Every pharmaceutical patent lawyer knows the frustration. A client asks how long drug patent protection lasts in Europe. The technically correct answer — 20 years from the filing date under the European Patent Convention — lands with a thud, because the real answer is far more complicated, commercially more important, and almost never 20 years in either direction.
A blockbuster drug might enjoy effective protection for 25 years through the Supplementary Protection Certificate (SPC) mechanism, an extension designed specifically to compensate pharmaceutical companies for the time lost during regulatory review. Or it might protect effectively for only 8 years if the compound patent was filed early in development and the regulatory approval process consumed most of the nominal term. Or protection might end at different times in Germany, France, Italy, Spain, and Poland — all nominally covered by the same European patent — because SPCs are granted and administered by individual national patent offices, each applying the same EU regulation with slightly different procedural results.
For any professional who needs to know when a drug loses exclusivity in specific European markets — whether you are modeling a generic launch opportunity, conducting M&A due diligence on a branded pharmaceutical asset, advising a biosimilar manufacturer, or tracking competitor patent positions — the 20-year headline is worse than useless. It creates false precision. This article corrects that.
What follows is a systematic account of every mechanism that extends or shortens European drug patent protection beyond the statutory baseline: Supplementary Protection Certificates, pediatric extensions, regulatory data exclusivity under the 8+2+1 framework, Orphan Drug Designation, and the Bolar experimental use exemption. It covers the 2023 Unitary Patent system that is reshaping European litigation strategy. It addresses what Brexit did to the UK’s patent framework. It walks through the SPC manufacturing waiver that changed competitive dynamics for European generics in 2019. And it uses documented case studies — Humira, Gleevec, Nexium — to show what these rules mean in practice.
Part One: The Statutory Foundation
The European Patent Convention: One Grant, Many Countries
The European Patent Convention (EPC), which entered into force in 1977, established the European Patent Office (EPO) as a centralized examination body [1]. An applicant can file a single European patent application with the EPO and, upon grant, receive a bundle of national patents valid in each EPC member state they designate. As of 2024, 39 countries are members of the EPC, including all 27 EU member states, plus the UK, Switzerland, Norway, Turkey, and others [2].
The critical conceptual point is that a European patent is not, in itself, a supranational right. At grant, it converts into a collection of national patents, each governed by national law for enforcement, validity challenges, and most procedural purposes. The EPO examines and grants; the national courts litigate and invalidate. This distinction has profound consequences for how protection actually works across markets, and it is why the question “how long does a European drug patent last” requires market-by-market analysis, not a single number.
The EPC sets 20 years from the filing date as the maximum patent term [3]. The filing date is the date the application was submitted to the EPO (or to a national patent office with subsequent European entry). For a drug that took 12 years to develop from compound identification to regulatory approval, a 20-year term that began ticking at the initial composition patent filing leaves roughly 8 years of patent-protected commercial life. That gap between the statutory term and practical commercial protection is the problem that SPCs were designed to address.
How EPO Examination Timelines Affect Practical Term
The EPO is not particularly fast. Average grant timelines from filing to grant have historically run 3 to 5 years for pharmaceutical patents, though this varies considerably by technology complexity and examination workload [4]. During that examination period, the applicant has provisional protection in designated member states (under most national laws), but full patent rights attach only at grant.
This prosecution timeline interacts with term calculations in a specific way: the 20-year clock runs from the filing date, not the grant date. So a patent filed in 2010 and granted in 2015 expires in 2030 regardless of when it was granted. The applicant gets five fewer years of enforceable patent rights due to examination delay — which is why the EPO has a patent term adjustment mechanism analogous to the USPTO’s PTA.
Under Rule 37(2) of the Implementing Regulations to the EPC, applicants can request accelerated examination. In pharmaceutical contexts, the “PACE” (Patent Prosecution Highway / Accelerated Examination) program allows applicants to request faster processing, with some applications moving to grant in under two years [5]. For a drug near regulatory approval, a six-month acceleration in grant timing can have meaningful commercial value.
National Validation: The Activation Step
Once the EPO grants a patent, the applicant must validate it in each designated country to activate protection. Validation involves paying national validation fees and, for many countries, filing a translation of the patent into the national language. Failure to validate in a country means the patent provides no protection there, regardless of the European grant.
Translation requirements have historically been a significant cost and administrative burden. The London Agreement of 2008, now adopted by 21 EPC member states including the UK (before Brexit), France, Germany, and the Netherlands, reduced translation requirements substantially for those countries [6]. But countries not party to the London Agreement — including Spain, Italy, Portugal, and most Central and Eastern European EPC states — still require full translations for validation.
The commercial implication is that pharmaceutical companies routinely choose not to validate European patents in every possible EPC member state. The decision involves a cost-benefit calculation: validation costs versus the commercial value of the market. A drug generating €50 million annually in Germany justifies validation costs easily. The same drug generating €2 million in Slovakia may not justify the cost of translation and annual renewal fees across the remaining patent term, particularly if the drug is approaching expiry anyway.
This means that for any specific drug, the geographic scope of actual patent protection in Europe frequently differs from the geographic scope of the original European patent grant. Analysts tracking competitive entry timelines need to verify validation status in specific target markets, not just confirm the existence of a European patent grant. Tools like DrugPatentWatch compile patent validation and SPC status data across EU member states for marketed pharmaceutical products, providing the multi-market view that individual national patent office searches cannot efficiently produce.
Part Two: Supplementary Protection Certificates — The Real Extension Mechanism
Why SPCs Exist
The SPC system was created by EU Council Regulation (EEC) No 1768/92, adopted in June 1992 [7]. The regulation’s preamble states the problem plainly: the period between filing a patent application for a new medicinal product and obtaining marketing authorization for it takes on average ten to twelve years, meaning that the period of effective protection under the patent is insufficient to cover the investment put into the research. The SPC compensates by extending effective protection.
The basic logic of the SPC is straightforward. Take the total time between the patent filing date and the first EU marketing authorization for the product. The SPC duration equals that period, minus five years, with a maximum SPC term of five years. This five-year ceiling reflects a policy judgment that the maximum total effective protection — patent term plus SPC — should be capped at 15 years from first marketing authorization.
Understanding this calculation concretely matters. If a compound patent is filed in January 2000, and the European Medicines Agency (EMA) grants the first EU marketing authorization in January 2012, the gap is 12 years. Subtract 5 years: the SPC runs for 7 years. But 7 exceeds the maximum of 5, so the SPC duration is capped at 5 years. The patent expires in January 2020 (20 years from filing). The SPC then runs until January 2025.
Alternatively, if that same compound patent is filed in January 2005 (earlier research phase, later patent strategy), and the marketing authorization still arrives in January 2012, the gap is 7 years. Subtract 5: the SPC runs for 2 years, from January 2020 (patent expiry) to January 2022. No cap applies. This is why drug companies and their IP counsel think carefully about the timing of patent filings relative to development timelines — a filing that is too early can produce a shorter SPC, while a filing that is too late may produce no valid patent at all if prior art is published in the interim.
The Five Conditions for SPC Eligibility
EU Regulation No 469/2009 (which consolidated and replaced the 1992 regulation) [8] specifies that an SPC application must meet five conditions:
The product must be protected by a basic patent in force — the “basic patent” being the compound, formulation, or process patent whose claims cover the product.
A valid authorization to place the product on the market must exist in the EU member state where the SPC application is filed.
The product must not have already been the subject of an SPC.
The marketing authorization cited must be the first authorization to place the product on the market as a medicinal product.
The SPC must be applied for within six months of either the marketing authorization date or the patent grant date, whichever is later.
Each of these conditions has generated substantial litigation at both national and European Court of Justice (CJEU) levels. The “first authorization” requirement in condition four has produced the most complex case law. The CJEU has consistently held that the relevant “first authorization” is the first authorization anywhere in the EU/EEA, not the first authorization in the specific member state where the SPC is filed [9]. This means that a drug approved in Germany in 2015 and in Bulgaria in 2018 gets its SPC duration calculated from the 2015 German authorization even when applying for an SPC in Bulgaria.
What Products Qualify: The “Product” Definition
One of the most litigated questions in European SPC law is what counts as a “product” for SPC purposes. The Regulation defines “product” as the active ingredient or combination of active ingredients of a medicinal product [10]. This sounds simple; it has generated over two decades of CJEU case law.
