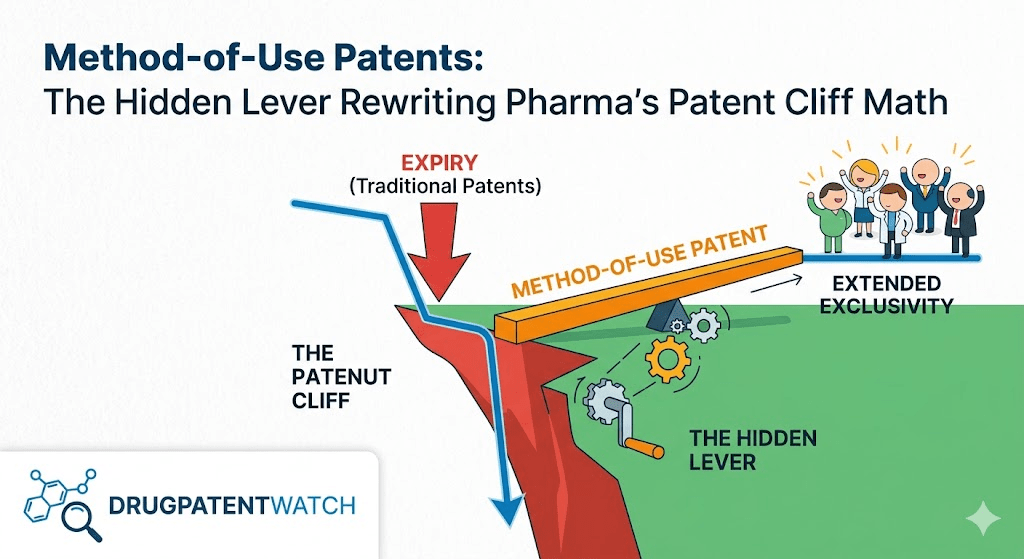
The composition-of-matter patent is the loudest piece of intellectual property a pharmaceutical company owns. It is also, increasingly, the least decisive. Between 2025 and 2030, an estimated $200 billion to $300 billion in branded pharmaceutical revenue will lose primary patent protection, with some projections reaching $400 billion by 2033 [1]. Yet the actual erosion curves on these assets rarely match the dramatic vertical drop that analyst slide decks predict. The reason is not regulatory delay, manufacturing complexity, or payer inertia, though those each play a role. The reason is the secondary patent layer, and within that layer, the method-of-use claim is doing more strategic work than any other instrument in the originator’s portfolio.
For investors, generic strategists, and IP counsel building risk models around the next wave of loss-of-exclusivity events, treating the composition patent expiration date as the cliff edge is a forecasting error. The cliff is not a date. It is a litigation surface, and that surface is shaped by method-of-use claims, induced infringement doctrine, and the narrowing protections available to generic challengers under Section viii of the Hatch-Waxman Act. The Supreme Court’s January 2026 grant of certiorari in Hikma v. Amarin, with accelerated oral argument set for April 27, 2026, is the clearest signal yet that the rules governing this surface are about to change [2]. The companies that read this correctly will protect billions in revenue. The ones that don’t will face a different curve than their models assumed.
Why the Composition Patent No Longer Tells the Whole Story
For two decades, pharmaceutical valuation models have used the primary composition-of-matter patent expiration as the central anchor for revenue forecasting. The logic was clean. A small molecule with a strong compound patent enjoys roughly 10 to 13 years of effective market exclusivity post-launch, after which generic entry triggers an 80% to 90% revenue loss within twelve months [3]. The cliff was vertical, predictable, and required no special analytical machinery to model.
That model is breaking down for two reasons. The first is biologics. The drugs driving the current cliff are not chemical pills. Keytruda, Opdivo, Stelara, Eylea, and Dupixent are biologic molecules manufactured from living cells, and their post-LOE revenue curves look nothing like the carvedilol or atorvastatin curves of the prior cycle. Biosimilar uptake is constrained by interchangeability designations, payer formulary inertia, prescriber switching reluctance, and the structural absence of an automatic substitution mechanism at the pharmacy counter. The result is what some analysts have started calling a ‘patent slope’ rather than a patent cliff [4].
The second reason, and the one this article is concerned with, is that the composition patent is no longer the only patent that matters. A 2012 empirical analysis of secondary pharmaceutical patenting found that independent method-of-use patents add an average of 7.4 years of nominal patent life beyond the primary composition claim, with a 95% confidence interval of 6.4 to 8.4 years [5]. Independent formulation patents add 6.5 years on average. Independent polymorph, salt, ester, and prodrug claims add another 6.3 years. For best-selling drugs, late-filed independent secondary patents are statistically more common than for the broader Orange Book population, which is consistent with what industry observers have long described as active lifecycle management.
Method-of-use patents do something the other secondary patents cannot. They survive generic entry. A formulation patent can be designed around. A polymorph patent can be challenged on obviousness grounds, often successfully. But a method-of-use patent tied to a specific FDA-approved indication, properly listed in the Orange Book, forces a generic challenger into one of two unattractive positions. The generic either accepts the patented indication and waits for expiration, or carves it out via a Section viii statement and accepts the litigation exposure that decision now carries.
The Statutory Architecture: Section viii and the Skinny Label
The Hatch-Waxman Act of 1984 created two mechanisms for generic challengers to address listed patents. The first, Paragraph IV certification under 21 U.S.C. § 355(j)(2)(A)(vii)(IV), is the well-known litigation pathway. A generic certifies that a listed patent is invalid, unenforceable, or not infringed, the brand has 45 days to sue, and a 30-month stay attaches. Paragraph IV is what most patent litigators spend their careers on.
The second mechanism, Section viii of 21 U.S.C. § 355(j)(2)(A)(viii), is structurally different and has, for most of its history, attracted far less attention. Under Section viii, a generic applicant certifies that it is not seeking approval for a method of use claimed in a listed patent. The generic carves the patented indication out of its proposed labeling. The remaining indications, those not covered by any listed method-of-use patent, become the basis for FDA approval. This is the ‘skinny label’ pathway. It was designed to do exactly one thing: let generic competition reach the market for unpatented indications years before the patented indication’s expiration date.
Consider how the math works on a typical multi-indication drug. A brand drug is approved for three indications. The compound patent expired in 2020. A method-of-use patent covering Indication C runs until 2028. Under Section viii, a generic can launch in 2020 with a skinny label covering Indications A and B, capturing whatever share of prescriptions are written for those uses. Indication C remains the brand’s exclusive territory until 2028. The brand loses some revenue. The generic gets early market entry. Patients get cheaper drugs for unpatented uses. Congress wrote it that way on purpose.
This worked, more or less, for nearly 40 years. Then came GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc., and the calculus changed.
The Carvedilol Decision and Its Aftermath
Carvedilol is a non-selective beta-blocker that GSK sold under the brand name Coreg. FDA initially approved it for hypertension and post-myocardial infarction left ventricular dysfunction, indications that GSK held compound and method-of-use patents on. Those patents Teva did not contest. The flashpoint was a third indication: congestive heart failure. GSK held U.S. Patent No. 5,760,069 (and a reissued version, RE40,000), which claimed a method of decreasing mortality in CHF patients by administering carvedilol in combination with certain other therapeutic agents [6].
Teva filed a Section viii statement, carved CHF out of its proposed labeling, and received FDA approval to market generic carvedilol for the unpatented indications only. The label was clean. The carve-out followed the statutory procedure. By every traditional metric, this was a textbook skinny label launch.
GSK sued for induced infringement under 35 U.S.C. § 271(b). The theory was not that Teva’s label instructed CHF treatment. The theory was that Teva’s press releases, marketing catalogs, sales force communications, and product references to Coreg equivalence functioned, in aggregate, as instructions to physicians to prescribe Teva’s generic for the patented CHF indication. A Delaware jury agreed and awarded GSK $235 million. The district court vacated the verdict on judgment as a matter of law. The Federal Circuit, in a 2020 panel decision later reaffirmed on rehearing in August 2021, reinstated the verdict [7]. Chief Judge Sharon Prost wrote a sharp dissent arguing that the majority opinion effectively nullified the skinny label pathway that Congress had created.
The Supreme Court declined to take the case in May 2023. GSK and Teva quietly settled in early 2024. The doctrine, however, survived.
Amarin v. Hikma and the Doctrine’s Expansion
In June 2024, the Federal Circuit issued its opinion in Amarin Pharma, Inc. v. Hikma Pharmaceuticals USA Inc., 104 F.4th 1370, reversing a district court dismissal of Amarin’s induced infringement claim against Hikma’s generic icosapent ethyl product [8]. Vascepa, Amarin’s branded version, is approved for two indications: severe hypertriglyceridemia (the SH indication, originally approved in 2012) and cardiovascular risk reduction in certain patient populations (the CV indication, approved in 2019 and protected by method-of-use patents). Hikma’s skinny label included only the SH indication.
The Federal Circuit found that Amarin had plausibly alleged induced infringement based on three factual patterns: Hikma’s press releases referred to the product as a ‘generic version’ of Vascepa; those press releases cited Vascepa total sales figures that included both indications; and Hikma’s website marketed the product within the broad therapeutic category of ‘Hypertriglyceridemia’ rather than the narrower SH indication that its label actually covered. The label itself, the court conceded, might not alone support inducement. The label combined with the public statements did.
