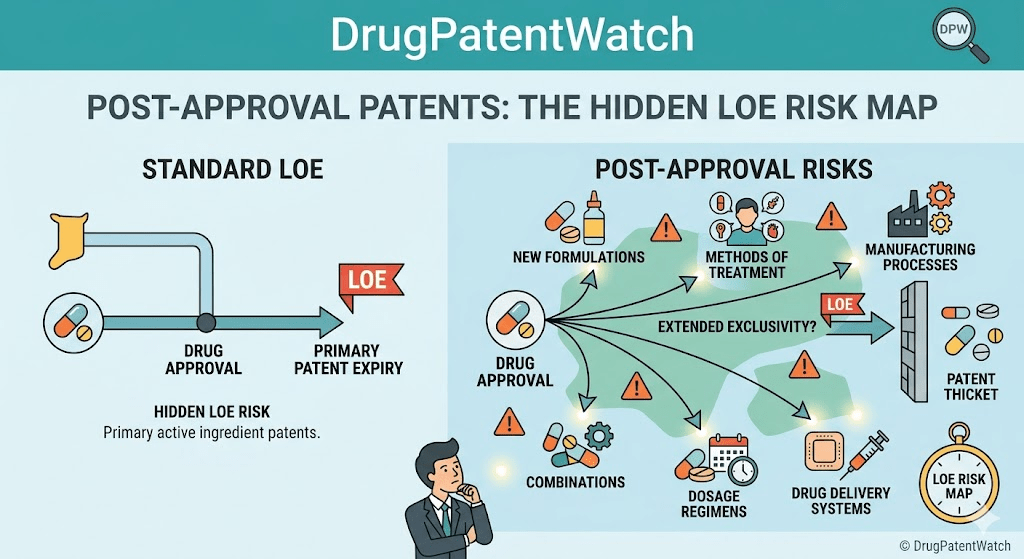
Most pharmaceutical IP work gets framed as a pre-approval problem. File the composition patent during discovery, secure the use claims around Phase II readouts, line up the method patents before NDA submission, and hand the asset to commercial teams to sell. That framing is wrong, or at least incomplete enough to cost real money. The patent estate that determines loss-of-exclusivity (LOE) timing, generic entry risk, biosimilar carve-out exposure, and product-hopping liability is built largely after approval, through supplemental filings, manufacturing changes, label updates, and reformulation decisions that most regulatory affairs teams treat as compliance work rather than competitive intelligence.
This article walks through how post-approval changes interact with patent strategy, what the regulatory pathways look like in practice, and where commercial teams, in-house counsel, and investors should be looking for risk and opportunity. It draws on Hatch-Waxman litigation, BPCIA biosimilar cases, FDA guidance documents, and the public patent record. Where useful, it points to tools such as DrugPatentWatch, IQVIA Pipeline Intelligence, and the FDA’s Orange Book that practitioners actually use for monitoring.
Why Post-Approval Changes Are a Patent Strategy, Not Paperwork
The regulatory file for a drug does not freeze at approval. Manufacturers submit hundreds of supplemental applications across the life of a major product, covering everything from a switch in excipient supplier to a new pediatric indication. Each of these supplements is a potential vehicle for adding patent protection, extending exclusivity, or, when handled poorly, forfeiting it.
Under 21 CFR 314.70, changes to an approved NDA fall into four tiers based on the level of FDA review required. The tiering looks bureaucratic on paper. In practice, it sets the cadence at which a sponsor can refresh its Orange Book listings, layer in new method-of-use claims, or trigger 30-month litigation stays under Hatch-Waxman [1].
What counts as a post-approval change under 21 CFR 314.70
FDA divides post-approval changes into prior approval supplements (PAS), changes being effected in 30 days (CBE-30), changes being effected immediately (CBE-0), and annual reportable changes. A PAS requires FDA sign-off before implementation. A CBE-30 can be implemented 30 days after FDA receipt unless the agency objects. A CBE-0 allows immediate distribution. An annual reportable change is documented retrospectively. The hierarchy maps roughly to risk: process-defining changes that affect identity, strength, quality, purity, or potency go through PAS; minor scale changes within validated ranges go in the annual report [1].
