent term consumed during regulatory review, but they differ in calculation and scope. US patent term extension under Hatch-Waxman restores up to five years of the patent term lost during FDA review, but the restored term plus the remaining patent term cannot exceed 14 years from the date of FDA approval. The calculation is based on time spent in clinical trials and FDA review. EU SPCs cover active ingredients (or combinations) in approved products, take effect at patent expiry, and run for the time between patent filing and first EU marketing authorization, minus five years, up to a maximum of five years of additional protection. The pediatric extension adds six months. Both systems can result in up to five and a half years of effective additional protection from patent expiry, but the specific calculation differs and can produce different outcomes for the same product depending on development timelines.
Q5: What specific analytical capabilities should a generic manufacturer’s regulatory intelligence function have to operate effectively in both the US and EU markets simultaneously?
A: Effective cross-jurisdictional patent intelligence requires several distinct capabilities working in integration. First, real-time tracking of Orange Book listings and changes, because brands add and remove patents, and new listings can trigger new Paragraph IV obligations even after an ANDA is filed. Second, comprehensive SPC monitoring across EU member states, because SPC expiry dates differ by country based on national filing histories and different interpretations of CJEU case law on SPC eligibility. Third, litigation surveillance covering both US ANDA-related patent cases and EU national court proceedings, including preliminary injunction applications. Fourth, data exclusivity timeline modeling that accounts for the US five-year/three-year distinction and the EU 8+2+1 framework for products approved in both markets. Fifth, active monitoring of patent office publications for divisional applications, continuation applications, and re-examinations that could create new IP obstacles around target molecules. Tools like DrugPatentWatch address the US side of this intelligence requirement comprehensively, but full cross-jurisdictional intelligence typically requires combining US-focused platforms with EU patent office databases, EMA public assessment reports, and national court monitoring services.
References
[1] Feldman, R., & Frondorf, E. (2018). Drug wars: How big pharma raises prices and keeps generics off the market. Cambridge University Press.
[2] Drug Price Competition and Patent Term Restoration Act of 1984, Pub. L. No. 98-417, 98 Stat. 1585 (1984).
[3] Food and Drug Administration. (2023). Approved drug products with therapeutic equivalence evaluations (43rd ed.). U.S. Department of Health and Human Services.
[4] 21 U.S.C. § 355(j)(2)(A)(vii) (2023).
[5] 21 U.S.C. § 355(j)(5)(B)(iii) (2023).
[6] Hemphill, C. S., & Sampat, B. N. (2012). Evergreening, patent challenges, and effective market life in pharmaceuticals. Journal of Health Economics, 31(2), 327-339.
[7] Federal Trade Commission. (2002). Generic drug entry prior to patent expiration: An FTC study. FTC.
[8] Medicare Prescription Drug, Improvement, and Modernization Act of 2003, Pub. L. No. 108-173, 117 Stat. 2066 (2003).
[9] Grabowski, H., Long, G., Mortimer, R., & Boyo, A. (2014). Updated trends in US brand-name and generic drug competition. Journal of Medical Economics, 17(1), 13-19.
[10] Wouters, O. J., McKee, M., & Luyten, J. (2020). Estimated research and development investment needed to bring a new medicine to market, 2009-2018. JAMA, 323(9), 844-853.
[11] Federal Trade Commission. (2010). Pay-for-delay: How drug company pay-offs cost consumers billions. FTC.
[12] FTC v. Actavis, Inc., 570 U.S. 136 (2013).
[13] DiMasi, J. A., Grabowski, H. G., & Hansen, R. W. (2016). Innovation in the pharmaceutical industry: New estimates of R&D costs. Journal of Health Economics, 47, 20-33.
[14] 21 U.S.C. § 355(c)(3)(E)(ii) (2023).
[15] Biologics Price Competition and Innovation Act of 2009, Pub. L. No. 111-148, 124 Stat. 804 (2010).
[16] Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001. Official Journal of the European Communities, L 311/67.
[17] Case C-316/95, Generics (UK) Ltd. v. Smith Kline and French Laboratories Ltd. [1997] ECR I-3929.
[18] Directive 2004/27/EC of the European Parliament and of the Council of 31 March 2004. Official Journal of the European Union, L 136/34.
[19] Directive 2001/83/EC, Article 10(6), as amended.
[20] Regulation (EC) No 469/2009 of the European Parliament and of the Council of 6 May 2009. Official Journal of the European Union, L 152/1.
[21] Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006. Official Journal of the European Union, L 378/1.
[22] European Commission. (2009). Pharmaceutical sector inquiry final report. DG Competition.
[23] Agreement on Trade-Related Aspects of Intellectual Property Rights, Apr. 15, 1994, Marrakesh Agreement Establishing the World Trade Organization, Annex 1C, 1869 U.N.T.S. 299.
[24] Sell, S. K. (2007). TRIPS-plus free trade agreements and access to medicines. Liverpool Law Review, 28(1), 41-75.
[25] Comprehensive Economic and Trade Agreement between Canada and the European Union. (2016). Official Journal of the European Union, L 11/23.
[26] European Commission. (2009). Pharmaceutical sector inquiry: Final report. Competition DG.