The central tension is between broad and narrow interpretations. A patent might cover a genus of thousands of related compounds. The specific compound approved as a drug might be explicitly disclosed in that genus patent, or might be covered only by the general structural formula. Does the SPC attach to the genus patent? The CJEU addressed this in Eli Lilly v. Human Genome Sciences [11], holding that an SPC can be granted on a patent that covers a product through a functional description — a claim that specifies the biological activity or mechanism of action rather than the specific molecule — provided that the product is specifically identifiable in light of all the information disclosed in the patent. This remains a contested area where national courts continue to apply the CJEU’s guidance with varying results.
Combination products present another layer of complexity. When a new drug is a combination of two active ingredients — one previously approved, one new — the question of which patent qualifies as the “basic patent” for the combination’s SPC has been the subject of extensive CJEU litigation. The Actavis v. Sanofi case [12] established that an SPC for a combination product requires that each ingredient of the combination be specified in the claims of the basic patent, not merely covered by one ingredient’s patent. This ruling eliminated SPC opportunities that some manufacturers had anticipated for combination products protected by single-ingredient patents.
Calculating the SPC: Step-by-Step
For any specific drug, the SPC calculation requires four data inputs:
The filing date of the basic patent (not the grant date, not the priority date — the actual EPO or national filing date for purposes of term calculation).
The grant date of the basic patent (relevant for the six-month application deadline).
The date of the first marketing authorization for the product in the EU/EEA.
The expiry date of the basic patent (20 years from filing, adjusted for any patent term corrections in the relevant jurisdiction).
The SPC duration formula under Article 13 of Regulation 469/2009: SPC duration = (Marketing Authorization Date – Patent Filing Date) – 5 years, subject to a maximum of 5 years.
The SPC begins running from the expiry date of the basic patent and provides the same rights as the patent itself — the right to exclude others from making, using, selling, and importing the product — for its duration.
A worked example: Compound X is the subject of a European patent filed April 15, 2001, expiring April 15, 2021. The first EU marketing authorization for Compound X is granted October 20, 2011. The interval between filing and authorization is 10 years and roughly 6 months, or about 10.5 years. Subtract 5: SPC duration is 5.5 years. Capped at 5 years. The SPC runs from April 15, 2021 to April 15, 2026. Total effective protection from first EU authorization to end of SPC: approximately 14.5 years.
This example illustrates why top-selling drugs in Europe rarely lose exclusivity at the 20-year patent expiry mark. SPCs routinely extend effective commercial protection by 3 to 5 years beyond patent expiry, and in cases where the approval timeline was long enough, a full 5-year SPC adds a meaningful commercial period that analysts who focus only on patent expiry dates will miss entirely.
The Pediatric Extension: Six Months More
EU Regulation No 1901/2006 on medicinal products for pediatric use created an incentive for pharmaceutical companies to conduct clinical trials in children: a six-month extension to the SPC [13]. The extension applies if the company completes and submits a Pediatric Investigation Plan (PIP) that has been approved by the EMA’s Pediatric Committee, and the pediatric studies are either reflected in the product label or noted as completed.
The extension applies even if the pediatric studies fail to demonstrate efficacy in children — the incentive is for conducting the research, not for finding positive results. This design has been criticized on policy grounds but is well-established legally. Practically, it means that a drug with a full 5-year SPC and a pediatric extension achieves total protection of 5 years and 6 months beyond the basic patent expiry, or approximately 15.5 years from first EU authorization.
The pediatric extension is administered by the same national patent offices that administer the underlying SPC, and must be applied for before the SPC expires. Companies that have completed PIPs and received positive opinions from the Pediatric Committee but fail to apply for the extension before SPC expiry lose it entirely — a procedural risk that is straightforward to track through patent office records.
For revenue modeling purposes, the pediatric extension is commercially material on blockbuster drugs. Six months of additional exclusivity on a drug generating €1 billion annually in Europe represents €500 million in additional protected revenue. Most major pharmaceutical companies pursue pediatric extensions aggressively for their top-selling products.
Part Three: The 8+2+1 Framework — Data Exclusivity Without Patent Protection
What Data Exclusivity Is and Is Not
Data exclusivity is frequently confused with patent protection. The two are legally distinct mechanisms that can overlap chronologically but operate through completely different pathways and provide different types of protection.
Patent protection is a property right. It allows the patent holder to exclude competitors from using the patented invention, regardless of whether competitors independently developed the same product. Infringement requires no use of the patent holder’s data; it requires only that the competitor’s product fall within the claims of the patent.
Data exclusivity is a regulatory barrier. EU Directive 2004/27/EC and Regulation 726/2004 established the “8+2+1” framework [14], which prevents generic drug manufacturers from relying on the originator’s clinical trial data to obtain marketing authorization for a specified period. The generic manufacturer can file an ANDA-equivalent application — called a Marketing Authorization Application (MAA) via the abridged procedure — from the day of originator approval, but the regulatory agency cannot approve it for 8 years (the “data exclusivity” period). After those 8 years, the agency can approve the application, but the generic cannot actually market the product until 10 years from the originator’s approval (the additional 2-year “market exclusivity” period). If the originator obtains a new therapeutic indication during the 8-year data exclusivity period that provides “significant clinical benefit,” an additional 1 year of market exclusivity is granted.
The practical effect is that the minimum period between an originator’s first EU approval and any generic’s legal market entry is 10 years, regardless of patent status. If no patents protect the drug, or if all patents are invalid, generics still cannot enter until the 10-year regulatory exclusivity expires.
How the 8+2+1 System Interacts With Patent Term
The interaction between the 8+2+1 framework and patent/SPC protection depends entirely on timing. Consider two scenarios:
In Scenario A, a drug is approved with 10 years of patent term remaining on its compound patent, plus a 4-year SPC. Total patent/SPC protection from approval: 14 years. Data exclusivity runs for 10 years from approval. Here, the patent/SPC framework dominates — data exclusivity expires before patent/SPC protection ends, so the binding constraint on generic entry is patent/SPC expiry.
In Scenario B, a drug is approved after most of its patent term has run, with only 6 years of patent term remaining and no SPC (because the regulatory approval took less than 5 years from patent filing). Data exclusivity runs 10 years from approval. Here, the data exclusivity framework dominates — the patent expires in year 6 after approval, but generics cannot enter until year 10. For the 4-year gap between patent expiry and data exclusivity expiry, the drug has protection against generic entry even though it has no patent.
This second scenario is more common than many analysts realize, particularly for drugs approved on accelerated regulatory pathways, drugs where the compound patent was filed late in development, and drugs in therapeutic areas where early-stage patents face crowded prior art that forces narrow claim scope and discourages earlier broad filings.
The “1” in “8+2+1” — the additional year for a new indication — is worth tracking separately. When an originator’s drug receives an additional approved indication during the data exclusivity period, and the new indication rests on clinical studies that provide “significant clinical benefit,” the total market exclusivity period extends to 11 years. This incremental year has been the subject of litigation over what “significant clinical benefit” requires, but it has been successfully obtained in documented cases and represents a material revenue extension for drugs with active clinical programs post-approval.
National vs. Centralized Procedure Data Exclusivity
The 8+2+1 framework applies uniformly across EU member states for drugs approved through the EMA’s centralized procedure, which is mandatory for biologics, advanced therapy medicinal products, and drugs for certain conditions, and optional for other new molecular entities [15]. For drugs approved through national procedures (e.g., the mutual recognition procedure or decentralized procedure), data exclusivity periods may be counted from the individual national authorization dates rather than from a single EU-wide date.
In practice, most major branded drugs use the centralized procedure, and their data exclusivity runs from the EMA’s centralized marketing authorization date simultaneously across all EU member states. This synchronized clock means that generics in all EU markets face the same data exclusivity expiry date for centrally-authorized products — a meaningful simplification compared to the market-by-market patent analysis required for SPC coverage.
Part Four: Orphan Drug Designation and Rare Disease Protection
Ten Years of Market Exclusivity for Rare Diseases
EU Regulation No 141/2000 on orphan medicinal products grants 10 years of market exclusivity to drugs approved for rare diseases (defined as conditions affecting fewer than 5 in 10,000 people in the EU) [16]. This orphan exclusivity prevents the EMA from accepting or approving marketing applications for similar medicinal products for the same indication for the duration of the exclusivity period.