Annual reportable changes that still affect IP
Sponsors sometimes assume that annual reportable changes have no IP implications because they do not require pre-approval. That is not always correct. A change in a stabilizer concentration or a tweak to a coating dispersion can fall within the claim scope of a newly issued formulation patent. If the patent issues during the post-approval period and reads on the marketed product, it must be listed in the Orange Book within 30 days of issuance under 21 USC 355(c)(2). Missing that window does not invalidate the patent, but it does affect the sponsor’s ability to invoke a 30-month stay against a later Paragraph IV challenger [2].
How CBE-30, CBE-0, and PAS differ
The practical difference between CBE-30 and PAS lies in commercial risk tolerance. A site transfer for a sole-source API often goes through PAS to avoid the regulatory uncertainty of a 30-day review that could be interrupted by an FDA hold. Container closure changes for a stable small-molecule oral solid often go through CBE-30. The patent intelligence angle is that PAS submissions correlate with new patent filings about twice as often as CBE-30 submissions in the data set tracked by DrugPatentWatch and similar services, because PAS is where sponsors are more likely to introduce process or formulation innovations worth patenting.
How Patent Intelligence Reduces Regulatory Risk During the Product Lifecycle
Patent intelligence in the post-approval phase has two jobs. First, it tracks the company’s own filings to ensure timely Orange Book listing and proper coordination with regulatory supplements. Second, it tracks competitors and generic challengers to surface early warning signals before Paragraph IV notices arrive.
Both jobs require linking three data streams that typically live in different systems: USPTO patent assignment and prosecution records, FDA supplement filings indexed in Drugs@FDA, and litigation dockets from PACER. Vendors including DrugPatentWatch, IQVIA, Clarivate Cortellis, and Patexia aggregate these feeds, with varying levels of structure around drug-patent linkage.
The Orange Book mechanics every commercial team should know
The Approved Drug Products with Therapeutic Equivalence Evaluations, known universally as the Orange Book, lists patents that claim the approved drug, its method of use, or the approved drug product. Process patents are not eligible for listing [3]. The listing process is sponsor-driven: FDA does not review the substantive merit of a listed patent, only its formal eligibility. That structure has produced both legitimate listings and aggressive ones, the latter drawing FTC enforcement attention.
When a new patent must be listed within 30 days of issuance
Under 21 USC 355(c)(2) and the implementing rule at 21 CFR 314.53, a sponsor with an approved NDA must submit patent information within 30 days of issuance of any patent that claims the approved drug or method of use. The clock runs from the date of issuance, not from the date the sponsor decides the patent matters. Late listing carries a specific consequence: a Paragraph IV ANDA filer who served notice before the late listing does not trigger a 30-month stay tied to that patent [2].
21 USC 355(b)(1) listing duties after approval
The original NDA listing duty under 355(b)(1) covers patents in existence at the time of NDA submission. The post-approval listing duty under 355(c)(2) is the operative provision for patents issued later. Confusion between these two provisions has produced real litigation losses, including cases where sponsors argued, unsuccessfully, that a method-of-use patent issued years after approval was not subject to the 30-day deadline because no corresponding sNDA was pending.
What Happens If a Patent Lists Late in the Orange Book
Short answer: A late-listed patent is still enforceable, but the sponsor loses the automatic 30-month stay against ANDA filers who served Paragraph IV notice before the listing. The patent can still be litigated, and the patentee can still seek a preliminary injunction, but the structural advantage Hatch-Waxman provides to timely listers is forfeited.