The decision matters for two reasons. First, it confirmed that GSK was not a one-off ruling tied to the peculiar facts of Teva’s reissued patent and the FDA’s later labeling intervention. Second, it lowered the bar. GSK v. Teva involved an explicit FDA-mandated relabeling that arguably negated the carve-out. Amarin v. Hikma involved a clean Section viii carve-out where the inducement theory rested entirely on non-label communications. Hikma had argued that the panel was ‘allowing inducement claims to proceed without any statement by Hikma encouraging the claimed methods.’ The Federal Circuit denied rehearing en banc [9].
The Supreme Court granted certiorari on January 16, 2026, with oral argument scheduled for April 27, 2026. The question presented is whether allegations that a generic drugmaker calls its product a ‘generic version’ and cites public information about the branded drug are sufficient to plead induced infringement when the label fully carves out the patented use. Whatever the Court decides, the doctrinal uncertainty has already done its work. Generic manufacturers spent 2024 and 2025 scrubbing websites, rewriting launch announcements, retraining sales forces, and recalibrating their launch economics on multi-indication drugs.
The Economics of the Method-of-Use Moat
The strategic value of a method-of-use patent does not come from preventing generic launch outright. The compound patent already did that. The value comes from controlling what share of the post-LOE market the generic can actually capture. If physicians prescribe a drug 60% of the time for an indication that remains under method-of-use protection and 40% for unpatented indications, a clean skinny label launch theoretically caps the generic at 40% of the volume. In practice, because pharmacy substitution happens at the molecule level rather than the indication level, the generic typically captures more than its share of the unpatented volume. But the brand retains a defensible commercial position the compound-only model would have predicted as lost.
Now overlay the induced infringement risk introduced by GSK and Amarin. The generic that launches with a clean skinny label still faces potential liability for the patented indication if its marketing behavior, sales force conduct, or public statements can be read as inducing prescribing for the carved-out use. Damages in such cases are not trivial. The Teva carvedilol jury verdict was $235 million on a drug whose primary patent had already expired. Plaintiff lawyers and brand counsel now treat the launch communications of every Section viii generic as evidence-gathering opportunities.
The practical effect is that generic challengers must either accept litigation exposure on the patented indication, launch with restricted marketing that limits their commercial penetration even on unpatented indications, or wait for the method-of-use patent to expire entirely. Each option transfers economic value to the brand. The first option transfers value through litigation cost and potential damages. The second transfers value through suppressed generic uptake. The third transfers value through extended de facto exclusivity.
For analysts running discounted cash flow models on brand pharmaceutical assets, this is not a minor adjustment. A blockbuster generating $5 billion per year that holds onto 30% of its volume for an additional three years past primary patent expiration represents $4.5 billion in gross revenue that traditional cliff models would have written off. Even adjusting for pricing erosion and payer pressure, the economic delta is substantial.
‘When present, independent method of use patents add 7.4 years (95% C.I.: 6.4 to 8.4 years) of patent life beyond primary composition claims. Late-filed independent secondary patents are more common for higher sales drugs, consistent with active lifecycle management of patent portfolios.’ — Kapczynski et al., PLoS ONE, 2012 [5]
How Originators Build Method-of-Use Portfolios
A method-of-use patent strategy is not a single filing decision. It is a multi-year discipline that runs from clinical development through post-approval lifecycle management. The strongest portfolios share four characteristics, each of which I’ll work through with reference to real-world examples.
Filing Discipline Tied to Clinical Milestones
The conventional wisdom that patent applications should be filed as early as possible runs counter to optimal method-of-use strategy. Early filing maximizes the time before the 20-year patent term begins eroding against pre-approval development time. Late filing, often tied to Phase II or Phase III data readouts, captures specific therapeutic effects, dosing regimens, or patient populations that only emerge from clinical evidence. The trade-off is term length against claim specificity and clinical defensibility.
Bristol-Myers Squibb’s management of Revlimid (lenalidomide), inherited through the Celgene acquisition, shows this discipline in action. The primary composition patent was supplemented by a portfolio of method-of-use and formulation patents that, combined with regulatory exclusivities and litigation settlements, extended effective exclusivity until 2026, well beyond what the primary patent alone would have supported [10]. The Revlimid post-LOE trajectory has tracked closer to a biologic curve than the small molecule curve its molecular weight would predict.
Indication Expansion as Patent Generation
The most defensible method-of-use claims come from new indications, not from reformulations of existing ones. Each new approved indication creates a potential filing opportunity that, if granted and properly listed in the Orange Book, extends the effective market exclusivity for that specific use. Drugs like Keytruda (pembrolizumab) have followed this template aggressively. Merck has pursued more than 30 approved indications across multiple tumor types, with each indication potentially supported by its own method-of-use patent portfolio. The result is a thicket that biosimilar manufacturers will navigate one indication at a time rather than as a single litigation event.
Sanofi’s Dupixent (dupilumab) provides a similar example. The drug is approved across atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyps, eosinophilic esophagitis, and prurigo nodularis, with method-of-use patents tied to several of these indications expiring on different dates between 2030 and 2031 [11]. The 2024 full-year reported Dupixent sales of €13.1 billion (approximately $15.5 billion) represented roughly 30% of Sanofi’s total revenue, and the staggered method-of-use patent expiration means biosimilar competition will be indication-specific rather than wholesale.
Dosing Regimen Claims
Method-of-use patents tied to specific dosing regimens, particularly extended-release schedules, weight-based dosing, or combination protocols, have proven durable against obviousness challenges when supported by clinical evidence. The mechanism is straightforward: if a generic cannot replicate the dosing instructions in its labeling without infringing a listed method-of-use patent, it cannot reach the prescribing physician with the same therapeutic profile. Even small differences in dosing language can create commercial distance that translates into preserved volume share for the brand.
Patient Population Claims
Some of the most strategically valuable method-of-use claims define specific patient populations rather than disease states. A claim covering ‘treating Disease X in patients with biomarker Y above threshold Z’ creates a narrow but defensible monopoly that may capture the majority of the commercially valuable prescribing volume even when the broader disease indication is unpatented. Vascepa’s CV indication patents follow this pattern. The SH indication, approved earlier and covering only patients with triglycerides above 500 mg/dL, was unpatented. The CV indication, covering a much larger patient population with triglycerides above 150 mg/dL plus established cardiovascular risk factors, was patented. The CV population represents the commercially dominant slice of icosapent ethyl prescribing volume.
The Generic Manufacturer’s Counter-Playbook
Generic strategists facing a method-of-use thicket have a limited toolkit, and the GSK and Amarin decisions have narrowed it further. Four approaches remain viable, each with distinct cost structures and risk profiles.
Direct Validity Challenge
The most aggressive approach is to challenge the method-of-use patent’s validity directly, typically through Paragraph IV certification followed by inter partes review (IPR) at the Patent Trial and Appeal Board. Method-of-use patents are particularly vulnerable to obviousness challenges when the underlying mechanism of action was disclosed in earlier publications or when the claimed therapeutic effect could be reasonably predicted from prior art. The economics of IPR favor generic challengers: filing costs run in the low millions, the standard is preponderance of the evidence rather than clear and convincing, and the institution rate, while declining in recent years, remains material.
The downside is timing. IPR proceedings can take 18 to 24 months, and parallel district court litigation extends the timeline further. For a generic targeting a drug with a method-of-use patent expiring in three years, the validity challenge may not resolve before the patent expires on its own. The strategic calculus is sharpest for patents with longer remaining terms.
Designed-Around Labels
The traditional Section viii skinny label remains available, but its execution requires far more discipline than it once did. The post-GSK and post-Amarin playbook for clean skinny label launches involves auditing every public communication for indication-neutral language, training sales forces on what they can and cannot say, avoiding total-revenue references that aggregate patented and unpatented indications, and resisting the marketing temptation to position the generic as a full substitute for the brand. Some generic manufacturers have begun adopting ‘limitation of use’ statements in their labels that explicitly disclaim the patented indication, going beyond what Section viii technically requires.
The cost is commercial penetration. A generic that markets aggressively as ‘generic Brand X’ captures more substitution volume than one that markets narrowly as ‘generic [molecule name] indicated for [SH/limited indication only].’ The legal protection of the latter approach reduces inducement exposure but cedes prescription share.
Wait-and-Launch
For some method-of-use patents with relatively short remaining terms, the economically rational approach is to simply wait. Generic launch costs include manufacturing scale-up, regulatory submission, sales infrastructure, and inventory. If the method-of-use patent expires within 12 to 18 months of the compound patent, the savings from waiting often exceed the gross profit from a constrained skinny label launch.
Authorized Generic Negotiation
A fourth approach involves negotiating an authorized generic license with the brand, often as part of a broader settlement of Paragraph IV litigation. The brand authorizes a specific generic to launch with a full label, including patented indications, in exchange for either delayed entry, royalty payments, or supply chain arrangements. Authorized generic launches account for a meaningful share of generic market entries and are particularly common where the method-of-use patent landscape would make a contested launch economically marginal.