[27] Ibid., p. 14.
[28] Ibid., pp. 195-202.
[29] Hess, B., Pfeiffer, T., & Schlosser, P. (2008). The Brussels I regulation (EC) no. 44/2001: Application and enforcement in the EU. Beck.
[30] Food and Drug Administration. (2023). Orange Book patent listing requirements for combination products and drug-device combinations: Final rule. 88 Fed. Reg. 42006.
[31] European Medicines Agency. (2022). Reflection paper on the regulatory requirements for the authorisation of a new therapeutic indication for a well-established substance. EMA/CHMP/509951/2006.
[32] European Commission. (2015). Notice to applicants: Medicinal products for human use, volume 2A. European Commission.
[33] Feldman, R. (2018). May your drug price be ever green. Journal of Law and the Biosciences, 5(3), 590-647.
[34] Wouters, O. J., Kanavos, P. G., & McKEE, M. (2017). Comparing generic drug markets in Europe and the United States: Prices, volumes, and spending. Milbank Quarterly, 95(3), 554-601.
[35] European Commission Joint Research Centre. (2018). The impact of generic and biosimilar entry on originator drug prices. JRC Technical Reports.
[36] Congressional Budget Office. (2019). Options for reducing the deficit: 2020 to 2029. CBO.
[37] Berndt, E. R., & Aitken, M. L. (2011). Brand loyalty, generic entry and price competition in pharmaceuticals in the quarter century after the 1984 Waxman-Hatch legislation. International Journal of the Economics of Business, 18(2), 177-201.
[38] Kanavos, P., Seeley, L., & Vandoros, S. (2009). Tender systems for outpatient pharmaceuticals in the European Union: Evidence from the Netherlands, Germany and Belgium. London School of Economics.
[39] Association for Accessible Medicines. (2023). Generic drug & biosimilar access & savings in the U.S.. AAM.
[40] European Generic and Biosimilar Medicines Association. (2023). The value of medicines: European generic and biosimilar medicines. Medicines for Europe.
[41] Anderson, G. F., Hussey, P., & Petrosyan, V. (2019). It’s still the prices, stupid: Why the US spends so much on health care, and a tribute to Uwe Reinhardt. Health Affairs, 38(1), 87-95.
[42] Federal Trade Commission. (2013). Authorized generic drugs: Short-term effects and long-term impact. FTC.
[45] Novartis AG v. Union of India, (2013) 6 SCC 1 (India).
[46] United States v. Ranbaxy Laboratories Ltd., No. 13-cr-00152 (D. Md. 2013).
[47] Medicare Prescription Drug, Improvement, and Modernization Act of 2003, § 1102, Pub. L. No. 108-173.
[48] Dylst, P., Vulto, A., & Simoens, S. (2012). The impact of originator brand medicine reference pricing systems in Europe. Expert Review of Pharmacoeconomics & Outcomes Research, 12(3), 333-344.
[49] 42 U.S.C. § 262(l) (2023).
[50] Lietzan, E. (2014). A new pathway for biosimilars. Food and Drug Law Journal, 69(1), 19.
[51] Sandoz Inc. v. Amgen Inc., 582 U.S. 1 (2017).
[52] European Medicines Agency. (2022). Biosimilar medicines: Overview. EMA.
[53] IQVIA Institute for Human Data Science. (2023). The use of medicines in the U.S. and Europe 2023. IQVIA.
[54] Medicines for Europe. (2023). Biosimilar medicines: European market statistics. Medicines for Europe.
[55] European Commission. (2023). Proposal for a directive on the union code relating to medicinal products for human use and repealing Directive 2001/83/EC. COM(2023) 192 final.
[56] Ibid., Article 80.
[57] European Federation of Pharmaceutical Industries and Associations. (2023). EFPIA position on the pharmaceutical legislation review. EFPIA.
[58] Creating and Restoring Equal Access to Equivalent Samples Act of 2019, Pub. L. No. 116-94, 133 Stat. 3186 (2019).
[59] Affordable Prescriptions for Patients Act of 2019, S. 1416, 116th Cong. (2019).
[60] World Trade Organization. (2001). Declaration on the TRIPS agreement and public health. WT/MIN(01)/DEC/2.
[61] Kessomboon, N., Limpananont, J., Kulsomboon, V., Udomaksorn, S., Eksaengsri, A., & Paothong, P. (2010). Impact on access to medicines from TRIPS-plus: A case study of Thailand. Southeast Asian Journal of Tropical Medicine and Public Health, 41(3), 667-677.
[62] International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2023). ICH guidelines. ICH.
[63] Agreement on a Unified Patent Court, Feb. 19, 2013, O.J. C 175/1 (2013).
[64] Regulation (EU) No 1257/2012 of the European Parliament and of the Council of 17 December 2012. Official Journal of the European Union, L 361/1.
[65] United States Patent and Trademark Office. (2023). Patent technology monitoring team statistics. USPTO.
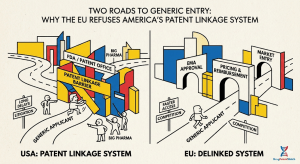



















")