The 10-year orphan exclusivity is distinct from both patent protection and data exclusivity. It runs from the date of marketing authorization. It is not reduced or modified by patent term. It is not the same as data exclusivity, though in practice both protections typically run simultaneously.
A drug with orphan designation therefore has a potential protection stack that includes:
Patent/SPC protection (up to 25.5 years from patent filing with full SPC and pediatric extension)
Data exclusivity (10 years from authorization under the 8+2+1 framework)
Orphan exclusivity (10 years from authorization)
Pediatric extension (additional 2 years for orphan drugs that complete a PIP, not 6 months as for non-orphan drugs)
The orphan pediatric extension is 2 years rather than 6 months, reflecting the higher difficulty of conducting pediatric studies in rare disease populations [17]. This extension applies to the orphan exclusivity period itself, not to a patent or SPC.
The policy rationale for the 10-year orphan exclusivity is to compensate for small market sizes that might otherwise make rare disease drug development economically unviable. The commercial reality is that some orphan drugs generate billions in annual revenue because they treat conditions that, while individually rare, affect enough patients globally to support premium pricing. Drugs like Spinraza (nusinersen), Hemlibra (emicizumab), and Orkambi (lumacaftor/ivacaftor) have demonstrated that orphan designation is compatible with very high commercial returns in Europe.
The orphan exclusivity can be reduced from 10 to 6 years if, after 5 years, it is established that the drug is sufficiently profitable that maintaining 10 years of exclusivity is no longer justified [18]. The EMA’s Committee for Orphan Medicinal Products (COMP) can review this status. In practice, the reduction from 10 to 6 years has been applied rarely, and the criteria for triggering the review have been applied narrowly.
Part Five: The Unified Patent System and the Unified Patent Court
What Changed in 2023
June 1, 2023 marked the entry into force of the Unitary Patent (UP) system and the opening of the Unified Patent Court (UPC) [19]. Both developments are the culmination of decades of EU-level efforts to create a more unified patent system across Europe, and both have material implications for pharmaceutical patent strategy.
Before the UP, every European patent that the EPO granted had to be individually validated in each national jurisdiction, enforced in national courts, and litigated under national procedural law. A patent holder facing infringement in Germany, France, and Italy needed three separate national lawsuits. A generic manufacturer challenging patent validity in multiple EU markets needed to file invalidity actions in each national court.
The Unitary Patent changes the first part of this picture. After receiving an EPO grant, applicants can now choose to request Unitary Patent status instead of (or in addition to) national validation in participating UP member states. A Unitary Patent is a single patent right valid across all participating UP states — currently 17 EU member states, with more expected to join [20]. It requires only a single set of renewal fees, no translation requirements (subject to a transitional regime), and is enforceable and potentially revocable through the UPC as a single right.
The UPC changes the second part. Patent infringement and revocation actions can now be filed centrally at the UPC, which has a Court of First Instance with both a Central Division (in Paris and Munich) and Local Divisions in individual member states, and a Court of Appeal in Luxembourg. A single UPC revocation action can invalidate a Unitary Patent across all UP states simultaneously.
Pharmaceutical Industry Response: The Opt-Out Mechanism
For pharmaceutical patent holders, the UPC’s central revocation capability is the most important — and most alarming — feature of the new system. A single successful revocation action at the UPC eliminates the patent across all UP states in a single proceeding. Under the old national system, a generic manufacturer needed to successfully invalidate a patent in each country separately, with varying claim construction standards and prior art arguments potentially reaching different conclusions in different courts.
Recognizing this risk, the UPC Agreement includes a 7-year transitional period during which patent holders can “opt out” existing European patents from UPC jurisdiction [21]. An opted-out patent remains subject only to national courts — the status quo ante — and cannot be centrally revoked through the UPC.
Major pharmaceutical companies pursued opt-out strategies aggressively. Pfizer, AstraZeneca, Johnson & Johnson, Roche, and other large pharma companies filed opt-out applications for thousands of their commercially important European patents in the days surrounding the UPC’s opening [22]. The logic: the UPC is an untested forum whose case law and procedural norms are being built from scratch. Exposing blockbuster drug patents to central revocation risk in an uncertain new court during the critical period before patent expiry is a risk that most brand pharmaceutical companies opted to avoid initially.
Generic and biosimilar manufacturers, by contrast, have generally been interested in using the UPC’s central revocation capability. The ability to challenge a patent across all UP states in a single action dramatically reduces the cost and complexity of multi-country patent challenges — exactly the kind of efficiency that benefits challengers facing the expense of simultaneous national invalidation actions.
The transitional period runs until June 2030. The balance between opt-outs and UPC-registered patents is expected to shift as pharmaceutical companies gain confidence in UPC case law and as the opt-out window closes for patents nearing expiry.
Early UPC Pharmaceutical Decisions
The UPC produced its first pharmaceutical decisions within months of opening. In July 2023, the UPC Court of First Instance (Hamburg Local Division) issued an injunction against a generic manufacturer in a pharmaceutical patent dispute — the first UPC injunction in a pharmaceutical case [23]. The speed of the injunction, granted in an expedited procedure within weeks of filing, demonstrated one of the UPC’s advertised advantages: faster interim relief compared to many national courts.
On the revocation side, the UPC Central Division received its first revocation actions in 2023 and 2024, and early decisions have been closely watched by patent practitioners across Europe. The UPC’s approach to claim construction, inventive step, and the use of prosecution history is being established through this early case law, and it will significantly shape pharmaceutical patent litigation strategy for the coming decades.
For portfolio valuation purposes, the UPC adds a new variable to European pharmaceutical patent risk assessment. Patents that have not been opted out are exposed to a new central revocation pathway. The litigation risk score for any such patent needs to account for the possibility that a single well-funded generic or biosimilar manufacturer could eliminate EU-wide protection through one UPC action.
Part Six: The UK After Brexit
UK Patent Protection: The Post-Brexit Framework
The United Kingdom’s departure from the EU on January 31, 2020, with the transition period ending December 31, 2020, severed UK pharmaceutical patent protection from the EU framework in two important respects: SPCs and regulatory exclusivity.
On patents themselves, the UK remains a member of the EPC, so EPO-granted European patents continue to be validated and enforceable in the UK exactly as before Brexit [24]. UK patent courts — the Patents Court and the Intellectual Property Enterprise Court (IPEC) within the English and Welsh courts, and the Court of Session in Scotland — continue to adjudicate pharmaceutical patent disputes under UK patent law, which mirrors EPC provisions and is implemented through the Patents Act 1977.
The UK is not participating in the Unified Patent Court or the Unitary Patent system. UK pharmaceutical patents remain under national UK jurisdiction exclusively [25]. This means the pre-UPC status quo applies in the UK: national filing/validation, national litigation, and no risk of central UPC revocation.
UK Supplementary Protection Certificates Post-Brexit
UK SPCs are now governed by domestic UK law rather than EU Regulation 469/2009. The Intellectual Property Office (IPO) administers UK SPC applications under retained EU law — the EU SPC regulations were incorporated into UK law at the point of Brexit and continue to apply domestically, though the UK can now amend this framework [26].
The practical implication for calculating UK SPC duration is that the calculation mirrors the EU approach: the reference “first authorization” is now the UK marketing authorization (granted by the Medicines and Healthcare products Regulatory Agency, or MHRA) rather than the first EEA authorization. For drugs that received EMA authorization before Brexit, the first MHRA authorization was typically the same date or very close (UK being an EU member state at the time of original authorization). For drugs approved after January 1, 2021, the MHRA grants independent UK authorizations, which may differ from EMA authorizations by months or even years depending on submission timing and review pace.
This divergence creates a new calculation complexity: a drug approved by the EMA in March 2022 and the MHRA in July 2022 will have an EU SPC calculated from the March date and a UK SPC calculated from the July date. The UK SPC will be 4 months shorter — not a trivial difference for a drug generating hundreds of millions in annual UK revenue.
The UK government has signaled willingness to reform its SPC framework, including proposals to create a “SPC manufacturing waiver” analogous to the EU’s 2019 amendment (discussed below) and to explore other mechanisms for making the UK an attractive jurisdiction for generic manufacturing and export. As of 2024, UK SPC reform is an active policy discussion with uncertain outcomes.
