The “patent death squad” label has haunted PTAB since 2012. The actual data tells a different story — one where the type of patent matters far more than the forum, where compound patents survive at near-perfect rates, and where the real carnage is concentrated in secondary method-of-use and formulation claims. This is a working intelligence brief for IP counsel, portfolio managers, biosimilar developers, and the institutional investors who need to price all of it correctly.
Why PTAB Exists — And Why It Isn’t Going Away
Congress created the Patent Trial and Appeal Board in 2012 as part of the Leahy-Smith America Invents Act, the most significant U.S. patent reform since 1952. The AIA’s central problem statement was specific: the pre-2012 system for challenging a granted patent was too slow, too expensive, and structurally biased toward incumbents. A federal district court fight over a single patent could cost both sides $5–10 million and take three to five years to reach a verdict. The existing administrative reexamination procedures were widely dismissed as toothless. The result was a quiet market for questionable patents — particularly in pharmaceuticals, where even a weak secondary patent could delay a biosimilar or generic competitor by years, adding billions to brand-drug revenues.
The AIA’s solution was a specialized administrative tribunal staffed by Administrative Patent Judges (APJs), each required by statute to hold advanced technical degrees and demonstrated expertise in patent law. Unlike a district court jury encountering a receptor-binding affinity dispute for the first time, an APJ panel adjudicating a monoclonal antibody claim will include judges who have prosecuted or litigated antibody patents. The PTAB’s mandate was to correct USPTO mistakes faster and cheaper than Article III courts, using a lower evidentiary standard — preponderance of the evidence rather than the “clear and convincing” bar that applies in district courts.
How Oil States and Arthrex Settled the Constitutional Debate
From 2012 through 2021, the PTAB operated under a persistent constitutional cloud. Challengers of issued patents — which courts have treated as a form of private property — argued that invalidation via an administrative process violated the Seventh Amendment right to a jury trial and Article III’s reservation of judicial power to life-tenured federal judges. The Supreme Court addressed this directly in Oil States Energy Services v. Greene’s Energy Group (2018), holding that because a patent is a “public franchise” created by government grant, Congress may constitutionally condition that grant on subsequent administrative review. The PTAB’s core mechanism survived.
The second challenge, resolved in United States v. Arthrex (2021), concerned the APJs themselves. The Court agreed that APJs wielded “unreviewable authority” that exceeded what is permissible for inferior officers under the Appointments Clause, but declined to dismantle the PTAB. Instead, it required that the USPTO Director retain discretion to review and modify APJ final decisions. This created what practitioners now call “Director Review” — a narrow but strategically important appellate mechanism in high-stakes cases, the implications of which are still developing in real time through the “settled expectations” doctrine discussed later in this report.
4%Orange Book patent petitions as % of PTAB total docket
73%Bio/pharma petition institution rate, FY2024
Why the ‘Death Squad’ Label Overstates the Pharmaceutical Risk
The headline statistics — 70% all-claims-invalidated rate across all technologies — are real, but they are driven almost entirely by the electrical and computer technology sector, which accounts for roughly 69% of all PTAB petitions. Pharmaceutical and biologic patents combined represent about 6% of the annual petition docket. The outcomes in that 6% differ sharply from the aggregate. A Ropes & Gray analysis comparing Orange Book patent outcomes across both PTAB and district court found that all-claims-invalidated decisions occurred in 23% of PTAB cases and 24% of district court cases. The gap essentially disappears.
This parity is not coincidental. It reflects the economic filtering that occurs before a pharmaceutical patent reaches the PTAB. Generic and biosimilar companies filing IPR petitions against high-revenue drugs have already run sophisticated claim-by-claim vulnerability analyses, typically supported by multiple expert declarations. The petitions that reach the institution threshold are not speculative or opportunistic — they are targeted assaults against the weakest patents in a brand’s portfolio. The institution rate for bio/pharma petitions hit 73% in FY2024, well above the overall 68%, which confirms that challengers are filing fewer but stronger petitions.
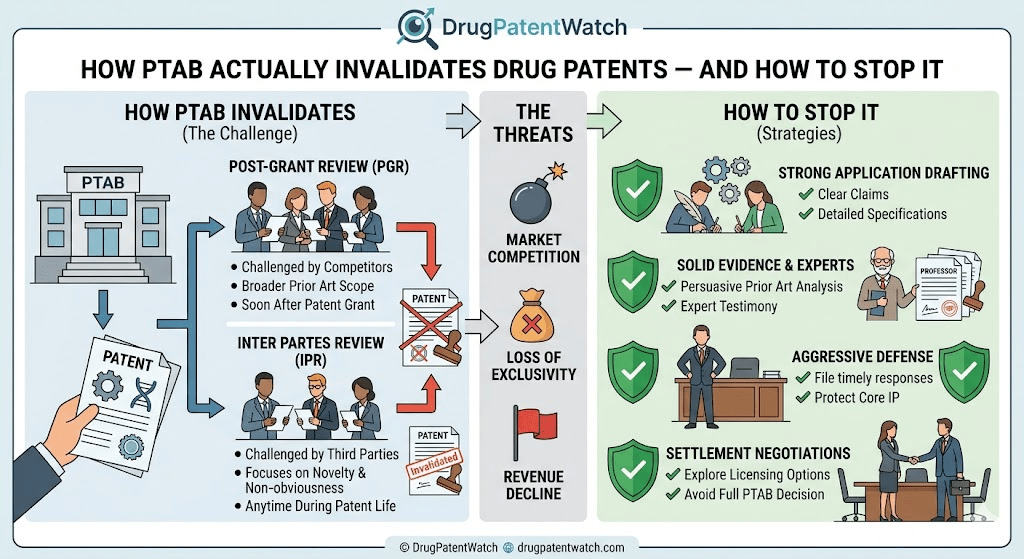
IPR vs. PGR: Which Mechanism Is More Dangerous for Your Portfolio
The AIA established two primary trial mechanisms: Inter Partes Review and Post-Grant Review. Both proceed before a three-APJ panel, both require a petition and institution decision, and both conclude in a Final Written Decision within approximately 12 to 18 months of institution. The strategic differences between them are consequential enough to drive entire patent prosecution strategies.