DrugPatentWatch and the Data Layer
The strategic frameworks above presume something that until relatively recently was difficult to achieve: comprehensive, current, accurate visibility into the Orange Book listings, patent claims, expiration dates, and litigation history of the drugs in question. Platforms like DrugPatentWatch have made this layer accessible to analysts who do not have in-house IP departments running their own dockets. For investors and strategic teams modeling LOE scenarios across multiple drugs, the ability to pull patent landscapes, identify method-of-use claims, track Paragraph IV filings, and monitor settlement activity in one workflow has changed how this work gets done.
What the data layer reveals, when used carefully, is that method-of-use patent strategy is not uniform across the industry. Some companies file aggressively and late. Others rely heavily on regulatory exclusivities. Some construct dense indication-by-indication thickets. Others concentrate on a small number of high-defensibility claims. The patterns are visible in the Orange Book if you know what to look for, and the predictive value of these patterns for post-LOE revenue trajectories is meaningful.
Case Study: Coreg and the Birth of the Modern Doctrine
The Coreg litigation deserves a closer look because it established the doctrinal framework that every subsequent skinny label dispute has operated within. Carvedilol’s compound patent expired in 2007. Teva had filed an ANDA in 2002 with a Paragraph IV certification on the CHF method-of-use patent. After the compound patent expired, Teva launched its generic with a skinny label that omitted the CHF indication, marketing only for hypertension and LVD.
The complicating factor was the patent that GSK held. The original CHF method-of-use patent had been reissued in 2008 (as RE40,000) with claims covering a method of decreasing mortality in CHF patients by administering carvedilol in combination with at least one other therapeutic agent (typically an ACE inhibitor or diuretic). The reissue narrowed the claim scope while preserving protection for the dominant clinical use pattern. FDA later directed Teva to add the CHF indication back to its label for safety reasons, creating a ‘full label’ phase during which Teva’s labeling expressly included the patented use.
The Federal Circuit’s 2020 panel decision, reaffirmed on rehearing in 2021, found inducement based on the totality of Teva’s conduct. Press releases describing the product as ‘AB-rated’ to Coreg, marketing catalogs that listed the product under therapeutic categories that included CHF prescribing, and witness testimony at trial confirming that Teva understood its generic would be substituted for Coreg in CHF patients. Judge Prost’s dissent was vigorous: the majority, she argued, had punished Teva for following the regulatory pathway Congress had created and effectively converted Section viii from a safe harbor into a litigation trap [12].
The case settled in early 2024 after the Supreme Court declined certiorari. The settlement terms were not disclosed. The damages award, originally $235 million, had been the subject of repeated procedural reversals. What remains is the doctrine: a generic manufacturer’s non-label conduct can sustain an induced infringement claim even when its label complies with Section viii.
Case Study: Vascepa and the Expansion of the Doctrine
The Vascepa litigation tested how far the GSK doctrine extended. Hikma’s skinny label was clean on its face. It covered only the SH indication, which was unpatented. Hikma did not engage in the kind of explicit relabeling that Teva had been forced into. The induced infringement theory rested entirely on what Amarin characterized as misleading non-label communications.
The specific factual hooks Amarin pleaded were illuminating. Hikma’s March 2020 press release announcing FDA approval referenced Vascepa’s $919 million in annual sales (a figure that included both SH and CV revenue). The company’s website described the product within the therapeutic category of ‘Hypertriglyceridemia’ broadly, which encompassed both severe and non-severe hypertriglyceridemia patients. Subsequent Hikma communications described the product as the ‘generic version’ of Vascepa rather than as a generic icosapent ethyl approved for the SH indication only.
The district court dismissed the case at the pleadings stage, finding that none of these allegations rose to the level of induced infringement. The Federal Circuit reversed, holding that the totality of allegations plausibly stated a claim sufficient to survive a motion to dismiss [13]. Hikma’s website disclaimer noting that the product was approved for fewer than all uses of Vascepa did not save the case, the court found, because the broader communications could plausibly be read by physicians as encouraging the patented use.
The Supreme Court’s grant of certiorari in January 2026 frames the issue precisely. The question is whether a complaint states a claim for induced infringement when the generic label fully carves out the patented use and the only alleged encouragement comes from product references and publicly available sales information. The accelerated schedule, with oral argument on April 27, 2026, and a decision expected before the Court’s summer recess, reflects the commercial stakes [14].
Case Study: Revlimid and the Combined Strategy
Revlimid (lenalidomide) is a multiple myeloma and myelodysplastic syndrome treatment that Celgene developed and BMS acquired in 2019. The primary composition patent on lenalidomide expired in 2019, but BMS extended effective exclusivity to 2026 through a combination of method-of-use patents, REMS-based distribution constraints, and settlement agreements with generic challengers that staggered authorized generic entry [15].
The Revlimid case is instructive because it shows how method-of-use patents work alongside non-patent exclusivities. The REMS (Risk Evaluation and Mitigation Strategy) program required for lenalidomide due to teratogenicity concerns created an additional barrier to generic entry that, combined with the patent thicket, allowed BMS to negotiate volume-limited generic settlements rather than face a vertical cliff. The post-2019 Revlimid revenue curve looks more like a biologic slope than a small molecule cliff, despite lenalidomide being a small molecule.
What Investors Should Be Modeling
For institutional investors building positions in branded pharmaceutical equities ahead of the 2026 to 2030 LOE cycle, the method-of-use patent analysis layer changes several modeling inputs. The traditional approach of using the primary patent expiration date as the cliff anchor and applying a generic erosion curve from historical analogs systematically underestimates revenue retention for drugs with strong method-of-use portfolios.
A more accurate model accounts for the following inputs separately: the primary composition patent expiration date, the latest method-of-use patent expiration date covering the dominant commercial indication, the litigation history and settlement patterns on similar drugs, the FDA’s pediatric exclusivity status, and any pending PTE (patent term extension) calculations. For biologics, the model should additionally account for biosimilar manufacturer pipeline status, BPCIA litigation patterns, and payer formulary positioning.
Consider Eliquis (apixaban). The drug generated approximately $13 billion in 2024 revenue [16]. Initial cliff projections placed primary patent expiration at 2026. Subsequent court rulings extended this to April 1, 2028, due to a composition patent with later expiration. Eliquis was also among the first 10 drugs subject to Inflation Reduction Act price negotiations, which creates a different revenue erosion mechanism running in parallel with patent expiration. The combined effect is that BMS and Pfizer face Eliquis revenue erosion from two distinct mechanisms (negotiation-driven price cuts beginning in 2026 and generic competition beginning in 2028), each with its own modeling characteristics.
Keytruda (pembrolizumab) presents a different profile. Merck generated $29.5 billion from the drug in 2024, with revenue projected to peak around $32.7 billion in 2026. The primary US compound patent expires in December 2028. Merck has pursued a Keytruda Qlex subcutaneous reformulation that analysts project could contribute $7 billion in sales by early 2032, plus more than 30 approved indications each potentially supported by method-of-use claims [17]. Biosimilar manufacturers including Amgen, Samsung Bioepis, and Bio-Thera Solutions are preparing entries, but the biosimilar pathway for monoclonal antibodies is not equivalent to small molecule ANDAs. The erosion curve will be a slope.
The Variables That Move the Most
Two variables move post-LOE revenue projections more than any others in the current cycle. The first is whether method-of-use patents cover the dominant clinical indication or only secondary indications. A method-of-use patent covering 60% of prescribing volume is materially different from one covering 15%. The second is the level of commercial sophistication the generic challenger brings to its launch communications. A sophisticated generic that prepares its launch materials with explicit attention to GSK and Amarin doctrine carries less litigation risk than one operating on pre-2021 assumptions, and the litigation risk profile directly affects the launch’s commercial reach.
The Regulatory Layer: Orange Book Listing and FDA’s Role
Method-of-use patents only achieve their full strategic value when properly listed in the Orange Book under 21 C.F.R. § 314.53. The listing requirements specify that the patent must claim ‘the drug for which the applicant submitted the application or which claims an approved method of using such drug.’ Orange Book listings are submitted by the new drug application holder, not reviewed substantively by FDA for patent validity or scope, and serve as the trigger for the Paragraph IV / Section viii framework.
FDA’s role in the skinny label process is procedural rather than substantive on patent questions. The agency confirms that a Section viii statement carves out the claimed method of use, ensures the resulting label is consistent with the unpatented indications, and approves the ANDA. FDA does not evaluate whether the carve-out adequately protects against induced infringement claims. That question is left to the courts.
The FTC has periodically raised concerns about Orange Book listings that arguably overreach, particularly device patents listed against drug-device combination products. A 2023 FTC enforcement action led to delistings of inhaler and EpiPen device patents that the agency characterized as not properly listable. The FTC’s position remains that Orange Book listings should be carefully scrutinized for both substantive accuracy and competitive impact. Whether this enforcement posture extends to method-of-use patents is unsettled, but the regulatory environment is more skeptical of broad listings than it was a decade ago.
The Inflation Reduction Act Wildcard
The IRA’s drug price negotiation provisions add a parallel pressure that interacts with method-of-use patent strategy in ways that are not yet fully understood. Drugs become eligible for negotiation 9 years after FDA approval for small molecules and 13 years for biologics, with negotiated prices taking effect 2 years after selection. The first 10 drugs were selected in 2023 with negotiated prices effective January 1, 2026. The second 15-drug list was selected in 2025.