UK Data Exclusivity: The 8+2+1 Equivalent
The UK retained the EU’s data exclusivity framework at the point of Brexit. UK marketing authorizations for new medicines receive 8 years of data exclusivity and 10 years of market exclusivity from the date of UK authorization, mirroring the EU 8+2+1 structure [27].
The “1” additional year for a new indication is also retained in UK law. Post-Brexit, the reference authorization for the UK exclusivity clock is the UK marketing authorization date, not the EMA authorization date. For drugs approved post-Brexit, this means separate exclusivity calculations for EU and UK markets.
The UK also retains the 10-year orphan drug exclusivity period for designated orphan medicines, with the Rare Disease Advisory Group advising the MHRA on orphan designation decisions.
Part Seven: Country-by-Country Variations in SPC Administration
Germany: The Largest European Pharmaceutical Market
Germany generates the largest pharmaceutical market revenue in Europe and, by extension, its SPC system receives the most commercial attention. German SPCs are granted by the German Patent and Trade Mark Office (Deutsches Patent- und Markenamt, DPMA) under EU Regulation 469/2009. Germany’s national courts — the Federal Patent Court (Bundespatentgericht) and the Düsseldorf, Munich, and Mannheim Regional Courts — have extensive pharmaceutical patent litigation experience.
Germany’s patent courts are known for strong preliminary injunction practice. When a patent holder files for a preliminary injunction against a generic launch in Germany, courts can and do grant ex parte injunctions within days, making German patent enforcement among the most patent-holder-favorable in Europe. The Mannheim and Düsseldorf courts in particular have reputations for prompt injunctive relief and relatively patent-holder-sympathetic claim construction.
Germany is also notable for the validity split: under German procedure, infringement actions are heard by the regional courts, while validity challenges are heard by the Federal Patent Court. This bifurcation means a generic manufacturer cannot raise invalidity as a defense in infringement proceedings — it must file a separate invalidity action at the Federal Patent Court, which typically takes 2-3 years to resolve. In the interim, a preliminary injunction can block the generic launch even if the patent is ultimately invalidated.
For SPC calculations specific to Germany, the DPMA applies the standard EU formula without significant national variation. German SPC applications must be filed within 6 months of the later of the marketing authorization date or the patent grant date. The six-month application window is strictly enforced — German courts have upheld rejections of SPC applications filed even days after the six-month deadline.
France: Centralized and Relatively Predictable
French SPCs are granted by the Institut National de la Propriété Industrielle (INPI). France has historically had relatively predictable SPC grant practice and a specialized IP court (the Tribunal Judiciaire de Paris for patent matters) with dedicated pharmaceutical patent expertise.
French courts apply the CJEU’s SPC case law carefully and have generally produced decisions consistent with the broader EU framework. France is a notable jurisdiction for patent linkage considerations: unlike the UK, France does not have a formal patent linkage system that delays drug approval pending patent litigation. Generic manufacturers can obtain French marketing authorizations during the patent/SPC period; they simply cannot launch until protection expires or is cleared.
France also deserves attention for its approach to the “product” definition in SPC law. French courts have in several cases taken positions on combination product SPCs and the basic patent requirement that, while broadly consistent with CJEU precedent, reflect national interpretive judgments. Companies with combination product SPC strategies in France should specifically analyze French CJEU interpretation decisions, not only the CJEU rulings themselves.
Italy and Spain: Translation Requirements and Strategic Implications
Italy and Spain are two of the largest pharmaceutical markets in Europe outside Germany and France. Both require full translations of European patents for national validation, as neither is party to the London Agreement. Both have substantial generic manufacturing industries with active interests in challenging pharmaceutical SPCs.
Italy’s SPC practice is administered by the Italian Patent and Trademark Office (UIBM). Italian SPC litigation has produced a number of nationally significant decisions, particularly around the “first authorization” requirement and what constitutes the relevant marketing authorization date when EU approvals involved procedural complications.
Spain’s SPC administration is handled by the Spanish Patent and Trademark Office (OEPM). Spain is notable for having its own domestic pharmaceutical market protection mechanism — the “data exclusivity” periods apply from the Spanish marketing authorization date for nationally authorized products, with the usual EU framework applying to centrally authorized products.
Both Italy and Spain are active venues for SPC challenge litigation. Generic manufacturers targeting blockbuster drug patents frequently file parallel SPC nullification actions in Italy, Spain, Germany, and France simultaneously, recognizing that a successful challenge in any market opens that specific geographic area to competition without affecting protection in other markets (since SPCs are national rights even for centrally authorized products).
The Netherlands: SPC Litigation Hub
The Netherlands has become a significant European pharmaceutical patent litigation hub for several reasons. The District Court of The Hague has a dedicated IP chamber with extensive pharmaceutical patent expertise. Dutch courts apply English-language patent documents without requiring Dutch translations, making the Netherlands an attractive venue for international pharmaceutical patent disputes.
The Netherlands is party to the London Agreement, so European patents validated in the Netherlands do not require Dutch translations. Dutch SPC applications are filed with the Netherlands Patent Office (NLO) following the standard EU formula.
A Dutch patent court decision in one SPC dispute does not bind courts in Germany, France, Italy, or other member states — but Dutch decisions on CJEU SPC questions carry practical persuasive weight because they frequently reach the CJEU through the Dutch Supreme Court’s active referral practice. Several landmark CJEU SPC decisions began as Dutch national court referrals.
Poland, Czech Republic, Hungary, and Central-Eastern European Markets
Central and Eastern European (CEE) EU member states represent growing pharmaceutical markets with distinct SPC administration practices. Poland, Czech Republic, and Hungary are the largest of these, and all three have domestic pharmaceutical industries with significant generic manufacturing capacity.
SPC durations in CEE countries are calculated using the same EU formula as Western European markets, but with an important historical modification. When EU accession countries joined the EU in 2004 and later, drugs approved before accession used the “first authorization in the EU” concept in a way that created transitional issues. The CJEU addressed several of these transitional cases in rulings that remain important for drugs whose EU commercial history predates 2004.
National patent offices in CEE countries receive SPC applications and apply EU Regulation 469/2009 as implemented in national law. Validation costs in CEE markets are lower than in Western Europe, but the generic manufacturing industry’s political strength in several CEE countries creates a policy environment more actively interested in limiting SPC term and challenging SPCs.
Part Eight: The Bolar Exemption — How Generics Prepare Before Expiry
The Experimental Use Exception in European Patent Law
European pharmaceutical patent law includes an experimental use exception that permits activities conducted for experimental purposes relating to the subject matter of a patented invention without constituting infringement [28]. This general experimental use exception under national patent laws exists across EPC member states, though its scope has historically varied.
The pharmaceutical-specific version — the Bolar exemption, named after the U.S. litigation that prompted similar provisions — allows generic and biosimilar manufacturers to conduct studies and trials necessary for obtaining marketing authorization, even while the patent/SPC protecting the reference product is still in force. Without this exception, a generic manufacturer could not begin clinical bioequivalence studies, regulatory data compilation, or manufacturing scale-up until the day patent/SPC protection expired — adding years to the time from patent expiry to actual generic market entry, and effectively extending the commercial monopoly.
EU Harmonization of the Bolar Exemption
The EU harmonized the Bolar exemption through Article 10(6) of Directive 2004/27/EC, which amended the earlier pharmaceutical directive and established a uniform framework across EU member states [29]. The directive’s Bolar provision permits “studies, tests and trials necessary for the application of [marketing authorization] requirements.” This language is broad enough to cover:
Bioequivalence studies comparing the generic to the reference product.
Pre-clinical studies required by regulatory guidelines.
Manufacturing development and scale-up activities.
Submission of regulatory dossiers and the preparation of market authorization applications.
What the Bolar exemption does not cover, in most EU member states, is commercial stockpiling. A generic manufacturer cannot manufacture large quantities of the drug during the patent period under the Bolar exemption with the intent of launching the day patent protection expires. Manufacturing must be proportionate to the regulatory activities being conducted.
Post-2021 Bolar Expansion: The SPC Manufacturing Waiver
The EU substantially expanded the Bolar framework through a 2019 amendment to the SPC Regulation — in force from July 1, 2019 — that created a specific “SPC manufacturing waiver” [30]. This waiver goes significantly further than the Bolar exemption.