Inter Partes Review: The Standard Instrument of Generic Entry Strategy
An IPR petition may be filed any time after nine months from a patent’s grant date, or after a PGR proceeding has terminated. Grounds for challenge are narrow: anticipation under 35 U.S.C. §102 and obviousness under 35 U.S.C. §103, but only through patents and printed publications. Subject matter eligibility challenges under §101, written description challenges under §112, and enablement challenges under §112 are all outside IPR’s scope. The institution standard is whether there is a “reasonable likelihood” that the petitioner would prevail on at least one challenged claim.
Estoppel is where IPR gets punishing. Once an IPR proceeds to a Final Written Decision, the petitioner — and all entities in privity with the petitioner — is barred from raising in any subsequent USPTO, district court, or ITC proceeding any invalidity ground that was “raised or reasonably could have been raised” during the IPR. A petitioner who loses an IPR on an obviousness argument cannot relitigate that argument in a Paragraph IV trial. The estoppel provisions are designed to force a single, well-prepared shot at the PTAB; a missed opportunity can permanently hobble a later infringement defense.
Post-Grant Review: The Nine-Month Window That Patent Owners Fear Most
PGR is available only for patents with an effective filing date of March 16, 2013 or later — patents subject to the AIA’s first-inventor-to-file system — and the petition window closes precisely nine months after grant or reissue. There are no second chances. A competitor who fails to file within this window permanently forfeits the ability to bring a §101, §112 written description, or §112 enablement challenge at the PTAB. These are often the most powerful grounds against broad biological patent claims.
The expanded scope is the PGR’s defining advantage. Every statutory basis for invalidity except best mode is available: abstract-idea ineligibility, lack of written description, non-enablement, indefiniteness, and prior art. The institution standard is slightly tougher than IPR — “more likely than not” that at least one challenged claim is unpatentable — but that bar has rarely deterred a well-resourced challenger who has done the analytical work.
For patent owners, the nine-month anniversary of a patent’s grant is a milestone. Surviving it without a PGR petition substantially de-risks the asset, locking future challengers into the narrower prior-art-only confines of an IPR. For biosimilar developers and generic filers, the nine-month window forces an immediate, aggressive analysis of any newly issued competitor patent with potential commercial significance. A real-time Orange Book and USPTO grant monitoring system — the kind of data infrastructure provided by DrugPatentWatch — is the operational requirement that makes this analysis possible at the scale needed.
Feature
Inter Partes Review (IPR)
Post-Grant Review (PGR)
Filing window
After 9 months from grant
Within 9 months of grant — hard cutoff
Patent eligibility
All patents, any filing date
AIA patents only (eff. date ≥ Mar 16, 2013)
Grounds for challenge
§102, §103 via patents/publications only
Any ground: §101, §102, §103, §112 (all sub-sections)
Institution standard
“Reasonable likelihood” of prevailing on 1+ claim
“More likely than not” 1+ claim is unpatentable
Discovery
Limited, mostly expert depositions
Broader, still narrower than district court
Timeline to FWD
12 months from institution (18 w/ extension)
12 months from institution (18 w/ extension)
Petition fees (USPTO)
$9,000 base (20 claims)
$12,000 base (20 claims)
Estoppel trigger
After Final Written Decision
After Final Written Decision (broader scope)
Strategic use case
Attacking older patents via prior art; running parallel to Hatch-Waxman
Front-loading attack against newly granted, functionally broad genus claims
What Makes the Patent Owner Preliminary Response the Most Important Filing in the Process
Between petition filing and institution, the patent owner has one opportunity to prevent the trial from starting: the Patent Owner Preliminary Response, or POPR. It is optional, but declining to file one in a pharmaceutical context is rarely advisable. A well-constructed POPR attacks the petitioner’s evidentiary and procedural foundation before the PTAB has any reason to proceed. The most effective POPRs focus on threshold vulnerabilities: proving a key prior art reference was not publicly accessible before the patent’s priority date, demonstrating the petitioner is time-barred under 35 U.S.C. §315(b), or showing the claimed motivation to combine fails on its own terms.
After Thryv, Inc. v. Click-to-Call Technologies (2020), the Supreme Court confirmed that the PTAB’s decision on statutory time bars is non-appealable. If the PTAB rules at institution that a petition is timely, that ruling cannot be challenged at the Federal Circuit. The POPR is therefore the only point at which a patent owner can raise a timeliness defense with any realistic chance of impact. Getting it wrong — or skipping it entirely — effectively hands the petitioner a critical procedural advantage that persists through the entire proceeding.
What the PTAB Data Actually Shows for Pharma: Compound vs. Formulation vs. Method-of-Use
Aggregate PTAB statistics obscure the pattern that matters most for pharmaceutical portfolio strategy: outcomes differ sharply by patent type. A Ropes & Gray analysis of Orange Book patents provides the clearest breakdown available. Compound patents — those protecting the active pharmaceutical ingredient itself — were not fully invalidated in a single Final Written Decision in the study period. Formulation patents, covering drug delivery systems and excipient combinations, were fully invalidated in 15% of cases. Method-of-use patents, which cover therapeutic indications and dosing regimens, faced the worst odds: all challenged claims were invalidated in 27% of final decisions.
The hierarchy makes intuitive sense. A compound patent protects a novel molecular entity. The structural novelty of a new chemical entity is difficult to establish through prior art alone, and the “lead compound” analysis required by case law means a petitioner must do more than point to a structurally similar molecule — they must show a motivated path from that specific molecule to the claimed invention with a reasonable expectation of success. Formulation patents are more exposed because the combination of known excipients with a known API often presents a cleaner obviousness case. Method-of-use patents are the most exposed because clinical use of a drug in a related indication is frequently disclosed in published literature, conference abstracts, or regulatory filings well before the priority date.
0%Compound patents fully invalidated at PTAB (Ropes & Gray dataset)
15%Formulation patents fully invalidated at PTAB
27%Method-of-use patents fully invalidated at PTAB
76%Overall “success rate” for Paragraph IV challengers (incl. settlements)
Biologic Patents at the PTAB: A Different Risk Profile Than Small Molecules
Biologic patents perform worse at the PTAB than small-molecule Orange Book patents by a measurable margin. Available data on biologic patent PTAB proceedings shows a 70% all-claims-invalidated rate in Final Written Decisions, compared to 45% for Orange Book patents. The institution rate for biologic challenges has historically been lower (around 55% cumulative), which suggests that the petitions that do get instituted tend to be particularly well-targeted.