For brands with strong method-of-use portfolios, the IRA creates a difficult trade-off. The same lifecycle management strategies that extend effective patent exclusivity also keep the drug in the negotiation-eligible pool longer. A drug whose composition patent expired five years ago but whose method-of-use patents continue to suppress generic competition will face IRA negotiation while still operating under quasi-monopoly conditions. The net revenue effect depends on the relative magnitudes of negotiated price cuts versus preserved volume share.
Some companies have begun adjusting their lifecycle strategies in response. The calculation now includes not just patent term extension but also IRA timing optimization. Drugs that face IRA negotiation in 2028 may benefit less from method-of-use patent extensions running into 2030 than from accelerated successor product launches that shift revenue to a new product cycle. The strategic calculus is more complex than it was three years ago.
Litigation Cost Asymmetry
One factor that does not show up cleanly in patent analytics platforms but matters enormously in practice is the litigation cost asymmetry between brand and generic manufacturers. For a blockbuster drug generating $5 billion per year ($13.7 million per day), even a modest delay in generic entry produces value that swamps any plausible litigation budget. Brand counsel routinely operate with effectively unlimited litigation resources where blockbuster drugs are concerned. The constraint is not cost but risk management [18].
Generic manufacturers face a different calculus. Generic gross margins on commodified molecules typically run in single-digit to low double-digit percentages. The cost of contesting a single method-of-use patent through Paragraph IV litigation, IPR, and potential appeals can exceed the discounted present value of the additional revenue captured by an at-risk launch. Generic challengers therefore tend to settle rather than litigate to final judgment on method-of-use patents, and the settlements often involve delayed entry, royalty payments, or volume-limited launches that preserve substantial brand value.
This asymmetry is structural and difficult to model from outside the parties’ confidential settlement positions. It is one reason that historical erosion curves are imperfect predictors of future LOE outcomes. The brand-generic settlement market is more sophisticated than it was a decade ago, and the settlements increasingly look like commercial arrangements rather than patent compromises.
The European Dimension
Method-of-use patent strategy in Europe operates under a partially different framework than the US approach. Supplementary Protection Certificates (SPCs) extend patent term by up to 5 years (with a 15-year-from-MA cap), and Article 11 of the SPC Regulation governs how method-of-use SPCs interact with generic entry. EU patent law has historically been more skeptical of pure Swiss-style claims and ‘second medical use’ patents than US doctrine, though European Patent Office practice has converged somewhat over the past decade.
The practical effect is that a drug with strong US method-of-use protection may face earlier generic competition in EU markets, particularly for indications added after initial marketing authorization. Multi-region launch planning therefore requires separate analysis of patent landscapes in each major market, and the regional revenue profiles can diverge meaningfully even for the same molecule.
What This Means for Deal Structures
The strategic value of method-of-use patents has reshaped how pharmaceutical M&A is priced. Acquirers of late-stage development assets increasingly require detailed method-of-use patent landscape analyses as part of due diligence. The valuation of acquired pipelines depends not just on the projected revenue profile of the lead indication but on the strength of method-of-use claims that may be filed in subsequent indications.
Royalty stream financing structures, including the synthetic royalty deals that have become more common in biotech financing, often allocate value differently across indications based on method-of-use patent positions. A royalty buyer that takes a percentage of total revenue receives different value from a royalty buyer that takes a percentage of revenue from a specific patented indication. The contracts now reflect this distinction more carefully than they did even five years ago.
Licensing transactions for follow-on indications have also adapted. The standard pharmaceutical licensing model historically tied royalties to net sales of the licensed product. Modern deals increasingly distinguish royalties for the patented indication from royalties for unpatented uses, with different rate structures and different durations. The structural complexity reflects the strategic reality that not all indications carry the same patent-driven economic value.
Reading the Tea Leaves on Hikma v. Amarin
Predicting Supreme Court outcomes is hazardous, but the questions presented in Hikma v. Amarin point to a likely narrowing of the Federal Circuit doctrine. The Court’s grant of certiorari, accelerated argument schedule, and the Solicitor General’s prior position in GSK v. Teva all suggest that at least some justices are concerned about the chilling effect on generic competition. The April 27, 2026 oral argument will provide better signals, but the directional probability is that the Court will tighten the pleading standard for induced infringement in Section viii cases.
Two scenarios bear consideration. In the first, the Court holds that a complaint requires some explicit statement encouraging the patented use, beyond mere references to the brand product or aggregated sales data. This would substantially weaken the doctrine and restore much of Section viii’s original protective force. Generic skinny label launches would carry significantly less litigation risk, and the post-LOE revenue retention for drugs with method-of-use patents would compress.
In the second scenario, the Court affirms the Federal Circuit but with limiting language that requires more than the bare allegations Amarin pleaded. This would preserve the doctrine while constraining its expansion. Generic launches would still require communications discipline, but the pleading bar would rise.
Either outcome changes the modeling assumptions for the 2026 to 2030 cliff. Analysts who are not building both scenarios into their LOE projections are working with incomplete information.
The Diligence Checklist
For investors, business development teams, and strategic analysts evaluating a branded pharmaceutical asset’s post-LOE trajectory, the method-of-use analysis should cover several specific items.
First, identify every Orange Book listing for the asset, distinguishing composition patents, formulation patents, method-of-use patents, and device patents. Note the listing date, expiration date, and the FDA-approved indication each patent purports to cover.
Second, evaluate the dominant clinical indication. Which indication accounts for the largest share of prescribing volume and revenue? Is that indication covered by an unexpired method-of-use patent listed in the Orange Book? If yes, the post-LOE revenue retention will materially exceed the small molecule historical average.
Third, review the Paragraph IV and Section viii filing history. Have generic challengers filed against the listed patents? Have any IPR proceedings been instituted? Have any settlements been reached? The litigation history is the best available predictor of future generic behavior.
Fourth, assess the breadth of the method-of-use claims relative to the prescribing pattern. A patent claim covering ‘a method of treating Disease X’ is broader than ‘a method of treating Disease X in patients with biomarker Y at concentration Z.’ Narrow patient population claims can be commercially dominant if the patient population represents the bulk of prescribing volume.
Fifth, consider the post-Hikma v. Amarin doctrinal scenario. Model both the strong doctrine case (in which generic skinny label launches carry meaningful litigation risk) and the weak doctrine case (in which Section viii operates closer to its original protective scope). The difference in projected revenue trajectories will inform position sizing and risk hedging.
Operational Realities for Brand Teams
Brand commercial teams preparing for LOE need to align several functions in ways that historical playbooks did not require. Legal, regulatory, sales, marketing, and investor relations all need to operate with consistent understanding of the method-of-use patent landscape and the implications for generic competitor behavior.
Sales force training should emphasize the indication-specific clinical evidence that supports prescribing for the patented use, not just product positioning relative to generic competitors. The clinical evidence is what physicians use to make prescribing decisions, and the prescribing decisions are what determine post-LOE revenue retention. Marketing materials should be reviewed not for generic-defense messaging (which can backfire) but for clear articulation of clinical value in the patented indication.
Investor relations communications about LOE planning should distinguish total revenue projections from indication-specific projections. Investors who model post-LOE revenue at the aggregate level miss the dynamics that method-of-use patents create at the indication level. Companies that can articulate their lifecycle strategy in indication-specific terms tend to receive better analyst coverage and more accurate consensus estimates.
Operational Realities for Generic Teams
Generic strategy teams operate under different constraints but face equally specific operational requirements. Launch communications must be drafted with explicit attention to the GSK and Amarin doctrines, even pending the Supreme Court’s decision. Press releases should describe the product by its molecule name and approved indication, not as a ‘generic version’ of the brand. Sales representatives should be trained to communicate the labeled indication only, with documented compliance monitoring.
Where indication-specific marketing is feasible, the generic should pursue it. Some generic manufacturers have begun developing differentiated marketing materials for each ANDA indication, which simultaneously addresses prescriber education needs and creates documentation that supports defenses against induced infringement claims. The cost of this approach is real, but it is less than the expected cost of induced infringement litigation on a contested launch.
Launch sequencing decisions should account for the method-of-use patent landscape rather than focusing solely on compound patent expirations. A two-stage launch (skinny label first, full label after method-of-use expiration) may produce better aggregate economics than a single-stage launch with aggressive marketing that triggers litigation.
The Anatomy of a Method-of-Use Claim
To understand why method-of-use claims behave so differently from composition claims in litigation, it helps to look at the actual language patent attorneys use. A typical composition claim reads: ‘A compound having the structure [chemical formula], or a pharmaceutically acceptable salt thereof.’ The claim defines a thing. Infringement turns on whether the accused product is or contains that thing. The analysis is largely chemical.
A method-of-use claim reads differently: ‘A method of treating [disease] in a patient, comprising administering to the patient a therapeutically effective amount of [compound].’ The claim defines an action. Infringement turns on whether someone, somewhere, performs that action using the accused product. The direct infringer is typically the prescribing physician or the administering nurse. The generic manufacturer is not directly performing the patented method. The manufacturer’s liability runs through 35 U.S.C. § 271(b), which provides that ‘whoever actively induces infringement of a patent shall be liable as an infringer.’