Trigger conditions for 30-month stays
The 30-month stay under 21 USC 355(j)(5)(B)(iii) requires four conditions: an NDA-listed patent, a Paragraph IV certification by the ANDA filer, notice to the NDA holder, and a patent infringement suit filed within 45 days of notice. Miss any condition and the stay does not attach. The stay blocks final FDA approval of the ANDA, not its acceptance or substantive review, which is a common point of confusion in commercial forecasting [4].
How late listings forfeit stay rights
The forfeiture rule is technical. If patent P is listed after ANDA A has been filed with a Paragraph IV certification to other already-listed patents, ANDA A is not required to certify to patent P. The sponsor’s late listing therefore gives it no leverage against ANDA A. This rule prevents NDA holders from layering in patents over time to repeatedly reset the 30-month clock against a single generic challenger.
sNDAs and Patent Listing: The Compliance Tripwire
Supplemental NDAs (sNDAs) are the most common vehicle for adding patents to the Orange Book post-approval. A sponsor seeking approval of a new indication will often file the use patent on the new indication contemporaneously with the sNDA. The interaction between the sNDA approval date, the patent issuance date, and the Orange Book listing date is where many sponsors lose stay rights.
When a labeling change creates a new method-of-use patent
A labeling change that adds a new indication, dosing regimen, or patient population can be the predicate for a new use patent. The patent must claim the use described in the approved label and must be filed in a way that aligns with the sNDA approval. The U.S. Supreme Court’s decision in Caraco v. Novo Nordisk (2012) addressed the related issue of how use codes in the Orange Book describe protected uses, and made clear that generic firms have a counterclaim right under 21 USC 355(j)(5)(C)(ii)(I) to correct overbroad use code listings [5].
Skinny labels and the GSK v. Teva carvedilol decision
The skinny label, formally known as a section viii statement under 21 USC 355(j)(2)(A)(viii), allows a generic filer to carve out a patented indication from its label and market the generic for unpatented indications only. The Federal Circuit’s decision in GlaxoSmithKline v. Teva Pharmaceuticals USA, involving carvedilol, found Teva liable for induced infringement despite a partial label carve-out, on the theory that Teva’s marketing materials and press releases encouraged off-label use of the carved-out indication [6]. The case has unsettled generic manufacturers’ confidence in the section viii pathway and changed how launch communications are drafted across the industry.
Reformulation as IP Strategy: The Suboxone, Namenda, and Doryx Cases
Reformulation is the most visible post-approval patent move. Switching from an immediate-release tablet to an extended-release capsule, or from a tablet to a sublingual film, can support a new patent estate, a new exclusivity period, and a marketing transition that erodes the generic substitution base. It can also draw antitrust enforcement.
Product hopping and antitrust risk
Three reformulation cases set the doctrinal landscape. Reckitt Benckiser’s transition of Suboxone from tablets to sublingual film drew DOJ and FTC scrutiny. Forest Laboratories’ planned withdrawal of Namenda IR in favor of Namenda XR was enjoined by the Second Circuit in New York v. Actavis. And Mylan’s antitrust suit against Warner Chilcott over Doryx (doxycycline) reformulations produced a Third Circuit decision in favor of the defendant, illustrating that not every reformulation is anticompetitive [7].
New York v. Actavis (Namenda) Second Circuit ruling
The Second Circuit affirmed a preliminary injunction in 2015 preventing Forest from withdrawing Namenda IR while Namenda XR took market share, on the theory that a hard switch coerced patients into the patented formulation before generic memantine could enter [8]. The decision distinguished hard switches, where the old product is removed, from soft switches, where both products remain available and physicians and patients choose. Soft switches generally survive antitrust scrutiny. Hard switches face heightened risk, especially in therapeutic categories where state substitution laws drive generic uptake.