Under the manufacturing waiver, a generic or biosimilar manufacturer can manufacture a product protected by an SPC (not just a patent, but an SPC specifically) in an EU member state for the purpose of export to third countries outside the EU where protection has expired or does not exist. The waiver also permits EU manufacturing for the purpose of stockpiling for EU market entry on the day the SPC expires (subject to notification requirements to the SPC holder).
The stockpiling provision is particularly significant. Under it, a generic manufacturer can build a 6-month supply of the drug during the last 6 months of the SPC period, ready for launch on day one of generic entry. This closes what had been a competitive disadvantage for EU-based generic manufacturers relative to manufacturers in countries without SPC systems, who could produce and stockpile freely.
The manufacturing waiver includes notification obligations: the generic manufacturer must notify the SPC holder of its intent to manufacture under the waiver using a standardized form, and must mark the products manufactured under the waiver with a specific “black box” logo to indicate their status. Violations of the waiver conditions constitute SPC infringement, maintaining the SPC holder’s enforcement rights against misuse.
For pharmaceutical portfolio valuation purposes, the manufacturing waiver changes the competitive entry dynamic at SPC expiry. The practical implication is that the “day one” generic entry that had been theoretically possible but practically delayed by supply chain lead times is now genuinely achievable for well-prepared EU generic manufacturers. Portfolio models that assume a soft landing period of several months after SPC expiry before meaningful generic penetration need to be updated for products where EU generic manufacturers have used the stockpiling provision.
Part Nine: European Biosimilar Patent Framework
How Biologic Patent Protection Differs From Small Molecules
Biologics — drugs derived from living cells, including monoclonal antibodies, recombinant proteins, and cell therapies — receive the same patent protection framework as small-molecule drugs: patent term (20 years from filing), SPC eligibility, and data exclusivity. The legal framework applies equally to biologics and small molecules.
What differs is the nature and breadth of the patent estates that typically protect biologics, and the regulatory exclusivity periods specifically applicable to biologics.
For small-molecule drugs, the primary protection usually comes from a single compound patent covering the active molecule. For biologics, the patent estate typically spans cell line patents (covering the specific producer cell line and its genetic modifications), manufacturing process patents (covering the fermentation, purification, and formulation processes), antibody sequence patents (covering the variable region sequences of monoclonal antibodies), and method-of-treatment patents.
This multi-layered patent structure means that biosimilar manufacturers face not a single patent but a portfolio of patents, any one of which might independently block market entry even after the primary compound or sequence patent expires.
EU Regulatory Exclusivity for Reference Biologics
Reference biologics approved through the EMA’s centralized procedure receive the same 8-year data exclusivity and 10-year market exclusivity as small-molecule drugs under the 8+2+1 framework. There is no separate, longer data exclusivity period for biologics in the EU — a notable contrast to the United States, where the BPCIA grants 12 years of data exclusivity for biologic reference products.
This EU/US difference has significant competitive implications. A biologic drug approved simultaneously by the EMA and FDA receives 10 years of EU market exclusivity but 12 years of US data exclusivity. The EU regulatory exclusivity clock runs out before the US one, making European markets potentially more attractive for biosimilar entry while US markets remain protected.
EU orphan biologics combine the 10-year orphan exclusivity with the 8+2+1 framework. The longer of the two periods applies as the binding constraint. For most orphan biologics, the 10-year orphan exclusivity is the binding constraint in any case, since it runs on the same timeline as the 10-year market exclusivity under 8+2+1.
The EU Biosimilar Approval Pathway
The EU established the world’s first biosimilar approval pathway with the Biologics Directive in 2003 and subsequent EMA guidelines, and the European biosimilar market is substantially more developed than the US market [31]. By 2023, the EU had approved over 80 biosimilar medicines, compared to roughly 40 in the United States, and EU biosimilar penetration rates in volume terms have consistently exceeded US penetration in comparable therapeutic categories.
The biosimilar approval pathway in the EU does not involve a patent dance mechanism equivalent to the US BPCIA. EU biosimilar applicants file their Marketing Authorization Applications with the EMA and must demonstrate biosimilarity through comparative analytical, non-clinical, and clinical studies. There is no formal statutory process for patent information exchange between the reference product holder and biosimilar applicant.
Patent disputes in EU biosimilar contexts are litigated as straightforward patent infringement actions in national courts (or now potentially the UPC), without the BPCIA’s structured pre-litigation information exchange. This simpler, more direct approach has been cited as one reason for Europe’s faster biosimilar market development compared to the United States.
Part Ten: Case Studies in European Pharmaceutical Patent Protection
Humira in Europe: Earlier Cliff, Different Dynamics
AbbVie’s Humira (adalimumab) illustrates the contrast between European and US biosimilar entry timing. In the United States, AbbVie’s continuation patent strategy delayed biosimilar entry until 2023. In Europe, the Humira biosimilar market launched in 2018 — five years earlier.
The European earlier entry reflected structural differences in the SPC and biosimilar frameworks. The core adalimumab patents in Europe had SPCs that expired before US counterparts, and AbbVie’s continuation strategy, which was executed extensively in the US patent system through additional filings, had less scope in the European prosecution environment, where claim scope standards and continuation practice differ from USPTO approaches.
By 2020, the European Humira biosimilar market included products from Sandoz (Hyrimoz), Biogen (Imraldi), Boehringer Ingelheim (Cyltezo), Samsung Bioepis (Imraldi), and others, competing aggressively on price and achieving substantial volume share in markets including Germany, the Netherlands, and the Nordic countries [32]. UK and French market biosimilar penetration followed different trajectories driven by national formulary and procurement approaches.
The Humira European case demonstrates how SPC expiry dates can diverge from US patent expiry dates and how the absence of the BPCIA patent dance produces faster actual generic/biosimilar entry even when the statutory exclusivity frameworks are broadly comparable.
Gleevec/Imatinib: The SPC Calculation Dispute
Novartis’s imatinib mesylate, sold as Gleevec in the US and Glivec in Europe, generated one of the most discussed SPC calculation disputes in European pharmaceutical patent history. The compound patent for imatinib was filed in the early 1990s, giving it a statutory term that extended into the 2010s. The question of which marketing authorization counted as the “first authorization” for SPC calculation purposes became central to the SPC duration in several EU member states.
The complexity arose because imatinib received initial approval in 2001 for chronic myeloid leukemia, but earlier clinical use had occurred under compassionate use frameworks and specials schemes that might have constituted earlier “authorization to place on the market” under some interpretations. If earlier compassionate use constituted the first authorization, the SPC duration would be shorter; if only the formal marketing authorization counted, the SPC duration would be longer.
National courts in several EU member states addressed this question with varying approaches before the CJEU’s case law on what constitutes the relevant first authorization was fully developed. The dispute illustrated how the SPC calculation, which appears mathematically simple, depends on factual and legal determinations about authorization dates that can be contested vigorously.
For generic manufacturers, imatinib’s European patent and SPC landscape was intensely analyzed using all available data sources. DrugPatentWatch’s integration of EU regulatory approval data with national SPC records provides exactly the kind of multi-market, multi-date view that imatinib’s complex authorization history required — a single national patent office record would have been insufficient for accurate SPC calculation across all EU markets.
AstraZeneca’s Esomeprazole: The Abuse of Dominance Connection
AstraZeneca’s Nexium (esomeprazole) SPC strategy generated not only pharmaceutical patent litigation but one of the most significant antitrust enforcement actions in European pharmaceutical history. The European Commission found that AstraZeneca had abused its dominant position by making misleading representations to patent offices in multiple EU member states about when the “first authorization” for regulatory exclusivity purposes occurred [33].
The specific facts involved AstraZeneca’s selective use of national technical (price) authorization dates versus EU-level marketing authorization dates to maximize SPC duration in different countries. By providing misleading information to the EPO and national patent offices about the relevant first authorization dates, AstraZeneca obtained longer SPCs than it was entitled to under EU law. The Commission fined AstraZeneca €60 million in 2005, and the decision was upheld by the Court of Justice of the EU in 2012.
The esomeprazole case established that patent/SPC prosecution activity can constitute an abuse of dominant position under EU competition law, and that pharmaceutical companies face antitrust liability not only for commercial conduct but for the manner in which they obtain and maintain IP rights. The case is now a mandatory reference point in any discussion of pharmaceutical patent strategy in Europe.
The practical implication for portfolio valuation: SPCs obtained through factual representations about marketing authorization dates need to be independently verified. An SPC built on an incorrectly determined “first authorization” date is vulnerable to challenge not only on SPC validity grounds but potentially as a competition law violation, with the possibility of competitor damages claims on top of invalidity.