This indirect liability structure is what makes method-of-use enforcement so different from composition enforcement. A composition patent infringement case asks: did the defendant make, use, sell, or import the patented compound? The answer is binary and verifiable through laboratory analysis. A method-of-use infringement case asks: did the defendant induce another party to perform the patented method? The answer turns on the defendant’s communications, marketing materials, and conduct in the marketplace. Witness testimony, document discovery, and expert opinion on what physicians would have understood the defendant’s communications to mean all become material.
The Federal Circuit summarized the inducement standard in DSU Medical Corp. v. JMS Co., 471 F.3d 1293 (Fed. Cir. 2006) (en banc): ‘inducement requires evidence of culpable conduct, directed to encouraging another’s infringement, not merely that the inducer had knowledge of the direct infringer’s activities.’ This standard sounds simple but operationalizes into a question that juries find genuinely difficult: did the defendant’s behavior, taken as a whole, encourage the patented use? The Coreg and Vascepa cases turned on exactly this question.
The Knowledge Element
A second wrinkle in inducement doctrine is the knowledge requirement. The Supreme Court held in Global-Tech Appliances, Inc. v. SEB S.A., 563 U.S. 754 (2011), that induced infringement requires knowledge that the induced acts constitute patent infringement. Willful blindness can satisfy this requirement, but mere knowledge of the patent is insufficient. The accused inducer must have known, or been willfully blind to, the fact that the induced conduct would infringe.
For generic manufacturers, this knowledge element is functionally satisfied by the Orange Book listing process. The brand lists the method-of-use patent, the generic files its ANDA with full awareness of the listing, and the generic’s Section viii statement explicitly references the listed patent. Knowledge of the patent and its claims is essentially conceded at the filing stage. The question that remains is whether the generic’s conduct encouraged the infringing use.
The Specific Intent Question
Beyond knowledge, inducement requires specific intent to cause infringement. Generic manufacturers typically argue that their intent in launching a skinny-labeled generic was to compete only for the unpatented indications. Brand plaintiffs counter that the manufacturer’s commercial reality requires capturing substitution volume across all approved uses, and that the manufacturer’s marketing conduct reflects this commercial reality even if internal compliance documents claim otherwise.
The evidentiary work in these cases tends to be granular. Sales call notes. Internal product positioning memoranda. Email exchanges between marketing and regulatory teams. Trade show presentation materials. The discovery burden alone can be substantial, and the cost asymmetry between brand and generic in this discovery becomes a strategic factor in how cases are negotiated.
Section viii Procedures in Practice
The mechanics of filing a Section viii statement deserve closer examination because the procedural details shape the legal exposure that follows. An ANDA applicant who concludes that a listed patent claims a method of use for which the applicant is not seeking approval must submit, in lieu of the patent certification required for unlisted method-of-use patents, a statement explaining that the method of use patent does not claim a use for which the applicant is seeking approval.
FDA’s regulations at 21 C.F.R. § 314.94(a)(12)(iii) require that the Section viii statement specifically identify the patent and the method-of-use claim being carved out. The applicant must also submit proposed labeling that omits the carved-out indication. FDA reviews the proposed labeling to ensure that the omission is consistent with safe and effective use for the remaining indications. The agency does not evaluate whether the carve-out adequately protects against induced infringement claims, which remain a matter for the courts.
The procedural sequence creates several inflection points where strategic decisions matter. The patent’s Orange Book listing language sets the boundary of what can be carved out. The proposed labeling defines the indications the generic will market. The Section viii statement creates a public record of the carve-out. And the subsequent commercial launch creates the documentary record that any future inducement case will mine for evidence.
The Use Code Issue
One technical detail that has produced significant litigation is the ‘use code’ that the brand submits when listing a method-of-use patent in the Orange Book. The use code is a brief narrative description of the patented method, and FDA does not independently verify its accuracy. If the use code is written broadly, the brand may effectively block carve-outs that the patent claims themselves would not support. If the use code is written narrowly, generic applicants have more room to carve.
The Supreme Court addressed this issue in Caraco Pharmaceutical Laboratories, Ltd. v. Novo Nordisk A/S, 566 U.S. 399 (2012), holding that an ANDA applicant can use a counterclaim under 21 U.S.C. § 355(j)(5)(C)(ii)(I) to compel a brand to correct an overbroad use code. The Caraco decision provides a procedural mechanism for challenging overreaching use codes, but the mechanism is cumbersome and the litigation cost is meaningful. Many use code disputes end in settlements that adjust the listing language rather than producing definitive judicial determinations of scope.
The Statement Itself
The Section viii statement is typically brief, identifying the patent and explaining the carve-out in a few paragraphs. The statement does not engage with the patent’s validity or scope beyond what is necessary to support the carve-out. This brevity is intentional. A more detailed engagement with patent scope could create estoppel issues in subsequent litigation. Generic applicants therefore tend to draft Section viii statements in language that tracks the patent’s claim language and the brand’s use code, identifying the precise boundaries of the carve-out without making substantive validity arguments.
The Federal Circuit’s Doctrinal Drift
The arc of Federal Circuit decisions on induced infringement of method-of-use patents reflects a doctrinal drift over roughly two decades. Earlier decisions, including Warner-Lambert Co. v. Apotex Corp., 316 F.3d 1348 (Fed. Cir. 2003), and Allergan, Inc. v. Alcon Laboratories, Inc., 324 F.3d 1322 (Fed. Cir. 2003), generally protected generic manufacturers who followed the Section viii procedure. The Warner-Lambert decision in particular held that a generic’s intent to obtain approval for unpatented uses, manifested through a Section viii statement, precluded inducement liability even when the brand argued that some off-label prescribing would inevitably occur.
The drift toward expanded inducement liability began with cases like AstraZeneca LP v. Apotex, Inc., 633 F.3d 1042 (Fed. Cir. 2010), where the Federal Circuit affirmed an inducement finding based on the generic’s proposed labeling for a budesonide product. By GSK v. Teva, the court had expanded the doctrine to encompass non-label communications. By Amarin v. Hikma, the court had held that allegations of inducement could survive a motion to dismiss based on press releases and product positioning alone, without any explicit statement encouraging the patented use.
The drift is not unidirectional. Several Federal Circuit decisions during the same period found in favor of generic defendants on inducement claims, and panel composition has produced inconsistent outcomes. But the trend line, particularly since 2020, has been toward greater inducement exposure for generic manufacturers using Section viii carve-outs. This is the trend that the Supreme Court will likely address in Hikma v. Amarin.
The Solicitor General’s Role
The federal government’s position in these cases has been consistent and influential. In GSK v. Teva, the Solicitor General’s brief, signed by the USPTO, HHS, and FDA, urged the Supreme Court to grant certiorari and reverse the Federal Circuit. The brief argued that the Federal Circuit’s expansion of inducement doctrine threatened the Section viii pathway and would harm generic competition. The Court declined to take the case, but the Solicitor General’s position became part of the legal record and has been cited in subsequent briefing.
For Hikma v. Amarin, the federal government’s position is again expected to favor narrowing the doctrine. The accelerated argument schedule and the breadth of amicus support on both sides reflect the perceived importance of the case to both branded and generic pharmaceutical sectors.
The Biosimilar Adaptation
Biosimilar manufacturers face a substantially different regulatory pathway than small molecule generics, but the method-of-use patent issues that have shaped small molecule competition increasingly apply to biologic competition as well. The Biologics Price Competition and Innovation Act (BPCIA), enacted as part of the Affordable Care Act in 2010, created the 351(k) pathway for biosimilar approval. The BPCIA includes its own version of the patent dance, which is structured somewhat differently from the Hatch-Waxman process but addresses analogous issues.
For method-of-use claims specifically, biosimilar applicants can pursue a process roughly analogous to Section viii carve-outs. The biosimilar can seek approval for fewer than all of the reference product’s approved indications, omitting indications covered by unexpired method-of-use patents. FDA has approved several biosimilars with indication-specific labeling, and the practice has become routine for biosimilar launches against multi-indication biologics.
The induced infringement risk extends to this practice. Biosimilar manufacturers face the same doctrinal exposure that small molecule generics face, and the early biosimilar launches have produced their own litigation history. Humira biosimilar launches against AbbVie’s adalimumab portfolio involved complex method-of-use patent landscapes across multiple inflammatory indications, and the settlements that emerged from this litigation generally involved staggered launch dates and indication-specific terms [19].
Keytruda and the Biologic Method-of-Use Thicket
Merck’s Keytruda represents the most aggressive example of a biologic method-of-use thicket currently in operation. With more than 30 approved indications across multiple tumor types, the drug has been the subject of a sustained patent generation strategy that has produced filings covering specific combinations, dosing regimens, patient populations defined by biomarker status, and combination protocols with other oncology drugs. The primary composition patent expires in December 2028, but the method-of-use patent portfolio extends well beyond that date for several of the most commercially significant indications.