Manufacturing Change Risk: When Supply Chain Moves Affect Exclusivity
Manufacturing changes rarely make IP headlines but they shape exclusivity in two ways. First, a site transfer or process change can be the predicate for a new process patent, which is not Orange Book listable but can support trade secret protection and ITC enforcement against imports. Second, a change in API source can shift which Drug Master File (DMF) supports the product, which affects what an ANDA filer can rely on for bioequivalence demonstration.
Site transfers, DMFs, and bioequivalence proofs
An ANDA filer must demonstrate that its product is bioequivalent to the reference listed drug (RLD) as currently marketed. If the innovator has changed its manufacturing site, formulation, or release specification post-approval, the ANDA filer must match the current state of the RLD. This can create timing leverage for the innovator: a CMC change implemented shortly before generic entry can complicate ANDA filings that referenced an earlier version of the product.
Why API source changes matter to ANDA filers
Many APIs have only two or three commercial-scale manufacturers globally. When an innovator shifts API sourcing through a Type II DMF amendment, it can either narrow or broaden the pool of available API for generic filers. Sponsors that hold equity stakes or supply agreements with the dominant API producer can use those arrangements to delay generic supply, though such arrangements have themselves drawn FTC attention in cases involving paroxetine and other small molecules.
Patent Term Extension vs Patent Term Adjustment: The Five-Year Question
Two patent-extension mechanisms operate in parallel and often get confused. Patent Term Extension (PTE) under 35 USC 156 compensates for time lost during FDA regulatory review and is available for one patent per approved drug. Patent Term Adjustment (PTA) under 35 USC 154(b) compensates for delays during USPTO prosecution and is calculated automatically at patent issuance. PTE can extend a patent by up to five years; PTA has no statutory cap [9].
35 USC 156 mechanics
PTE eligibility requires that the patent claims the approved drug, a method of use, or a method of manufacture; that the regulatory review period included at least some delay caused by FDA; and that the patentee elects PTE within 60 days of approval. The PTE term equals half the testing phase plus the full review phase, capped at five years and limited so that the total post-approval patent life cannot exceed 14 years.
When PTE choice locks in LOE
The PTE election is irrevocable and applies to a single patent. Sponsors with multiple eligible patents must choose, and the choice has long-tail consequences. A composition patent extended via PTE typically provides broader exclusivity than a method-of-use patent, because the latter is vulnerable to skinny-label carve-outs. Sponsors that elected PTE on use patents thinking they would also rely on listed formulation patents have sometimes been surprised when the formulation patents were found invalid or were carved around by generic filers.
Paragraph IV Litigation: Timing, Tactics, and Settlement Patterns
Paragraph IV certification is the procedural trigger for most branded-generic patent litigation in the United States. When an ANDA filer certifies that an Orange Book-listed patent is invalid, unenforceable, or will not be infringed by the generic product, it must give notice to the patent holder. The patent holder then has 45 days to sue.
“In 2022, brand-name drugs lost an average of 75% of their unit sales within 12 months of generic entry, with first-to-file Paragraph IV winners capturing the largest share during the 180-day exclusivity period.” — IQVIA Institute for Human Data Science, U.S. generic drug spending and use, 2023 [10]
The 45-day window after Paragraph IV notice
The 45-day window is jurisdictional in a practical sense. A timely-filed suit triggers the automatic 30-month stay. A late-filed suit can still proceed but does not trigger the stay, which can mean the ANDA receives final approval and the generic launches at-risk while the case proceeds. The branded company facing an at-risk launch faces a difficult choice: seek a preliminary injunction (high standard, including likelihood of success on the merits and irreparable harm) or accept the launch and pursue damages.
Reverse payment settlements after FTC v. Actavis
The Supreme Court’s 2013 decision in FTC v. Actavis held that reverse payment patent settlements, in which a branded firm pays the generic challenger to delay entry, can violate antitrust law and are evaluated under a rule-of-reason analysis [11]. The decision did not categorically prohibit such settlements but raised litigation risk substantially. Industry settlement structures shifted toward early-entry dates with no cash payment, side deals on co-promotion or supply agreements that the parties characterize as having independent value, and acceleration triggers tied to other generic entries.