The vulnerability of biologic patents at trial reflects the fundamental tension in functional claiming. Broad antibody claims, CAR-T constructs, and receptor-targeting biologics are routinely drafted using functional language — “an antibody that binds antigen X” — rather than structural definitions. This approach is pragmatic: at the time of filing, the full structural space of viable antibodies may not be characterized. But post-Juno and post-Amgen, the PTAB and the Federal Circuit have demanded that the specification support the claimed functional scope through either representative structural examples or identified common structural features. Broad functional claims without that support are prime candidates for §112 attack, particularly through PGR in the nine-month window.
Why Settlement Rates in Pharma PTAB Cases Are Lower Than Average — and What That Signals
Across all technologies, roughly 32% of instituted PTAB trials terminate through settlement before a Final Written Decision. For Orange Book and biologic patents, settlement rates are lower — in the 17–29% range depending on the study. This divergence has a straightforward commercial explanation: Hatch-Waxman and BPCIA cases are usually resolved not by settlement agreements that end the PTAB proceeding, but by broader commercial negotiations that restructure the competitive landscape. When AbbVie settled with Amgen, Sandoz, and Coherus over Humira biosimilar entry, those deals involved royalty structures, authorized biosimilar provisions, and agreed market-entry dates — the kind of transactional complexity that doesn’t reduce to a simple IPR settlement agreement. The PTAB proceeding often continues or is withdrawn as part of a broader commercial package.
“Traditional thinking is that patents are far more likely to be killed at the PTAB than in district court. The data shows 23% of PTAB Orange Book decisions result in full invalidation — compared to 24% in district court.”
— Ropes & Gray analysis of Orange Book patent outcomes
The Humira Patent Thicket: What AbbVie Got Right, What It Got Wrong, and Why It Still Won
AbbVie’s defense of adalimumab (Humira) is the defining case study in pharmaceutical patent strategy of the last decade. The core composition-of-matter patent on adalimumab expired in 2016. By that point AbbVie had assembled a portfolio of more than 100 secondary patents — covering subcutaneous formulations, dosing regimens, manufacturing processes, autoinjector devices, and specific treatment protocols for rheumatoid arthritis, Crohn’s disease, and psoriasis. The strategy was explicitly attrition-based: make the cost and uncertainty of challenging the full thicket prohibitive enough that biosimilar developers would prefer a negotiated settlement to a prolonged legal campaign.
Coherus’s Successful Obviousness Attack on AbbVie’s Dosing Regimen Patents
Coherus Biosciences filed IPR petitions against AbbVie’s ‘135, ‘680, and ‘987 patents, which covered the 40 mg biweekly subcutaneous dosing regimen for rheumatoid arthritis. The invalidity argument was an obviousness combination: Kempeni disclosed biweekly intravenous administration of Humira; van de Putte disclosed subcutaneous administration. Coherus argued a skilled artisan would have been motivated to combine the two references to arrive at a more convenient, self-administrable subcutaneous regimen, with a reasonable expectation of success given the known tolerability of subcutaneous biologics.
AbbVie’s primary secondary consideration argument was commercial success — Humira had by that point generated roughly $14 billion in annual global revenue. But the PTAB rejected the nexus. In separate IPR proceedings defending its formulation patents, AbbVie had argued that Humira’s commercial success was attributable to its proprietary stable liquid formulation, not its dosing regimen. Coherus introduced those prior arguments as evidence. The PTAB found that it was impossible to determine whether commercial success was driven by the dosing regimen or the formulation, and the secondary-consideration argument collapsed. The lesson for any multi-patent defense: every argument made to save one patent must be consistent with every argument made to save another. Cross-portfolio argument consistency is a litigation risk management problem, not just a legal drafting problem.
How AbbVie Stopped Sandoz Cold — Without Winning on the Merits
When Sandoz challenged AbbVie’s ‘216 and ‘100 patents, the PTAB denied institution — not because the prior art was insufficient on its face, but because Sandoz failed to prove that its primary reference was actually prior art. Sandoz’s case relied heavily on the Humira package insert as evidence of the prior art state. AbbVie’s POPR pointed to language in an FDA approval letter indicating that Humira “will be marketed,” suggesting the product was not publicly accessible at the time Sandoz alleged. The Board agreed. Without that reference, the obviousness argument had no foundation.
This outcome illustrates the POPR’s leverage. AbbVie did not need to win the substantive question of whether the dosing regimen was obvious. It needed to establish that the most important piece of Sandoz’s evidence did not qualify as prior art. The evidentiary threshold for public accessibility — a question of fact — turned a potentially meritorious challenge into a non-starter at the institution stage.
Why AbbVie’s Thicket Strategy Still Worked Despite Losing Individual Battles
Between 2016 and 2023, Amgen, Sandoz, Mylan, Coherus, and several other biosimilar developers all reached settlement agreements with AbbVie, agreeing to delay U.S. Humira biosimilar launches. The commercial terms varied, but the common thread was that the cost and timeline uncertainty of challenging more than 100 patents — even with a high win rate on individual IPRs — made negotiated entry economically superior to full litigation exposure. AbbVie’s estimated cost to a challenger of exhausting the full portfolio exceeded $700 million in legal fees and IPR petition costs. The thicket strategy does not require winning every battle. It requires making the war expensive enough that settlement is the rational choice.
The competitive result: Humira biosimilars entered the U.S. market in 2023, roughly seven years after the core compound patent expired. The delay cost payers an estimated $10–15 billion in foregone biosimilar savings by some analyses. AbbVie’s U.S. Humira franchise generated approximately $200 billion in cumulative revenue from 2003 through 2023. The IP strategy is inseparable from the financial outcome.
Why Keytruda, Ozempic, and Dupixent Are the Next Humira-Scale IP Battles
The next generation of patent thicket defenses — and the most commercially significant PTAB exposure — sits with the drugs currently generating $10–30 billion in annual revenue: pembrolizumab (Keytruda), semaglutide (Ozempic/Wegovy), dupilumab (Dupixent), nivolumab (Opdivo), and ustekinumab (Stelara). Each combines immense revenue exposure with secondary patent portfolios that are already under challenge.