Biosimilar manufacturers preparing pembrolizumab biosimilar launches face a strategic puzzle. Each major indication has its own patent landscape. A biosimilar approved for, say, non-small cell lung cancer second-line treatment may be unable to market for first-line treatment or for combination protocols without facing method-of-use exposure. The launch sequencing decisions therefore become indication-specific, and the aggregate biosimilar opportunity is meaningfully smaller than a simple compound patent analysis would suggest.
Opdivo and Bristol-Myers Squibb’s Position
Bristol-Myers Squibb’s Opdivo (nivolumab) faces loss of exclusivity around the same window as Keytruda, with composition patent expiration also in 2028. The drug generated approximately $9 billion in 2024 revenue. BMS has pursued a similar indication-by-indication patent generation strategy, though the Opdivo portfolio is somewhat smaller in absolute terms than Keytruda’s. The simultaneous LOE on Eliquis (April 2028) and Opdivo creates the most concentrated revenue exposure of any major pharmaceutical company, with the BMS growth gap reportedly running to $38 billion across the 2026 to 2030 window.
The strategic response has emphasized both pipeline development (including new molecular entities for cardiovascular and oncology indications) and lifecycle management across the existing portfolio. The method-of-use patent layer for Opdivo specifically remains a material component of the post-LOE revenue retention scenario, particularly for combination protocols with other oncology agents.
Cross-Border Considerations
Method-of-use patent strategy operates differently in major non-US markets, and global pharmaceutical companies must coordinate their portfolio strategies across jurisdictions. The European framework, the Japanese system, and several emerging market jurisdictions each impose distinct constraints and opportunities.
European Patent Office Practice
The EPO has historically been receptive to second medical use claims, including Swiss-style claims (‘use of [compound] for the manufacture of a medicament for treating [disease]’) and EPC 2000 purpose-limited product claims (‘[compound] for use in treating [disease]’). The Enlarged Board of Appeal’s decision in G 2/08 confirmed that purpose-limited product claims can cover new dosing regimens for known compounds, expanding the scope of protectable secondary uses.
National implementation, however, varies. The UK Supreme Court’s decision in Warner-Lambert Co. LLC v. Generics (UK) Ltd [2018] UKSC 56 limited the enforceability of second medical use patents against generic manufacturers who carve out the patented indication, holding that the manufacturer’s intent and the actual prescribing pattern in the marketplace both matter. German courts have generally been more favorable to brand patentees in similar disputes, particularly through the Düsseldorf and Munich patent litigation venues.
The European Unified Patent Court, which became operational in 2023, has begun producing its own jurisprudence on these issues. The UPC’s early decisions on method-of-use claims have been mixed, and the court’s eventual position on the interaction between Section viii-equivalent procedures and induced infringement remains unsettled.
Japanese Patent Office Practice
Japan permits method-of-use claims for pharmaceutical inventions, though the procedural mechanisms for generic carve-outs differ from US Section viii practice. The Japanese Patent Office has historically been receptive to broad method claims tied to specific therapeutic effects, and the post-grant opposition mechanisms available to generic challengers are less aggressive than US IPR proceedings. The result is generally stronger nominal protection for method-of-use claims in Japan than in the US, though the practical enforcement landscape varies by case.
Emerging Markets
Several emerging market jurisdictions, including India and Brazil, have historically been skeptical of method-of-use patents, particularly Swiss-style claims. India’s Patent Act explicitly excludes ‘the mere discovery of a new form of a known substance’ from patentability under Section 3(d), and Indian courts have applied this provision to limit method-of-use patent enforcement in several high-profile cases. Brazilian patent practice similarly imposes stricter standards on method-of-use claims than US or European practice.
For pharmaceutical companies modeling global revenue from method-of-use protected indications, these jurisdictional differences mean that the US and Europe typically account for the bulk of the patent-driven economic value, while emerging markets contribute less. Generic manufacturers based in these jurisdictions, including major Indian manufacturers like Sun Pharma, Cipla, and Dr. Reddy’s, often face fewer constraints in their home markets than they do when launching in the US or EU.
The Authorized Generic Phenomenon
One increasingly common response to method-of-use patent thickets is the authorized generic launch. An authorized generic is a generic version of a branded drug marketed under license from the brand, typically with the same FDA-approved label, often through a subsidiary or contract manufacturing arrangement. Authorized generics differ from standard generics in two important respects: they can launch with full labels including patented indications, and they typically launch on terms negotiated through settlement of underlying patent litigation.
The economic logic of authorized generic settlements is straightforward for both parties. The brand captures additional revenue from generic competition that would otherwise go to an independent challenger. The generic launches earlier than litigation would have permitted, with reduced legal risk. The combined economics often exceed what either party would have achieved through contested litigation.
Authorized generic launches have become particularly common for drugs with complex method-of-use patent landscapes where independent generic launches would face significant induced infringement exposure. The first-filer Paragraph IV applicant typically negotiates an authorized generic arrangement as part of its settlement, which protects the 180-day exclusivity period that would otherwise be at risk.
FTC Scrutiny of Reverse Payment Settlements
The Federal Trade Commission has historically scrutinized authorized generic and reverse payment settlements for potential antitrust concerns. The Supreme Court’s decision in FTC v. Actavis, Inc., 570 U.S. 136 (2013), held that large reverse payments from brand to generic could violate antitrust law under a rule of reason analysis. The decision triggered a wave of FTC enforcement actions and private antitrust litigation that has shaped how settlements are structured.
Modern settlements typically avoid the explicit reverse payment structure that drew Actavis scrutiny. Authorized generic arrangements, royalty-bearing licenses, and similar structures can achieve similar economic outcomes while complying with post-Actavis legal standards. The FTC continues to monitor these settlements, and major pharmaceutical companies file annual reports with the agency detailing their patent settlements under the Medicare Modernization Act.
Lifecycle Management and the Successor Product Strategy
For drugs with strong method-of-use patent portfolios approaching LOE, the most effective overall strategy often involves not just patent term extension but also successor product launches that shift revenue to new product cycles less affected by patent expiration. The classic example is AstraZeneca’s successor strategy for Prilosec (omeprazole), which shifted prescribing to Nexium (esomeprazole) ahead of Prilosec’s generic entry. The strategy required substantial commercial investment but preserved meaningful revenue across the franchise.
Modern successor product strategies have become more sophisticated. The successor product is typically a new molecular entity with improved efficacy, tolerability, or dosing characteristics, supported by clinical evidence that justifies switching from the original product. The successor’s patent portfolio starts fresh, providing 20 years of nominal term plus any applicable extensions. Method-of-use claims on the successor product can be filed throughout its development, building a thicket that mirrors the strategy used on the original.
The economic logic of the successor strategy depends on several factors. The successor must provide enough clinical differentiation to support switching at scale. The commercial transition must be timed to capture demand before generic competition on the original product erodes pricing across the franchise. The clinical evidence supporting the successor must be strong enough to satisfy payer formulary committees, which have become increasingly skeptical of successor products that appear to be lifecycle management without meaningful clinical improvement.
The Keytruda Qlex Example
Merck’s Keytruda Qlex subcutaneous reformulation is a current example of successor strategy in action. The reformulation provides administration convenience advantages over the original intravenous pembrolizumab, supported by clinical data demonstrating comparable efficacy and pharmacokinetic profiles. Analyst projections suggest the reformulation could contribute $7 billion in sales by early 2032, providing meaningful offset to the IV pembrolizumab erosion that will follow biosimilar entry [17].
The Keytruda Qlex strategy depends on physician and patient adoption of the subcutaneous formulation, which Merck has pursued through clinical guidelines, payer contracting, and direct prescriber outreach. The method-of-use patent portfolio on the subcutaneous formulation provides extended exclusivity beyond the IV formulation’s expiration, but the commercial success of the strategy ultimately depends on whether prescribers and patients view the subcutaneous administration as preferable to the IV alternative once biosimilar IV pembrolizumab is available at significantly lower cost.
The Hatch-Waxman Settlement Market
Settlement of Paragraph IV and method-of-use patent litigation has evolved into a sophisticated market with its own conventions, expectations, and pricing dynamics. The annual volume of settled Paragraph IV cases runs into the hundreds, and the structural patterns in these settlements provide useful signals for analysts modeling specific LOE events.
The typical settlement provides for generic entry on a specific date, often coinciding with or shortly before the expiration of one or more listed patents. Some settlements include royalty payments, supply chain arrangements, or other consideration. Others involve only the entry date and associated terms. The settlement records filed with the FTC under the Medicare Modernization Act provide partial transparency into these arrangements, though the financial terms are often redacted.
Analysts attempting to model future generic entry dates can use the settlement history of similar drugs as a baseline. Drugs with strong method-of-use patent positions and ongoing IRA negotiation exposure tend to settle on terms that defer generic entry past the composition patent expiration, sometimes by several years. Drugs with weak secondary patent positions settle closer to the composition patent expiration date.
The 180-Day Exclusivity Calculation
The Hatch-Waxman Act provides a 180-day generic exclusivity period for the first ANDA applicant to file a Paragraph IV certification. This exclusivity prevents subsequent generic applicants from launching during the 180-day window following the first filer’s commercial launch. The economic value of 180-day exclusivity can be substantial, particularly for blockbuster drugs.