How DrugPatentWatch tracks Paragraph IV filings
Paragraph IV certifications are publicly disclosed by FDA on a delayed basis, and ANDA filer identities are often inferred from litigation filings rather than published directly. DrugPatentWatch and similar services build the timeline by combining FDA’s monthly Paragraph IV list, PACER docket entries, and SEC filings from generic firms that disclose ANDA submissions in 10-K and 10-Q reports. For commercial forecasting, the timeline matters more than the identity of the filer: a first-filer’s 180-day exclusivity, the 30-month stay expiration, and the projected trial date are the inputs to LOE risk models.
Biosimilars and the BPCIA Patent Dance
The Biologics Price Competition and Innovation Act (BPCIA) of 2010 created an abbreviated approval pathway for biosimilars and a parallel patent litigation framework that differs from Hatch-Waxman in several ways. Biologics are listed in the Purple Book rather than the Orange Book. The Purple Book records reference product exclusivity (12 years from first approval) but does not list patents.
Amgen v. Sandoz and the “patent dance” procedure
The Supreme Court’s 2017 decision in Amgen v. Sandoz held that the BPCIA’s information exchange procedure, often called the patent dance, is not enforceable by federal injunction [12]. A biosimilar applicant can opt out of the dance, accepting the consequence that the reference product sponsor can sue immediately on any patent rather than going through the staged disclosure process. The decision changed the strategic calculus: biosimilar applicants now routinely consider opt-out as a way to compress litigation timelines and force the sponsor to disclose its patent estate earlier.
Humira’s 130+ patent estate as a case study
AbbVie’s Humira (adalimumab) is the standard reference for how a biologic sponsor can build a post-approval patent estate. Over the course of more than two decades of marketing, AbbVie obtained and asserted more than 130 patents covering formulation, dosing regimens, manufacturing processes, and methods of treatment across multiple indications. Biosimilar developers including Amgen, Boehringer Ingelheim, Sandoz, and Pfizer reached settlements that staggered U.S. entry from January 2023 onward [13]. The Humira pattern illustrates how successive post-approval filings on dosing, formulation, and manufacturing can extend effective exclusivity well beyond the composition patent expiration, even without a single litigation win on the merits.
Forecast: LOE Risk Maps Through 2030
The next several years feature a concentrated set of LOEs across cardiology, oncology, immunology, and metabolic disease. Commercial forecasts depend on resolving three questions for each asset: when do the listed composition and use patents expire, what is the status of pending Paragraph IV or BPCIA litigation, and what post-approval changes might shift the LOE date.
Revlimid, Eliquis, Xarelto LOE timelines
Product
Sponsor
Class
U.S. LOE landscape
Revlimid (lenalidomide)
Bristol-Myers Squibb (Celgene)
Immunomodulator
Volume-limited generic entries under settlement from March 2022; uncapped generic entry from January 2026 [14]
Eliquis (apixaban)
BMS / Pfizer
DOAC anticoagulant
Composition patent challenges resolved in favor of BMS; generic entry expected around 2028 absent settlement
Xarelto (rivaroxaban)
Janssen / Bayer
DOAC anticoagulant
Composition expiry in 2024; method patents and pediatric exclusivity extend select formulations
Stelara (ustekinumab)
Janssen
IL-12/23 biologic
Biosimilar launches staggered from 2025 onward under settlements with Amgen, Alvotech, others
Entresto (sacubitril/valsartan)
Novartis
HF combination
Composition patent expiry mid-2025; combination claims and pediatric data extend select markets
How patent expiration cascades hit pricing
The pricing effect of LOE varies by therapeutic class and channel mix. Small molecules with high generic substitution rates lose 70 to 90 percent of branded unit volume within two years. Biologics with biosimilar competition see slower erosion, with biosimilar share typically reaching 30 to 50 percent within three years of first biosimilar launch in the U.S., per IQVIA data. Products subject to Medicare price negotiation under the Inflation Reduction Act face a separate price reduction mechanism that operates independently of patent status, which changes how sponsors model net pricing for negotiated products.