Semaglutide’s Lead Compound Defense and What the Mylan IPR Reveals
Novo Nordisk’s semaglutide patents are the most-watched pharmaceutical IP matter in the GLP-1 space. In Mylan Pharmaceuticals v. Novo Nordisk, Mylan filed an IPR against patents covering semaglutide’s structure, arguing obviousness from liraglutide as a lead compound. The PTAB agreed on the lead compound selection — liraglutide is the structurally closest commercially approved GLP-1 analogue and was the natural starting point. But institution was denied because Mylan failed to adequately explain why a skilled artisan would have been motivated to make three separate and specific structural modifications to liraglutide — the C-18 fatty diacid linker, the Aib substitution at position 8, and the specific amino acid substitutions for protease resistance — all simultaneously and with a reasonable expectation of achieving semaglutide’s superior half-life and potency profile.
This outcome shows both the strength and the vulnerability of the lead compound framework. It is a powerful shield against weak IPR petitions that simply point to a similar compound. It is less effective when a petitioner can cite published literature showing that each individual modification was known, motivated, and achievable. As additional semaglutide IPR petitions are filed by generic manufacturers seeking market entry ahead of the primary patent expiry — currently projected around 2032 for core composition claims — the question of whether compound petitioners can build a more complete motivation-to-combine record will define the competitive landscape for the entire GLP-1 class.
How Pembrolizumab’s Patent Portfolio Compares to Adalimumab’s
Merck’s pembrolizumab (Keytruda) generated approximately $25 billion in global revenue in 2023, making it the world’s top-selling drug by revenue. The core composition patent on the antibody itself is expected to expire in the U.S. around 2028. The secondary portfolio — covering specific PD-1 binding epitopes, combination uses with chemotherapy, specific dosing regimens (including the FDA-approved 200 mg flat dose), and manufacturing process patents — is substantially smaller than the Humira thicket but still represents years of potential delay for biosimilar developers.
The comparison to adalimumab is instructive but imperfect. Pembrolizumab is a monoclonal antibody biologic, subject to the BPCIA’s biosimilar interchangeability pathway rather than Hatch-Waxman. The patent dispute mechanism under the BPCIA — the “patent dance” — has different timing and disclosure requirements. Biosimilar applicants for pembrolizumab will need to assess not just IPR exposure but also the reference product exclusivity (12 years from first approval in 2014) and the available PTAB grounds for any newly issued formulation or combination-use patents that fall within the nine-month PGR window.
Which Drugs Face the Largest Revenue Cliff Through 2030
The revenue at risk from patent expiry through 2030 — across branded drugs facing their first loss of exclusivity — is estimated at over $200 billion in cumulative global sales. The table below identifies drugs with the highest near-term IP exposure and the specific patent vulnerability profile that determines their PTAB risk.
Drug (Active Ingredient)
Manufacturer
Peak Annual Revenue (Approx.)
U.S. LOE Window
Primary PTAB Vulnerability
Challenger Status
Humira (adalimumab)
AbbVie
$21B
2023 (entered)
Method-of-use, formulation
Multiple biosimilars launched
Keytruda (pembrolizumab)
Merck
$25B
2028
Combination use, dosing regimen
Pre-filing analysis underway
Ozempic/Wegovy (semaglutide)
Novo Nordisk
$21B
~2031–2032
Compound (lead cpd. defense active), formulation
Mylan IPR denied institution; others expected
Dupixent (dupilumab)
Regeneron / Sanofi
$14B
2031+
Antibody genus claim (§112 risk), indication expansion
BPCIA patent dance imminent
Stelara (ustekinumab)
J&J / Janssen
$10B
2023 (entered)
Core patent expired; biosimilar market developing
Amgen, Fresenius launched 2023
Eliquis (apixaban)
BMS / Pfizer
$12B
2026–2031 (tiered)
Formulation, pediatric exclusivity
Multiple Paragraph IV ANDA filers
Jardiance (empagliflozin)
BI / Eli Lilly
$8B
2025–2029
Method-of-use (HF indication), compound expiry
Generic ANDA filed
Why §112 Written Description and Enablement Are Now the Primary Weapons Against Biologic Patents
The Federal Circuit’s 2021 ruling in Juno Therapeutics v. Kite Pharma and the Supreme Court’s 2023 decision in Amgen v. Sanofi have together recalibrated what it means to hold a valid broad biologic patent claim. In Juno, the court invalidated a foundational CAR-T cell therapy patent covering a chimeric antigen receptor with a “binding element” defined solely by its functional target — CD19 — rather than by structural parameters. The specification disclosed two working examples. The court held that two examples do not support a claim to the full universe of binding elements capable of targeting CD19, absent structural commonalities or other guidance enabling a skilled artisan to identify members of the claimed genus.
In Amgen, the Supreme Court invalidated Amgen’s broadly claimed antibody patents covering all antibodies that bind to a specific PCSK9 region and block LDL receptor binding. The specification described roughly 26 specific antibodies out of a functional genus the court estimated could encompass millions of compounds. The holding — “the more one claims, the more one must enable” — is now the governing principle for biologic genus claims. It applies with equal force at the PTAB in PGR proceedings, where §112 challenges are available in the nine-month window immediately following patent grant.
What Amgen v. Sanofi Means for Dupilumab and Future Antibody Portfolio Strategy
Dupilumab (Dupixent) is the world’s top-selling biologic by number of approved indications as of 2026, with FDA approvals spanning atopic dermatitis, asthma, CRS with nasal polyps, prurigo nodularis, eosinophilic esophagitis, and COPD. The breadth of the indication portfolio creates secondary patent exposure across method-of-use claims for each indication. Post-Amgen, any broad antibody genus claim in the Dupixent portfolio that covers the IL-4 receptor alpha subunit binding class without sufficient structural examples is vulnerable to a §112 enablement challenge in a PGR. Regeneron and Sanofi’s prosecution strategy for Dupixent’s newer indications — filed after March 2013 and therefore PGR-eligible — will determine how exposed the secondary portfolio is to this attack vector.