The exclusivity calculation interacts with method-of-use patents in complex ways. The first filer must obtain final FDA approval and launch its product, or risk forfeiting exclusivity through one of several statutory triggers. For drugs with method-of-use patent issues, the first filer’s launch decision becomes complicated. Launching with a skinny label preserves exclusivity but exposes the manufacturer to inducement risk. Delaying launch may forfeit exclusivity. The strategic calculation can drive specific settlement terms that adjust around these timing considerations.
Patent Term Extension Strategy
Beyond method-of-use claims, the most important supplementary mechanism for extending effective exclusivity is the patent term extension (PTE) available under 35 U.S.C. § 156. PTE compensates patent holders for regulatory delays that consume patent term during clinical development and FDA review. The maximum extension is five years, with an overall cap of 14 years of post-approval exclusivity.
PTE strategy interacts with method-of-use claims in important ways. A PTE applied to a method-of-use patent extends only the specific patent and the specific indication it covers. A PTE applied to a composition patent extends the broader claim scope. Choosing which patent to apply PTE to (only one PTE per regulatory approval) is therefore a strategic decision that affects the post-LOE landscape.
For drugs where the dominant commercial value comes from a specific patented indication, applying PTE to the relevant method-of-use patent can produce better economic outcomes than applying it to the broader composition patent. The composition patent may not be the constraint that determines effective exclusivity if the method-of-use patent runs longer with the extension. Analysts modeling post-LOE revenue trajectories should examine PTE decisions as part of the broader patent landscape analysis.
The European SPC Analog
The European Supplementary Protection Certificate (SPC) regime provides analogous patent term extension in EU markets. SPCs extend patent term by up to five years (with a 15-year-from-MA cap), and the European Court of Justice has developed an extensive jurisprudence on the interaction between SPCs and method-of-use claims. The basic principle is that an SPC derived from a method-of-use patent protects the same product as authorized in the marketing authorization, not just the specific method of use claimed.
This interpretation, established in cases including Medeva BV v. Comptroller General of Patents (C-322/10) and subsequent decisions, creates a broader scope of protection for method-of-use-derived SPCs than the underlying patent claims might suggest. For global pharmaceutical companies, this means that EU method-of-use SPC strategy can produce different effective protection scope than US method-of-use PTE strategy on the same drug.
The Role of Pediatric Exclusivity
The Best Pharmaceuticals for Children Act (BPCA) provides an additional six months of market exclusivity to manufacturers who conduct pediatric studies in response to a Written Request from FDA. The six-month period is not standalone; it attaches to and extends every existing patent and regulatory exclusivity for the drug. For a blockbuster, this single extension can be worth hundreds of millions of dollars.
The pediatric exclusivity mechanism interacts with method-of-use patents in a particularly favorable way for brand manufacturers. The six-month extension applies to every Orange Book-listed patent, including method-of-use patents covering specific indications. A method-of-use patent that would otherwise expire in December 2027 is extended to June 2028. This stacking effect can shift the LOE date for the dominant commercial indication by enough to materially affect revenue projections.
Pediatric exclusivity strategy requires planning. The Written Request from FDA must be obtained, the pediatric studies must be conducted (often in disease areas where the drug is not commonly used in children), and the data must be submitted within the required timelines. The mechanics are well-established but operationally demanding. For drugs where the underlying patent portfolio supports continued post-LOE revenue generation, the pediatric exclusivity extension provides additional time to capture that revenue.
The Orphan Drug Overlay
The Orphan Drug Act provides seven years of market exclusivity for drugs approved for rare diseases (defined as conditions affecting fewer than 200,000 patients in the US). Orphan exclusivity is non-patent regulatory exclusivity, but it interacts with method-of-use patent strategy in important ways. A drug initially approved for an orphan indication can build a method-of-use patent portfolio around that indication, then expand to non-orphan indications with additional method-of-use protection layered on top.
The combination of orphan exclusivity and method-of-use patents has produced some of the most durable revenue franchises in pharmaceuticals. Drugs that started in narrow rare disease populations and expanded to broader uses often retain meaningful exclusivity well beyond the original orphan exclusivity period, particularly when the expanded indications are covered by their own method-of-use claims.
The Disease-Modifying Treatment Pattern
A particular pattern that has emerged in recent years involves disease-modifying treatments initially approved for narrow patient populations, then expanded to earlier-stage disease through additional clinical trials. The earlier-stage indications often represent the larger commercial opportunity, and method-of-use patents tied to specific disease stages or biomarker profiles can extend effective exclusivity for the commercially dominant population well past the primary patent expiration.
This pattern has been particularly important in oncology, immunology, and rare metabolic diseases. The strategic calculation involves balancing the development cost of new indication studies against the patent term value that successful approval creates. For drugs where the underlying composition patent has limited remaining life, indication expansion paired with method-of-use patent filings can substantially extend the economic franchise.
Reading the Orange Book
For analysts conducting their own method-of-use patent analyses, the Orange Book is the primary source document. The FDA publishes the Orange Book electronically as the ‘Approved Drug Products with Therapeutic Equivalence Evaluations,’ updated daily with patent listings, regulatory exclusivities, and ANDA approvals. Reading the Orange Book effectively requires understanding several technical conventions.
Each listed drug product has its own Orange Book record, identified by the NDA holder, the application number, and the approved indications. Listed patents are identified by patent number, expiration date, drug substance flag, drug product flag, method-of-use flag, and use code (for method-of-use patents). The use code provides a brief narrative description of the patented method, and analysts comparing the use code to the patent’s actual claims can sometimes identify scope discrepancies that affect carve-out strategy.
Pediatric exclusivity flags indicate whether the six-month BPCA extension applies to the listed patent. Regulatory exclusivity records (NCE, new chemical entity; ODE, orphan drug exclusivity; NPP, new patient population; and others) appear separately and have their own expiration dates. The interaction between patent expirations and regulatory exclusivities determines the practical generic entry date, and the combination is often non-obvious without careful reading.
The Patent Information Form
NDA holders submit Form FDA 3542 (or 3542a for newly approved drugs) to list patents in the Orange Book. The form requires the patent number, expiration date, identification of the patent as drug substance, drug product, or method of use, and the use code for method-of-use patents. FDA does not verify the substantive accuracy of these submissions. The listing is essentially self-certified by the NDA holder, subject to penalties for false statements.
The self-certification structure has produced occasional disputes over whether specific patents are properly listable. Device patents on drug-device combination products have been a particular source of FTC enforcement attention. The agency’s 2023 action against several inhaler and EpiPen device patent listings prompted delistings and has produced ongoing scrutiny of listing accuracy. Whether this scrutiny extends to method-of-use patents remains to be seen, but the regulatory environment has shifted toward greater oversight of listing practices.
The Investor’s Decision Framework
For institutional investors building positions in branded pharmaceutical equities or biosimilar manufacturers, the method-of-use analysis informs several specific decisions. Position sizing on companies facing LOE events should account for the method-of-use patent strength of the affected drugs, not just the composition patent expiration timing. Companies with stronger secondary patent positions deserve higher post-LOE revenue assumptions in valuation models.
Hedging strategies should distinguish drugs where the post-LOE revenue retention is structurally supported by method-of-use patents from drugs where the retention depends primarily on biosimilar pathway delays. The hedging instruments and the timing of their deployment may differ between these scenarios.
Generic and biosimilar manufacturer equity positions should be evaluated against the specific drugs in the manufacturer’s pipeline and the method-of-use patent landscapes on those drugs. A generic manufacturer focused on drugs with weak secondary patents will produce different revenue trajectories than one focused on drugs with complex method-of-use thickets.
Long-short pair trades in the pharmaceutical space increasingly require this granular analysis. The simple long-brand, short-generic trade ahead of LOE events misses much of what now drives relative performance. Brand companies with strong method-of-use portfolios may outperform on LOE timing, while generic manufacturers facing inducement risk may underperform their volume-based projections.
What Comes Next
The next 18 months will produce several developments that should reshape how the industry thinks about method-of-use patents and skinny labels. The Supreme Court’s decision in Hikma v. Amarin is the most significant, with implications that extend well beyond the Vascepa franchise. Several other cases currently in district court litigation will produce additional Federal Circuit decisions that clarify the contours of the doctrine.
The first IRA-negotiated prices took effect on January 1, 2026, with the second cohort coming in 2027. The interaction between negotiated pricing and method-of-use patent-driven exclusivity extension will become clearer as data on actual revenue trajectories accumulates. Some of the early IRA-affected drugs (Eliquis, Xarelto, Januvia, Jardiance, Farxiga) have meaningful method-of-use patent positions that will test whether negotiation and patent strategy work together or against each other.
Biosimilar launches against the major biologic patents (Keytruda in 2028, Opdivo in 2028, Stelara already in 2025) will provide real-world tests of how method-of-use patent strategies translate from small molecule precedents to biologic franchises. The biosimilar manufacturers preparing these launches are sophisticated and well-funded, and their launch strategies will be informed by the same doctrinal developments that have reshaped generic small molecule launches.
Key Takeaways
The composition-of-matter patent is no longer the most strategically important patent in a branded pharmaceutical portfolio. Method-of-use claims, properly filed and properly listed in the Orange Book, extend effective market exclusivity by 6 to 8 years on average and shape the post-LOE revenue trajectory more than any other intellectual property layer.