Part Eleven: Practical Valuation Tools and Data Infrastructure
Mapping SPC Status Across EU Markets
A pharmaceutical patent portfolio analysis covering EU markets must address SPC status at the national level for each major market. This is substantially more complex than US patent analysis, where a single USPTO record and a single Orange Book cover the relevant national territory.
The minimum data required for each product in each EU member state:
The filing date and grant date of the basic patent(s).
The date of the first EU/national marketing authorization relevant to that member state.
The SPC application date, grant date, and expiry date as recorded by the national patent office.
Any pediatric extension application and its status.
Any SPC nullification proceedings before national courts or, post-2023, the UPC.
Assembling this data manually across even five major EU markets (Germany, France, Italy, Spain, UK) for a portfolio of twenty drugs requires dozens of database queries and translation of several national patent office records. This is where structured pharmaceutical IP data services provide genuine analytical leverage.
DrugPatentWatch maintains SPC expiry data for marketed drugs across major EU member states, integrated with its US Orange Book tracking. For a due diligence analyst examining a European pharmaceutical acquisition, the ability to pull national SPC status across markets through a single interface rather than individually querying the DPMA, INPI, UIBM, OEPM, and UK IPO databases is a meaningful time and accuracy advantage. The cross-referencing of SPC data with ANDA-equivalent application data for EU generic markets provides the competitive entry timeline view that portfolio valuation requires.
Building the European Patent and SPC Expiry Timeline
For any drug or portfolio subject to European valuation, the analyst should construct what practitioners call the “IP cliff calendar” — a chronological timeline showing when each layer of protection expires in each major market. For a typical EU blockbuster drug, this timeline might look like:
Month 0: Patent expiry in Germany (compound patent, SPC did not qualify due to short review period).
Month 4: Patent expiry in France (same compound patent, validated 4 months later in French records due to administrative timing).
Month 8: SPC expiry in Spain (SPC calculated from Spanish national authorization date, which was 8 months after EMA authorization for this product).
Month 14: SPC expiry in Germany (SPC calculated from EMA authorization date, which was earlier than Spanish authorization).
Month 30: SPC expiry with pediatric extension in France (France SPC plus 6-month pediatric extension).
Month 36: SPC expiry with pediatric extension in Germany.
The cliff is not a single event in a single month. It is a sequence of events across markets, each with its own competitive entry implications. A generic manufacturer entering at month 0 in Germany gains a German market foothold but faces SPC-protected competition prevention in Spain until month 8. A portfolio model that treats “SPC expiry” as a single date misrepresents the competitive dynamics.
Monitoring EPC Opposition Proceedings
EPC oppositions — filed with the EPO within 9 months of patent grant — provide a centralized mechanism for challenging the validity of a European patent before it is validated into national patents. A successful EPO opposition that revokes the patent eliminates it across all EPC member states simultaneously.
The opposition rate for pharmaceutical patents at the EPO has historically run between 8 and 15 percent of patents granted, substantially higher than the overall EPO opposition rate [34]. This reflects the financial stakes in pharmaceutical patent validity and the organized monitoring programs that generic manufacturers maintain.
EPO opposition records are publicly accessible through the European Patent Register and through commercial patent analytics platforms. DrugPatentWatch cross-references EPO opposition data with marketed drug records where applicable, providing a signal of which compound patents face organized generic or third-party challenge at the grant stage. A European patent subject to opposition is a patent under active validity challenge, and its SPC — if one was applied for — faces the risk that the underlying basic patent may be revoked, eliminating the SPC as well.
Part Twelve: Competitive Intelligence on European Generic Entry
Understanding the EMA’s Centralized Generic Approval Pathway
EU generic marketing authorization applications use the Article 10 abridged procedure under Directive 2001/83/EC [35]. Under this procedure, a generic applicant demonstrates that its product is bioequivalent to the reference product, relying on the reference product’s clinical data without needing to generate its own. The EMA or national agencies grant approval based on demonstrated pharmaceutical equivalence, bioequivalence, and the same therapeutic indication.
Unlike the US Hatch-Waxman system, there is no formal “first filer” exclusivity incentive in the EU. The first generic applicant to receive EU marketing authorization does not receive a period of exclusivity before other generics can enter. All generic applicants whose products meet bioequivalence standards can receive authorization simultaneously, and the EU generic market tends to see rapid multi-brand entry upon exclusivity expiry.
This absence of first-filer exclusivity removes a key element of the US competitive dynamic from EU analysis. In the US, tracking the specific Paragraph IV first filer matters because that filer receives 180 days of generic exclusivity. In the EU, tracking the pipeline of generic applications matters for understanding competitive intensity at expiry, but no single filer has a protected window.
Tracking EU Generic Pipeline Through National Databases
EU member states’ national competent authorities maintain registers of marketing authorization applications and approvals. Germany’s Federal Institute for Drugs and Medical Devices (BfArM), France’s ANSM, and the EMA for centrally authorized products each publish information about pending applications with varying degrees of specificity.
Unlike the FDA’s ANDA database, EU national drug application pipelines are not uniformly transparent. The EMA publishes lists of applications under evaluation for centrally authorized products, but the full list of pending generic applications is not always publicly accessible in the same structured format as FDA ANDA records. Generic manufacturers in Europe do not face the same public disclosure requirements as US ANDA filers regarding patent certifications.
This comparative opacity means that EU competitive entry timeline analysis depends more heavily on inference from available signals — SPC expiry dates, known generic manufacturers’ pipeline disclosures in their own regulatory filings, and marketing authorization approval records — than on the direct pipeline visibility that the US system provides.
Part Thirteen: Policy Reform Landscape
EU Pharmaceutical Legislation Revision
The European Commission published a comprehensive proposal to revise the EU’s pharmaceutical legislation in April 2023 — the most significant reform of EU pharmaceutical IP frameworks in two decades [36]. The proposal includes changes to data exclusivity periods, regulatory incentive structures, and SPC administration.
Key proposals relevant to pharmaceutical patent protection include:
A modification to the 8+2+1 data exclusivity framework that would allow the 10-year market exclusivity period to be extended to 12 years if manufacturers meet specific criteria — including manufacturing at least 20 percent of their product in the EU and launching in all EU member states within 2 years of first authorization.
Conversely, a reduction of the standard 10-year market exclusivity to 8 years for manufacturers that do not launch in all EU member states within the required period, effectively creating a “use it or lose it” additional 2 years of exclusivity tied to pan-EU market access.
Changes to orphan drug exclusivity that would modify when and how the 6-year reduction from 10 years can be triggered, responding to concerns about high orphan drug pricing.
Proposals for a reformed SPC regulation, including potential centralization of SPC grant through the EUIPO rather than national patent offices, which would eliminate national-level SPC calculation variations.
These proposals were under negotiation in the European Parliament and Council through 2024, with final outcomes uncertain. For portfolio valuation purposes, the proposed legislative changes would, if enacted, materially alter the exclusivity stacking calculations for products in development. Analysts should model both the current framework and the proposed changes as scenario inputs.
The SPC Centralization Proposal
The most structurally significant SPC proposal in the 2023 pharmaceutical legislation revision is the move toward centralized SPC grant through the EU Intellectual Property Office (EUIPO) [37]. Under the current system, an applicant for pan-EU SPC protection must file separate applications at each of 27 national patent offices — paying 27 sets of national fees, meeting 27 national procedural deadlines, and managing 27 separate administrative processes. The same EU regulation applies in each country, but national patent offices interpret and apply it with subtle variations, as the case law discussed throughout this article demonstrates.
A centralized “unitary SPC” — issued by EUIPO and effective across all EU member states as a single IP right — would eliminate this administrative fragmentation and the national variation in SPC calculation. It would also, as with the Unitary Patent, create a centralized invalidity mechanism through which a single successful challenge could eliminate EU-wide SPC protection.
The competitive dynamics of centralized SPC invalidity closely parallel the UPC analysis: brand pharmaceutical companies view centralized invalidity as a risk, while generic manufacturers view it as an opportunity cost reducer. Whether the administrative savings justify the concentrated invalidity risk is a calculation that varies by product, SPC duration, and the specific prior art landscape.