How to Draft Biologic Patent Claims That Survive Post-Amgen PTAB Scrutiny
The practical response to Juno and Amgen is not to abandon broad functional claiming entirely, but to build an evidentiary record in the specification that makes §112 challenges structurally difficult to mount. This means filing with a representative sample of working examples that spans the claimed functional space, identifying the structural features common to the claimed genus that predict function, and including data showing that a skilled artisan can reliably screen candidates within the genus without undue experimentation. When a specification includes 50 antibody examples with characterized CDR sequences alongside a structure-function analysis, a PGR petition arguing inadequate written description faces a much harder evidentiary burden than when the specification includes two examples and a broad functional claim.
For drugs already approved and under BPCIA or Hatch-Waxman challenge, the prosecution history is fixed. The strategic response shifts to litigation: building the expert record on “possession” of the full claimed scope, documenting the state of the art at the priority date to establish what a person of ordinary skill would have been able to synthesize given the specification’s teachings, and identifying any limitations in the challenger’s PGR petition that can be attacked in the POPR.
How Pharmaceutical Evergreening Works — and Where PTAB Creates the Friction
Evergreening refers to the set of patent prosecution and regulatory strategies that extend effective market exclusivity beyond the expiry of a core composition patent. The canonical mechanism is filing patents on new formulations, new salts or polymorphs, new dosing regimens, new combinations, pediatric indications, and new delivery devices in the years after the primary compound patent is granted. When each of these secondary patents is listed in the FDA Orange Book (for small molecules) or relied upon in the BPCIA exclusivity period (for biologics), it creates additional legal barriers to generic or biosimilar entry that can cumulatively extend effective exclusivity by five to fifteen years past the original compound expiry.
PTAB’s most direct impact on evergreening is through the elevated IPR institution rates and Final Written Decision invalidation rates for secondary patent types. Method-of-use and formulation patents — the primary instruments of evergreening — face the highest PTAB invalidation rates. A well-funded generic company can systematically dismantle the outer layers of a patent thicket through serial IPR petitions, each targeting a different secondary patent, at a combined cost that is still substantially lower than the cost of losing years of market exclusivity.
The Role of FDA Orange Book Listing Strategy in PTAB Exposure
Not all secondary patents are Orange Book-listed. The FDA’s Orange Book lists patents that claim the approved drug product or an approved method of using it. A patent on a manufacturing process may not qualify for listing even if it confers substantial competitive protection. Patents that are not Orange Book-listed are less likely to be challenged through Paragraph IV ANDA litigation but are equally vulnerable to IPR challenge by any party at any time — including competitors building toward a different regulatory pathway, such as a 505(b)(2) NDA.
The decision of which patents to list is itself a strategic call. Listing a patent triggers the 30-month stay of ANDA approval upon a Paragraph IV certification, which is commercially valuable. But listing also makes the patent a visible target for IPR and Paragraph IV litigation. Patents at the margin of Orange Book eligibility may be better held in reserve as unlisted IP that provides manufacturing or formulation protection without advertising their existence to a potential challenger conducting prior art analysis.
Pediatric Exclusivity and Patent Term Extension: The Regulatory Moats That Survive PTAB
Two regulatory tools provide exclusivity that PTAB cannot invalidate. Patent term extension (PTE) under the Hatch-Waxman Act allows brand manufacturers to recover up to five years of patent term lost during FDA regulatory review, subject to a post-extension cap of 14 years from NDA approval. This extension applies to a single patent per approved drug and cannot be challenged at the PTAB on the merits of the extension — only its eligibility can be contested. Separately, pediatric exclusivity granted under the BPCA adds six months to the end of applicable exclusivities, including any listed patent protection period, as an incentive for pediatric studies.
These tools are outside the scope of PTAB challenge. A drug manufacturer who secures both PTE and pediatric exclusivity on a core compound patent can push effective U.S. exclusivity years past the nominal expiry date regardless of what happens to secondary patents at the PTAB. For investor analysis, tracking PTE applications and pediatric study completion dates is as important as tracking the secondary patent portfolio itself.
What Investors Are Watching: Translating PTAB Litigation Into Revenue Risk Models
For equity analysts and portfolio managers covering pharmaceutical and biotech companies, PTAB litigation is a material financial event that most standard revenue models underweight. A single IPR denial of institution is worth — in terms of extended exclusivity value — hundreds of millions to billions of dollars in net present value for high-revenue drugs. A Final Written Decision invalidating a key method-of-use patent can accelerate generic or biosimilar entry by two to four years, with corresponding erosion to a brand’s revenue trajectory that typically reaches 80–90% within 24 months of first generic entry for solid oral dosage forms, and 30–60% within three years of first biosimilar launch for biologics.
How to Read PTAB Dockets as Leading Indicators of Revenue Cliff Timing
The PTAB’s patent trial docket is public. Each IPR and PGR petition filed against an Orange Book-listed patent is a declared intent by a well-resourced challenger to seek earlier market entry. The institution decision, typically issued within six months of petition filing, is the first material data point: denial of institution on all grounds means the primary patent has cleared the most acute near-term threat. Institution on even a subset of claims means the litigation is active and the Final Written Decision — due within 12 to 18 months — will resolve the challenge or escalate it to the Federal Circuit.
Investors who track petition filing dates, institution decisions, and Final Written Decision outcomes in real time can build a probabilistic timeline of exclusivity erosion with substantially more precision than analysts relying only on nominal patent expiry dates. A drug with a compound patent expiring in 2031 but five instituted IPR proceedings against secondary patents — with trial timelines running through 2027 — carries a materially different revenue risk profile than a drug with the same compound expiry and a clean PTAB docket.
How Paragraph IV Certification Strategy Interacts with PTAB Timing
Under the Hatch-Waxman Act, the first generic company to file an ANDA with a Paragraph IV certification against an Orange Book-listed patent earns 180 days of generic market exclusivity — a prize worth hundreds of millions of dollars for a major small-molecule drug. This “first-filer exclusivity” incentive drives aggressive, early Paragraph IV certification strategies by generic companies. The interaction with PTAB is structural: a generic company that files both a Paragraph IV ANDA and an IPR petition against the same patent is pursuing two parallel challenges. If the IPR results in invalidation, the Paragraph IV case is resolved administratively. If the PTAB grants institution but the FWD is not issued within the 30-month stay period, the ANDA filer may launch at-risk or wait for the PTAB outcome depending on its risk calculus.