The Section viii skinny label pathway, designed by Congress to enable early generic entry on unpatented indications, has been narrowed by the Federal Circuit’s GSK v. Teva and Amarin v. Hikma decisions. Generic manufacturers using skinny labels now face induced infringement exposure based not just on label content but on press releases, sales force communications, and product positioning.
The Supreme Court’s grant of certiorari in Hikma v. Amarin, with oral argument set for April 27, 2026, will likely produce a decision before the Court’s summer recess that either narrows or affirms the Federal Circuit doctrine. Either outcome materially affects post-LOE revenue projections for drugs with method-of-use patent portfolios.
For analysts modeling the 2026 to 2030 patent cliff, traditional erosion curves based on primary patent expiration dates systematically underestimate revenue retention for drugs with strong method-of-use protection. Indication-level analysis, not aggregate revenue analysis, produces better forecasts.
Generic manufacturers operating in this environment must align their launch communications, sales force training, and marketing materials with the post-GSK and post-Amarin doctrinal landscape. The economic cost of communications discipline is real but smaller than the expected cost of induced infringement litigation.
Brand companies with sophisticated lifecycle management programs are using method-of-use patents in combination with regulatory exclusivities, REMS programs, formulation patents, and authorized generic settlements to convert what historical models predicted as vertical cliffs into slopes that preserve material revenue beyond primary patent expiration.
The IRA’s drug price negotiation provisions add a parallel mechanism of revenue compression that interacts with method-of-use patent strategy in ways that are still being worked out. Drugs with strong method-of-use portfolios may face IRA negotiation while still operating under quasi-monopoly conditions, producing a different net economic outcome than either mechanism alone would predict.
Platforms that aggregate Orange Book data, Paragraph IV filings, and litigation history, including DrugPatentWatch, have made indication-level analysis accessible to a broader population of analysts and investors than was the case a decade ago. The data is available. The interpretive frameworks are catching up.
FAQ
Q1: How long do method-of-use patents typically extend effective exclusivity beyond the primary composition patent?
Empirical research on US pharmaceutical patents found that independent method-of-use patents add an average of 7.4 years of nominal patent life beyond the primary composition claim, with a 95% confidence interval of 6.4 to 8.4 years [5]. Independent formulation patents add 6.5 years on average, and independent polymorph, salt, ester, and prodrug claims add another 6.3 years. The actual market impact depends on whether the method-of-use patent covers the dominant prescribing indication and whether the generic challenger pursues a Section viii carve-out, validity challenge, or settlement.
Q2: What is a skinny label, and why has it become legally risky?
A skinny label is a generic drug label that omits one or more indications covered by a still-valid method-of-use patent, approved under Section viii of the Hatch-Waxman Act. Historically, skinny labels were a routine mechanism for early generic entry on unpatented indications. The Federal Circuit’s decisions in GSK v. Teva (2020/2021) and Amarin v. Hikma (2024) found that generic manufacturers can be liable for induced infringement based on press releases, marketing materials, and product communications even when the label itself complies with the Section viii carve-out. The Supreme Court is reviewing the doctrine in Hikma v. Amarin, with oral argument scheduled for April 27, 2026.
Q3: How should investors adjust LOE revenue models for drugs with strong method-of-use patent portfolios?
Traditional models that apply small molecule erosion curves (80% to 90% revenue loss in year one) to drugs with strong method-of-use protection systematically underestimate revenue retention. A more accurate approach analyzes revenue at the indication level, identifies which indications are covered by unexpired method-of-use patents, and models separate erosion trajectories for patented and unpatented indications. The aggregate revenue trajectory then reflects the weighted average of these indication-specific curves. For biologics, additional adjustments for biosimilar pathway timing, interchangeability designation, and payer formulary behavior are also necessary.
Q4: What role does DrugPatentWatch play in this analytical workflow?
DrugPatentWatch and similar platforms aggregate Orange Book patent listings, Paragraph IV certifications, ANDA filings, and patent expiration data into a workflow accessible to analysts who do not maintain in-house IP litigation tracking. For investors and business development teams modeling LOE scenarios across multiple drugs, the platform provides the data layer that supports indication-level analysis. The platforms do not substitute for substantive legal analysis of patent validity or infringement risk, but they make the underlying data accessible in a workable form.
Q5: How does the Inflation Reduction Act’s drug price negotiation interact with method-of-use patent strategy?
The IRA’s negotiation provisions apply to drugs 9 years post-approval for small molecules and 13 years post-approval for biologics, with negotiated prices taking effect 2 years after selection. Drugs with strong method-of-use patent portfolios may extend effective exclusivity well beyond these thresholds, which means they face IRA negotiation while still operating under reduced competitive pressure. The net revenue effect depends on the magnitude of negotiated price cuts versus the magnitude of preserved volume share. Some manufacturers have begun adjusting lifecycle strategies to account for this interaction, including accelerated successor product launches that shift revenue to new product cycles less affected by negotiation timing.
References
[1] Alcimed. (2025, October 27). Patent cliff: What strategies can help biopharma stay competitive? https://www.alcimed.com/en/insights/patent-cliff/
[2] Duane Morris LLP. (2026, January). U.S. Supreme Court grants certiorari in Hikma v. Amarin, placing ‘skinny label’ inducement in focus. https://www.duanemorris.com/alerts/us_supreme_court_grants_certiorari_hikma_amarin_placing_skinny_label_inducement_focus_0126.html
[3] DrugPatentWatch. (2026, February 7). Kill the patent cliff: How to turn a $400 billion revenue loss into a competitive edge. https://www.drugpatentwatch.com/blog/kill-the-patent-cliff-how-to-turn-a-400-billion-revenue-loss-into-a-competitive-edge/
[4] BioPharma Dive. (2023, February 21). Big pharma’s looming threat: A patent cliff of ‘tectonic magnitude.’ https://www.biopharmadive.com/news/pharma-patent-cliff-biologic-drugs-humira-keytruda/642660/
[5] Kapczynski, A., Park, C., & Sampat, B. (2012). Polymorphs and prodrugs and salts (oh my!): An empirical analysis of ‘secondary’ pharmaceutical patents. PLoS ONE, 7(12), e49470. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3515607/
[6] Gibson, J. (2024). Generic drugs and the struggle to compete: The role of skinny labels. PMC. https://pmc.ncbi.nlm.nih.gov/articles/PMC11963906/
[7] GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc., 7 F.4th 1320 (Fed. Cir. 2021).
[8] Amarin Pharma, Inc. v. Hikma Pharmaceuticals USA Inc., 104 F.4th 1370 (Fed. Cir. 2024). https://www.cafc.uscourts.gov/opinions-orders/23-1169.OPINION.6-25-2024_2339226.pdf
[9] Crowell & Moring LLP. (2025, April 10). Hikma and amici curiae ask Supreme Court to revisit induced infringement by generic ‘skinny labels.’ https://www.crowell.com/en/insights/client-alerts/hikma-and-amici-curiae-ask-supreme-court-to-revisit-induced-infringement-by-generic-skinny-labels
[10] DrugPatentWatch. (2025, July 20). The power of patience: Delaying patents to enhance pharma market exclusivity. https://www.drugpatentwatch.com/blog/the-power-of-patience-delaying-patents-to-enhance-pharma-market-exclusivity/
[11] Labiotech. (2026, March 23). The next pharma patent cliff: How 2026-2032 will reshape revenue. https://www.labiotech.eu/best-biotech/pharma-patent-cliff/
[12] IPWatchdog. (2024, July 25). Call off Chicken Little: The sky is not falling for skinny labeling after GSK v. Teva. https://ipwatchdog.com/2024/07/25/call-off-chicken-little-sky-not-falling-skinny-labeling-gsk-v-teva/
[13] DLA Piper. (2024, September 19). Amarin v. Hikma: Defining the limits of protection that skinny labels afford. https://www.dlapiper.com/en-us/insights/publications/synthesis/2024/amarin-v-hikma-defining-the-limits-of-protection-that-skinny-labels-afford
[14] IPWatchdog. (2026, April 29). Justices voice concern that upholding CAFC’s Hikma ‘skinny label’ ruling will harm generics industry. https://ipwatchdog.com/2026/04/29/justices-voice-concern-upholding-cafcs-hikma-ruling-will-harm-generics-industry/
[15] DrugPatentWatch. (2025, November 10). The patent playbook: 7 key strategies pharma uses to extend market exclusivity. https://www.drugpatentwatch.com/blog/the-patent-playbook-7-key-strategies-pharma-uses-to-extend-market-exclusivity/
[16] DeepCeutix. (2026, February 2). $300 billion in pharma revenue loses patent protection by 2030. https://deepceutix.com/insights/patent-cliff-reformulation
[17] PharmaVoice. (2026, January 30). How big pharma is navigating a $300 billion patent cliff. https://www.pharmavoice.com/news/big-pharma-navigating-patent-cliff-300-billion-jnj-merck-abbvie/810915/
[18] DrugPatentWatch. (2026, January 18). Maximizing pharmaceutical patent longevity: A mechanistic and strategic guide to IP term extension and lifecycle fortification. https://www.drugpatentwatch.com/blog/how-long-do-drug-patents-last/