Part Fourteen: Putting It All Together — The European IP Cliff Valuation Model
Step One: Mapping the Protection Stack
For any European pharmaceutical asset under valuation, the first analytical step assembles the complete protection stack for each major EU market and the UK:
The compound/composition patent: filing date, national validation status in each market, applicable SPC, SPC pediatric extension, expiry dates in each country.
Secondary patents: formulation patents, method-of-treatment patents, dosage patents, manufacturing process patents — with coverage status mapped to the marketed product’s actual commercial embodiment.
Regulatory data exclusivity: 8-year data exclusivity and 10-year market exclusivity dates by product, calculated from the relevant first authorization date (EMA for centrally authorized products, national authorization for nationally authorized products).
Orphan drug exclusivity: 10-year period from authorization, plus potential 2-year pediatric extension, for designated orphan products.
Step Two: Revenue-Weighting the Market Portfolio
Not all EU markets contribute equally to a drug’s EU revenue. Germany and France together typically represent 40-50 percent of Western European pharmaceutical revenue for most therapeutic categories. Italy and Spain add another 25-30 percent. The UK, while outside the EU, is often the second-largest single market after Germany for major drugs.
Weight each market’s SPC/exclusivity expiry date by that market’s revenue contribution. The revenue-weighted average exclusivity expiry date is a more accurate single-date representation of “when exclusivity expires” than any individual country’s expiry date. The earliest expiry date identifies where generic entry risk is concentrated; the revenue-weighted average identifies the expected value of the exclusivity portfolio.
Step Three: Probability-Weighting for Challenge Risk
European pharmaceutical patent and SPC challenge risk differs mechanically from US risk but is analytically comparable. Key risk factors include:
EPO opposition history: Has the basic patent been subject to EPO opposition? If so, did it survive? A patent that survived a central EPO opposition has stronger presumptive validity than one that has never been challenged.
National court SPC nullification history: Have any SPC nullification actions been filed in major markets? What were the outcomes?
UPC exposure: Has the patent been opted out of UPC jurisdiction? If not, is it exposed to central revocation risk?
CJEU referral risk: Are there unresolved CJEU questions about the SPC’s validity — particularly around the “product” definition, “basic patent” requirement, or “first authorization” date — that create uncertainty about the SPC’s legal foundation?
Each risk factor can be translated into a probability discount on the SPC’s remaining commercial value, using the same probability-weighted DCF approach described for US portfolios.
Step Four: Competitive Entry Timeline Modeling
Model the competitive entry timeline from the supply chain perspective, accounting for the SPC manufacturing waiver:
Determine which EU-based generic/biosimilar manufacturers have announced pipeline products for this drug.
Estimate when those manufacturers would apply for the SPC manufacturing waiver to begin production and stockpiling.
Model the Day 1 competitive intensity on SPC expiry, reflecting the number of manufacturers with authorized products and supply ready for launch.
Apply the appropriate European generic penetration curve — which differs from the US curve in reaching a lower price equilibrium but potentially a faster volume shift, particularly for drugs where institutional procurement (hospital formularies, national insurance reimbursement lists) drives rapid substitution.
Key Takeaways
A European drug patent’s statutory 20-year term is almost never the operative exclusivity period. SPCs, pediatric extensions, regulatory data exclusivity, and orphan drug exclusivity all modify the actual protection window substantially.
Supplementary Protection Certificates are the most commercially important extension mechanism. They run from compound patent expiry for a period equal to the time from patent filing to first EU marketing authorization minus 5 years, subject to a 5-year maximum. A full SPC plus pediatric extension provides up to 5 years and 6 months of additional protection beyond the patent.
The 8+2+1 regulatory data exclusivity framework provides 10 years of market exclusivity from first EU authorization, regardless of patent status. For drugs where patents expire before the 10-year mark, data exclusivity operates as the binding constraint on generic entry.
Orphan drug designation adds 10 years of market exclusivity from authorization, with a 2-year pediatric extension for orphan products with completed PIPs. Drugs combining orphan exclusivity with strong patent/SPC portfolios can achieve effective protection exceeding 20 years from market authorization.
SPCs are national rights administered by individual national patent offices, despite being governed by a single EU regulation. SPC durations can and do differ across EU member states for the same drug product, primarily because the “first authorization” date varies between centrally and nationally authorized products, and because national patent offices apply the EU regulation with interpretive differences.
The Unified Patent Court, operational since June 2023, creates a central revocation pathway that can eliminate a Unitary Patent across all participating EU member states in a single action. Most major pharmaceutical companies opted pharmaceutical patents out of UPC jurisdiction during the transitional period, but this position is expected to evolve as UPC case law develops.
Brexit severed UK SPC and data exclusivity calculations from EU reference dates. UK SPCs now reference MHRA authorization dates, which may differ from EMA authorization dates by months or years for post-2020 approvals.
The 2019 SPC manufacturing waiver allows EU generic manufacturers to produce and stockpile SPC-protected products during the last 6 months of SPC term for EU market entry on Day 1. Portfolio models that assume a soft landing period after SPC expiry need updating for drugs where major EU generic manufacturers have used this provision.
European biosimilar entry occurs without the BPCIA patent dance mechanism, producing faster actual biosimilar market entry relative to the US, as demonstrated by Humira’s 2018 European biosimilar launch compared to 2023 US biosimilar entry.
Proposed EU pharmaceutical legislation (2023 Commission proposal) would modify data exclusivity periods and potentially centralize SPC grant through EUIPO. Analysts modeling European pharmaceutical assets beyond a 5-year horizon should build regulatory scenario analysis into their models.
FAQ
Q1: Can a company hold both a European patent and a Unitary Patent on the same drug simultaneously?
A1: No. A Unitary Patent and national European patents for the same invention in the same territory are mutually exclusive. When an applicant requests Unitary Patent effect after EPO grant, that converts the European patent into a single unitary right across participating UP member states, and the applicant cannot also validate the same patent as national patents in those countries. The applicant can, however, combine a Unitary Patent for UP member states with national validations in EPC member states that are not part of the UP system — Switzerland, Norway, Turkey, and others. This hybrid structure is legitimate and allows comprehensive geographic coverage while the UP member states are covered centrally. For pharmaceutical companies managing blockbuster drug patent portfolios, the combination of Unitary Patent plus national validations in non-UP states (plus a separate UK patent) covers the major European revenue markets in a single coordinated strategy.
Q2: If a generic manufacturer successfully invalidates an SPC in Germany, does that affect the SPC in France?
A2: No. SPCs are national IP rights, and a German court decision invalidating a German SPC has no direct legal effect on French, Italian, Spanish, or other national SPCs for the same product. Each country’s SPC must be challenged and invalidated separately in that country’s national courts. This is a fundamental structural feature of the current EU SPC system that the proposed centralized SPC regulation would change — a central EUIPO SPC invalidated through a central mechanism would fall across all participating member states simultaneously. Under current law, a generic manufacturer seeking to clear the market across multiple EU member states must file parallel SPC nullification actions in each major market, typically simultaneously or in close succession. The outcomes can and do differ: a German court might uphold an SPC that a French court simultaneously invalidates, if the legal questions being decided involve national procedural differences or different applications of CJEU precedent. This market-by-market independence is why coordinated multi-jurisdiction SPC challenge strategies require careful sequencing and jurisdiction selection.
Q3: How does a compulsory license affect pharmaceutical patent and SPC protection in Europe?
A3: Compulsory licensing of pharmaceutical patents is provided for in the TRIPS Agreement and implemented in national patent laws of EPC member states, but it has been invoked rarely for commercial pharmaceutical products in Europe. Most European compulsory licensing provisions require specific findings of national emergency, public non-commercial use, or anti-competitive conduct before a compulsory license is granted. Germany, France, Italy, and the UK each have compulsory licensing provisions in their national patent laws. The COVID-19 pandemic reignited discussion of compulsory licensing for vaccines and therapeutics, with several EU member states examining their legal authority but ultimately not exercising it for COVID-19 products. For SPC protection specifically, a compulsory license does not invalidate the SPC — it provides a government-authorized right to use the patented/SPC-protected product under terms set by the licensing authority (typically including royalty payments to the rights holder) without the rights holder’s consent. The rights holder retains the SPC but cannot enforce it against the compulsory licensee. For portfolio valuation purposes, compulsory licensing risk is generally categorized as a tail risk for EU pharmaceutical assets, with the exception of products in therapeutic areas that have become subject to political pricing pressure and public access debates.