Post-Thryv, the brand manufacturer’s ability to challenge the timing of that parallel IPR is significantly constrained. Once the PTAB rules that a petition is timely — even if the brand believes the petitioner’s one-year statutory bar has run — that ruling is final and non-appealable. Pharmaceutical companies whose drugs are subject to active ANDA litigation must now treat every IPR petition filed by an ANDA filer as a presumptively timely challenge and build their defense accordingly, rather than relying on a time-bar argument to terminate the proceeding.
Revenue at Risk: Key Patent Expiry and Litigation Milestones Through 2032
Para. IV resolved via settlement; multiple generics
70–85% erosion (small molecule)
Dupixent (dupilumab)
~$9B U.S.
2031+
Pre-filing; BPCIA patent dance not yet triggered
Slower erosion; interchangeability barrier
Xarelto (rivaroxaban)
~$5B U.S.
2024 (entered)
Para. IV resolved; generic competition active
60–75% erosion underway
The PREVAIL Act: How Proposed Legislation Would Reshape the IPR Risk Calculus
The Promoting and Respecting Economically Vital American Innovation Leadership (PREVAIL) Act represents the most significant push to reform PTAB procedures since the AIA itself. Its core provisions would shift the forum in ways that benefit patent owners substantially and impose new restrictions on challenger access.
The most commercially significant provision: raising the burden of proof from preponderance of the evidence to the same “clear and convincing evidence” standard used in district courts. This change alone would be expected to reduce institution rates and Final Written Decision invalidation rates substantially, narrowing the gap between PTAB and district court outcomes that currently makes the PTAB the forum of choice for well-funded generic and biosimilar challengers. Combined with a proposed standing requirement — limiting IPR petitioner eligibility to parties who have been sued for or threatened with infringement — the Act would effectively close the PTAB to non-practicing entities, activist investors, and the kind of Kyle Bass-style short-and-challenge campaigns that briefly emerged in 2015–2016.
What the PREVAIL Act Would Mean for Generic Entry Timelines
If passed in its current form, the PREVAIL Act’s forum-election provision — requiring a challenger to choose between PTAB and district court — would eliminate the parallel dual-track strategy that currently allows Paragraph IV filers to run simultaneous Hatch-Waxman litigation and IPR challenges. This is significant for smaller generic manufacturers who have used the PTAB’s lower cost and faster timeline as a mechanism to force settlements with brand companies that might otherwise use litigation expense as a delay tactic. Removing the parallel-track option would increase the effective cost of challenging a pharmaceutical patent, potentially extending average exclusivity periods for drugs with defensible but not bulletproof secondary patent portfolios.
The pharmaceutical industry’s two primary lobbying blocs are on opposite sides. PhRMA and the Biotechnology Innovation Organization (BIO) support the Act’s patent-strengthening provisions. The Association for Accessible Medicines (AAM), which represents generic manufacturers, and biosimilar industry organizations oppose provisions that would restrict PTAB access and raise the invalidity bar. The legislation’s fate is uncertain, but its progress signals congressional willingness to revisit the AIA’s balance — and creates near-term uncertainty for both brand and generic IP strategy.
The ‘Settled Expectations’ Doctrine: A New USPTO Director Discretion Wildcard
Separately from the PREVAIL Act, USPTO Director Review has begun to introduce a new concept into PTAB institution decisions: the “settled expectations” doctrine. The Director has issued several decisions denying IPR institution against older patents on the grounds that the patent owner has a reasonable, settled expectation in the patent’s validity given the passage of time without challenge. This doctrine has no explicit basis in the AIA statute — it represents an exercise of the discretionary institution authority confirmed by Arthrex — and its contours are still being defined through case-by-case application.
For patent owners holding older assets in the portfolio, this doctrine offers a potential new defensive argument in the POPR. For challengers, it creates a time pressure that is separate from and potentially more aggressive than the statutory one-year bar in §315(b): a petitioner who waits too long to challenge a patent may face a discretionary denial even if the petition is technically timely. The practical implication is that competitive intelligence on patent vulnerability should trigger challenge decisions earlier rather than later, and that monitoring USPTO Director Review decisions is now a required component of PTAB litigation intelligence.
Kyle Bass and the Coalition for Affordable Drugs: What the Activist Investor IPR Campaign Reveals About PTAB’s Limits
Between 2015 and 2016, hedge fund manager Kyle Bass and his Coalition for Affordable Drugs (CFAD) filed approximately 40 IPR petitions against pharmaceutical company patents while publicly maintaining short positions in the corresponding stocks. The strategy’s logic was explicit: use the threat of patent invalidation to create stock price volatility that could be monetized through short positions, while arguing publicly that the campaign served a public interest by targeting questionable pharmaceutical patents and reducing drug prices.
The PTAB declined to sanction Bass or terminate his petitions on abuse-of-process grounds, ruling that the statute places no restriction on a petitioner’s financial motive. CFAD’s petitions achieved institution at above-average rates and won invalidations in several cases that reached final decisions. But the campaign’s financial returns were mixed at best. Early petitions against companies like Celgene, Acorda Therapeutics, and Jazz Pharmaceuticals produced significant short-term stock price drops. Later filings saw diminished market impact as investors priced in the PTAB’s mixed outcomes for brand pharmaceutical patents and recognized the statistical resilience of compound patents.
The episode identified the outer boundary of PTAB as a financial instrument: it works as a challenge mechanism when the underlying patent is genuinely weak. It does not work as a standalone stock-manipulation tool when the challenged patents are defensible. The campaign also accelerated the legislative push that eventually produced the PREVAIL Act’s proposed standing requirement, which would explicitly prohibit non-practicing entity petitioners without infringement exposure from filing IPR petitions.