Q4: What happens to an EU SPC if the underlying basic patent is partially invalidated through an EPO opposition?
A4: If an EPO opposition partially invalidates a European patent — for example, if the opposition deletes one of three independent claims but leaves two others in force — the SPC remains valid if the surviving claims still cover the protected product. An SPC is linked to a “product” (the active ingredient) and the “basic patent” that protects that product. If the surviving claims of the patent still cover the product, the SPC foundation remains intact. If the opposition revokes the patent entirely, the SPC falls with it, since Article 15 of Regulation 469/2009 provides that the SPC lapses if the basic patent lapses or is revoked. This is why monitoring EPO opposition proceedings against basic patents is a critical component of SPC risk assessment. A patent subject to a full EPO opposition — particularly one where the prior art cited in the opposition directly threatens the claims covering the commercial product — should be flagged as high-risk in any SPC portfolio analysis. The practical recommendation for pharmaceutical patent holders facing EPO oppositions on basic patents underlying commercially important SPCs is to ensure that all national SPCs have been applied for and granted before the opposition proceeding concludes, and to actively participate in the opposition with the full scope of the commercial stakes in mind.
Q5: How do European pharmaceutical patent and SPC databases compare to US sources like DrugPatentWatch, and where are the data gaps?
A5: The European patent data landscape is more fragmented than the US system’s centralized FDA Orange Book plus USPTO structure. The EMA publishes the European Public Assessment Reports (EPARs) for centrally authorized products, which contain regulatory approval dates and some patent-related information, but EPARs do not systematically compile SPC status across member states. The EPO’s European Patent Register provides prosecution history, grant records, and opposition status for European patents. Individual national patent offices — DPMA (Germany), INPI (France), IPO (UK), and others — maintain SPC registers that must be individually queried in each country. The practical data gaps include: standardized SPC expiry data across all 27 EU member states in a single searchable format, real-time tracking of national SPC nullification proceedings filed in multiple jurisdictions simultaneously, and integrated views linking EMA approval dates to national SPC calculations across markets. DrugPatentWatch addresses a portion of this gap for the markets it covers, providing integrated patent, SPC, and regulatory exclusivity data that reduces the time required for multi-market European patent analysis. Commercial patent analytics platforms like Derwent Innovation and Orbit Intelligence provide international patent family data including European counterparts and EP opposition records. Comprehensive EU SPC coverage across all 27 member states and the UK still requires combining multiple data sources, which is why the proposed EU pharmaceutical legislation’s SPC centralization provision — if enacted — would represent a genuine analytical improvement for both rights holders and competitive intelligence analysts.
Sources
[1] European Patent Office. (2023). The European Patent Convention. EPO. https://www.epo.org/law-practice/legal-texts/epc.html
[2] European Patent Office. (2024). Member states of the European Patent Organisation. EPO. https://www.epo.org/about-us/foundation/member-states.html
[3] European Patent Convention, Art. 63. (2023). Duration of the European patent. European Patent Office.
[4] European Patent Office. (2023). EPO Annual Report 2022: Statistics and trends. EPO Publications.
[5] European Patent Office. (2024). PACE program: Accelerated prosecution and examination. EPO. https://www.epo.org/applying/online-services/pace.html
[6] European Patent Office. (2008). London Agreement: Agreement on the application of Article 65 EPC. EPO.
[7] Council Regulation (EEC) No 1768/92 of 18 June 1992 concerning the creation of a supplementary protection certificate for medicinal products. Official Journal of the European Communities L 182, 1-5.
[8] Regulation (EC) No 469/2009 of the European Parliament and of the Council of 6 May 2009 concerning the supplementary protection certificate for medicinal products. Official Journal of the European Union L 152, 1-10.
[9] Court of Justice of the European Union. (2012). Case C-130/11, Neurim Pharmaceuticals v. Comptroller-General of Patents. ECLI:EU:C:2012:489.
[10] Regulation (EC) No 469/2009, Art. 1(b). Definition of “product.” Official Journal of the European Union.
[11] Court of Justice of the European Union. (2013). Case C-493/12, Eli Lilly and Company Ltd v. Human Genome Sciences Inc. ECLI:EU:C:2013:835.
[12] Court of Justice of the European Union. (2013). Case C-443/12, Actavis Group PTC EHF and Actavis UK Limited v. Sanofi. ECLI:EU:C:2013:833.
[13] Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use. Official Journal of the European Union L 378, 1-19.
[14] Directive 2004/27/EC of the European Parliament and of the Council of 31 March 2004 amending Directive 2001/83/EC on the Community code relating to medicinal products for human use. Official Journal of the European Union L 136, 34-57.
[15] European Medicines Agency. (2023). Centralised procedure. EMA. https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/centralised-procedure
[16] Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products. Official Journal of the European Communities L 18, 1-5.
[17] Regulation (EC) No 1901/2006, Art. 37. Paediatric extension for orphan medicinal products. Official Journal of the European Union.
[18] Regulation (EC) No 141/2000, Art. 8(2). Reduction of orphan exclusivity period. Official Journal of the European Communities.
[19] Unified Patent Court Agreement (UPCA), OJ EU C 175, 20.6.2013. Entry into force: June 1, 2023.
[20] Unified Patent Court. (2023). Participating member states in the Unitary Patent system. UPC. https://www.unified-patent-court.org
[21] Unified Patent Court Agreement, Art. 83. Transitional regime and opt-out. OJ EU C 175.
[22] Millien, R. (2023). The UPC opt-out rush: What major pharmaceutical companies chose and why. IAM Magazine, (July 2023).
[23] Unified Patent Court, Local Division Hamburg. (2023). Case UPC_CFI_1/2023 (First UPC provisional injunction in pharmaceuticals). UPC Registry.
[24] Intellectual Property Office. (2020). Patents after Brexit. UK IPO. https://www.gov.uk/government/publications/patents-after-brexit
[25] Intellectual Property Office. (2023). Unitary Patent and Unified Patent Court: UK position. UK IPO. https://www.gov.uk/government/publications/unitary-patent-and-upc-uk-position
[26] Intellectual Property Office. (2021). Supplementary Protection Certificates (SPCs) after the UK’s exit from the EU. UK IPO.
[27] Human Medicines Regulations 2012 (SI 2012/1916), as amended. Data exclusivity provisions for UK marketing authorisations.
[28] European Patent Convention, Art. 27(b). Rights conferred by European patents: Experimental use. EPO.
[29] Directive 2004/27/EC, Art. 10(6). Bolar exemption for medicinal product marketing authorisations. Official Journal of the European Union.
[30] Regulation (EU) 2019/933 of the European Parliament and of the Council of 20 May 2019 amending Regulation (EC) No 469/2009 concerning the supplementary protection certificate for medicinal products (SPC manufacturing waiver). Official Journal of the European Union L 153, 1-10.
[31] European Medicines Agency. (2023). Biosimilar medicines: Overview of EMA’s biosimilar approval record. EMA. https://www.ema.europa.eu/en/human-regulatory/overview/biosimilar-medicines-overview
[32] IQVIA Institute for Human Data Science. (2022). Biosimilars in Europe: Development and use across different European countries. IQVIA.
[33] Commission Decision of 15 June 2005 relating to a proceeding under Article 82 of the EC Treaty and Article 54 of the EEA Agreement against AstraZeneca AB and AstraZeneca plc (Case COMP/A.37.507/F3 — AstraZeneca). Official Journal of the European Union L 332, 24-124.
[34] European Patent Office. (2023). EPO statistics on opposition proceedings: pharmaceutical patents. EPO Annual Report 2022.
[35] Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use, Art. 10. Abridged marketing authorization procedure. Official Journal of the European Communities L 311.
[36] European Commission. (2023, April 26). Proposal for a Regulation of the European Parliament and of the Council on the Union code relating to medicinal products for human use (COM(2023) 193 final). Publications Office of the European Union.
[37] European Commission. (2023, April 26). Proposal for a Regulation of the European Parliament and of the Council on the supplementary protection certificate for medicinal products (COM(2023) 222 final). Publications Office of the European Union.
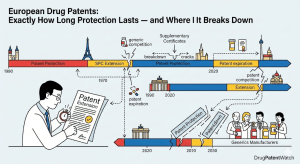















")