The PTAB Defense Playbook: Proactive and Reactive Strategies for Pharmaceutical Patent Owners
Why Prosecution Is the First Line of PTAB Defense
The single most durable PTAB defense is a patent that was drafted and prosecuted with challenge in mind. Every argument made before the USPTO examiner — every claim amendment, every declaration, every response — becomes part of the prosecution history that a PTAB petitioner will read in forensic detail. Inconsistencies between prosecution arguments and litigation arguments are the primary mechanism by which credible secondary consideration defenses collapse, as AbbVie discovered with Coherus.
Proactive prosecution strategy means conducting exhaustive prior art searches before filing, not after. It means drafting independent claims that are specific enough to be defensible but broad enough to provide commercial protection. It means building a specification with sufficient working examples to survive a §112 written description attack — a requirement that has become more demanding in the post-Juno, post-Amgen environment. And it means maintaining a portfolio-wide argument consistency protocol, so that the commercial success nexus argument used to defend a dosing regimen patent is never in tension with the argument used to defend a formulation patent for the same drug.
The Multi-Layer Patent Thicket: How to Build It Without Overextending
A multi-layer pharmaceutical patent thicket is not simply filing as many secondary patents as possible. It is structuring a portfolio such that each additional patent layer adds genuine legal complexity for a challenger while remaining scientifically defensible. The most effective thickets have the following properties: the patents are staggered in time so that their expiry dates do not cluster; each patent addresses a distinct technical contribution (formulation stability, delivery mechanism, therapeutic regimen, manufacturing step); the portfolio includes patents in multiple jurisdictions where the biosimilar or generic developer will need to seek regulatory approval; and each patent is supported by a specification that can withstand a §112 challenge in a nine-month PGR window.
The risk of over-extension is real. Filing broad, poorly supported patents to inflate the apparent size of the thicket can create liabilities: a PGR victory by a challenger on a §112 enablement ground, if widely covered in trade press, signals to other potential challengers that the patent owner’s portfolio has weak points. The strategic value of the thicket depends in part on the perception that each patent represents a genuine, independent barrier. Junk patents undermine that perception and can destabilize the entire defensive structure.
Secondary Considerations: How to Build a Commercial Success Nexus That Survives PTAB Scrutiny
Evidence of secondary considerations of non-obviousness — commercial success, long-felt unmet need, failure of others, unexpected results — can be decisive in an IPR final decision. But the PTAB consistently demands a clear nexus between the commercial success and the specific features of the claimed invention. The Humira/Coherus failure is the canonical example of nexus destruction through inconsistent portfolio arguments.
To build a defensible nexus, the commercial success documentation should begin years before a PTAB proceeding is contemplated. Annual reports, physician surveys, market research reports, and sales data should be archived with annotations tying specific commercial performance to specific patented features. If Humira’s adoption by rheumatologists was driven by both the biweekly subcutaneous dosing convenience and the formulation’s stability at room temperature, those contributions need to be documented separately and consistently from the first year of commercialization. A fact witness who can testify credibly about why physicians chose Humira over competing biologics — and which patented features drove that choice — is more valuable than any collection of sales charts.
The PTAB Offense Playbook: How Generic and Biosimilar Companies Identify and Target Vulnerable Patents
How Competitive Intelligence Determines Which Patents to Challenge
No rational generic or biosimilar company challenges every patent in a competitor’s portfolio. The decision process begins with a commercial threshold: does the patent block entry for a drug with sufficient revenue to justify the $500,000–$1,000,000 cost of a fully litigated IPR? For drugs generating over $1 billion in annual U.S. revenue, the answer is almost always yes. Below $500 million, the economics become more sensitive to the probability of success.
Once commercial threshold is established, the vulnerability analysis begins. Claim-by-claim analysis, review of the prosecution history, comparison of claims to prior art published before the priority date, and expert evaluation of the specification’s written description support collectively produce a probability-weighted risk assessment. The most actionable targets are method-of-use patents with prior art from clinical trials, conference presentations, or regulatory submissions that predates the priority date; formulation patents where the specific excipient combination was suggested in published pharmaceutical formulation literature; and newly granted patents within the PGR window that use broad functional claiming without extensive structural examples in the specification.
How Biosimilar Launch Timing Is Affected by PTAB Outcomes
Under the BPCIA, a biosimilar applicant and the reference product sponsor engage in the “patent dance” — a structured exchange of patent lists and infringement contentions that determines which patents will be litigated before launch. PTAB challenges against BPCIA-listed patents can proceed in parallel with the patent dance litigation, potentially invalidating patents that the brand relies on in the infringement suit before that suit is resolved. The interaction is complex: a biosimilar developer who wins an IPR against a key reference product patent during the patent dance litigation can shift the commercial negotiation substantially, as the brand manufacturer loses one of its principal legal barriers.
The timing dynamics create a strategic window. A biosimilar developer who files an IPR petition 12 months before the anticipated BPCIA litigation will have an institution decision — and potentially a Final Written Decision — before the brand’s infringement trial begins. If the FWD invalidates a claim the brand is relying on in district court, the district court litigation loses a pillar. Coordinating PTAB timing with BPCIA litigation scheduling is one of the primary tactical decisions in biosimilar entry strategy.
Common Investor and IP Team Questions About PTAB Pharmaceutical Patent Risk
What is the single most important metric for assessing a drug’s PTAB risk?
Patent type is more predictive than any other single variable. Compound patents protecting novel molecular entities have a near-zero empirical invalidation rate at the PTAB. Method-of-use patents covering dosing regimens and indications have a 27% full-invalidation rate. The starting question for any PTAB risk assessment is: what type of patent is being challenged, and what does the prosecution history look like? Revenue level is the second most important variable — higher-revenue drugs attract more motivated, better-resourced challengers.
Can a patent that survived a PTAB challenge be challenged again?
IPR estoppel prevents a petitioner (and its real parties in interest) from raising grounds that were raised or reasonably could have been raised in an IPR that proceeded to a Final Written Decision. But a different petitioner — one not in privity with the original challenger — faces no such bar. Serial petitions by different entities against the same patent are a known strategy, particularly in competitive pharmaceutical markets where multiple generic companies are pursuing ANDA approval simultaneously. The PTAB’s discretion to deny serial petitions that raise substantially similar arguments provides some protection, but it is not absolute.
How does the FDA’s Orange Book listing interact with PTAB petition filing strategy?
Orange Book listing is the primary signal that a patent blocks ANDA market entry and therefore warrants the economic investment of an IPR challenge. Unlisted patents — including process patents, device patents, and formulation patents that don’t meet the Orange Book eligibility criteria — can still be challenged by IPR, but the challenger has less immediate commercial incentive unless the patent is also asserted in a separate infringement action. Monitoring Orange Book updates for newly listed patents against high-revenue drugs is a required activity for any generic company doing proactive competitive IP intelligence.
What happens to a drug’s revenue trajectory after loss of exclusivity?
Outcomes differ significantly between small molecules and biologics. For branded small-molecule drugs facing first generic entry, price erosion is typically severe: the brand retains 10–30% of volume within 12 months and 5–15% within 24 months, with prices falling 70–90% from brand levels. Biologics face slower but still material erosion. FDA-approved biosimilars generally capture 30–60% of the originator’s U.S. market share within three years, with additional erosion as interchangeable biosimilars enter the market. The Humira biosimilar market provides the current reference case: two years after first biosimilar launch in 2023, Humira retained a smaller-than-expected volume share, while biosimilar competition drove net price reductions that exceeded analyst consensus forecasts.
How does the PREVAIL Act’s proposed standing requirement differ from current PTAB access rules?
Currently, any person other than the patent owner may file an IPR petition, regardless of whether they face any patent infringement exposure. This has enabled activist investor campaigns, academic institution challenges, and non-practicing entity petitions. The PREVAIL Act would limit petitioner eligibility to parties who have been served with an infringement complaint or who have a reasonable apprehension of suit. For pharmaceutical companies, the practical impact is limited — generic and biosimilar companies filing Paragraph IV ANDAs routinely face infringement complaints within the 45-day window following ANDA acceptance, establishing clear standing. The provision would primarily affect non-traditional challengers with no regulatory filing pending.
What is biosimilar interchangeability and why does it matter for post-exclusivity revenue models?
Biosimilar interchangeability is an FDA designation indicating that a biosimilar may be substituted for the reference biologic at the pharmacy level without physician intervention, following the same automatic substitution model that applies to small-molecule generics. Achieving interchangeability requires additional clinical switching studies demonstrating that alternating between the reference product and the biosimilar does not produce greater risk than using the reference product alone. Only a small subset of approved biosimilars carry interchangeability designation, which limits pharmacy-level substitution and slows market share erosion relative to the small-molecule generic model. Investors modeling post-LOE revenue for biologics should track interchangeability application status as a key variable in market share erosion rate assumptions.
Key Takeaways
01Compound patents protecting novel molecular entities have not been fully invalidated in a single PTAB Final Written Decision in the available dataset. Method-of-use patents are invalidated in full 27% of the time. The patent type — not the forum — is the primary risk variable.
02The PTAB’s institution rate for bio/pharma petitions reached 73% in FY2024, above the all-technology average, confirming that pharmaceutical petitions are fewer but stronger. Any petition received by a brand company should be treated as a serious, well-researched challenge.
03Post-Amgen and post-Juno, broad functional genus claims in biologic patents without robust structural support in the specification face existential §112 risk in PGR proceedings. Drafting practice must change accordingly.
04The Patent Owner Preliminary Response is the most consequential single filing in a PTAB proceeding. After Thryv, it is also the only opportunity to raise a time-bar defense. Missing it or filing it weakly permanently cedes leverage.
05Commercial success secondary consideration arguments are powerful — unless they are inconsistent with arguments made in other PTAB proceedings defending other patents in the same portfolio. Argument consistency across the portfolio is a risk management requirement.
06The PREVAIL Act, if enacted, would raise the invalidity burden to clear and convincing evidence, require standing, and force a forum election. It would increase the effective cost of patent challenges and extend average exclusivity for drugs with defensible secondary portfolios.
07The USPTO Director’s “settled expectations” doctrine adds a new time pressure for challengers independent of the statutory one-year bar, and a new defensive argument for patent owners in the POPR. Both sides need to monitor its development in real time.
Investment Strategy: How to Price PTAB Litigation Into Pharma Equity and Credit Positions
PTAB litigation is a binary risk event that standard DCF models handle poorly. A drug generating $5 billion annually does not simply “have a 30% probability of losing two years of exclusivity” — the actual outcome is either that a patent survives and exclusivity continues as modeled, or that it is invalidated and the revenue trajectory shifts abruptly. Building PTAB outcomes into pharma financial models requires a scenario-weighted approach with explicit assumptions about patent type, institution probability, FWD invalidation probability, and settlement likelihood, each of which can be estimated with reasonable precision using available PTAB and Hatch-Waxman litigation data.
For equity positions in innovator companies, the primary metric is revenue-at-risk from actively challenged secondary patents in the two to four years before projected loss of exclusivity. For positions in generic and biosimilar companies, the relevant metrics are the probability of successful ANDA or 351(k) approval combined with the probability of prevailing in or settling patent litigation before commercial launch. The 76% overall Paragraph IV challenger success rate — when including favorable settlements — is a useful prior, but it must be adjusted for patent type (method-of-use challenges succeed more frequently), drug revenue level (higher revenue produces more determined brand defense), and the specific facts of the pending PTAB or district court proceedings.
Credit investors in pharmaceutical company debt should focus on the same metrics with particular attention to maturity laddering: a credit maturing in 2028 for a company whose primary revenue drug faces compound patent expiry in 2028 and active secondary patent challenges at the PTAB is a materially different risk from a credit with the same maturity and no active PTAB proceedings. The PTAB docket, updated daily, is a real-time monitor of the IP risk embedded in pharmaceutical credit positions.
This report was compiled from public USPTO PTAB trial statistics, Federal Circuit and Supreme Court opinions, Ropes & Gray, Mintz, and Fish & Richardson published analyses, DrugPatentWatch database, Congressional Research Service publications, and industry financial disclosures. All revenue figures are approximate and based on publicly reported financial results. Patent expiry projections reflect current Orange Book and USPTO data and are subject to change based on litigation outcomes, term adjustments, and regulatory exclusivity decisions. This is an analytical intelligence brief, not legal or investment advice.







")


