How to Build a Post-Approval Patent Monitoring Program
A working monitoring program for post-approval patent activity has four components: a data layer that links FDA supplements to USPTO filings, a litigation feed that surfaces Paragraph IV and BPCIA actions, a competitive intelligence layer that tracks generic and biosimilar pipelines, and a governance process that connects legal, regulatory, and commercial decision-making.
Inputs from PAIR, FDA, and litigation dockets
USPTO’s Public PAIR system provides patent prosecution history. FDA’s Drugs@FDA and the Orange Book provide approval and listing data. PACER provides docket information for federal litigation. Vendors including DrugPatentWatch, Patexia, Clarivate Cortellis, and Lex Machina aggregate and normalize these feeds with varying levels of pharma-specific structure. The choice of vendor turns on the analyst’s needs: portfolio-level LOE forecasting favors DrugPatentWatch’s drug-patent linkage; litigation analytics favors Lex Machina’s docket coverage; biosimilar tracking favors Cortellis or Evaluate Pharma.
Internal coordination across legal, regulatory, and commercial
The most common organizational failure is treating patent listing as a legal task disconnected from the regulatory supplement process. The fix is procedural: every PAS, sNDA, or CBE-30 submission should trigger a checklist review by IP counsel for new patent filing opportunities and Orange Book listing obligations. Commercial forecasting should consume the patent calendar as a primary input rather than receiving it as an annual update. Reviewers who treat the patent file as a static deliverable miss the dynamic interactions that determine LOE.
Comparison Table: Hatch-Waxman vs BPCIA Patent Litigation
Feature
Hatch-Waxman (small molecules)
BPCIA (biologics)
Patent listing
Orange Book; sponsor lists composition, formulation, use
Purple Book lists exclusivity but not patents
Challenger filing
ANDA with Paragraph IV certification
Biosimilar application (351(k)) with optional patent dance
Notice trigger
Paragraph IV notice to NDA holder
Notice of commercial marketing 180 days pre-launch
Stay mechanism
30-month automatic stay on FDA approval
No automatic stay; injunction must be sought
First-filer reward
180 days of generic exclusivity
Interchangeable exclusivity, narrower scope
Approval pathway
21 USC 355(j)
42 USC 262(k)
Why Citizen Petitions Sit Inside Post-Approval Patent Strategy
Citizen petitions under 21 CFR 10.30 allow any party to request FDA action, including delay of generic approval based on technical or scientific concerns. Branded sponsors have used citizen petitions to raise bioequivalence, manufacturing, or labeling issues shortly before generic approval. Congress responded with section 505(q) of the Food, Drug, and Cosmetic Act, requiring FDA to take final action on petitions affecting ANDA approvals within 150 days and to refer petitions that appear to be primarily for delay to the FTC [15].
Section 505(q) constraints on petition timing
FDA has applied 505(q) to reject citizen petitions that raise concerns the sponsor could have raised earlier in the approval cycle. The implication for post-approval patent strategy is that delay-motivated petitions filed close to projected generic launch face a much lower probability of changing FDA’s review timeline than they did before the 2007 amendments. Petitions that raise legitimate scientific concerns earlier in the cycle remain viable but require coordination with the regulatory affairs function rather than being a freestanding legal lever.
What This Means for Generic and Biosimilar Strategy Teams
From the challenger side, the post-approval patent landscape creates both opportunities and traps. Late-listed patents may not trigger a 30-month stay against an early ANDA filer, which is a structural advantage. Use codes that overreach can be narrowed through a section viii statement plus a counterclaim under Caraco. Reformulation by the brand should be tracked as a signal that hard-switch antitrust theories may be available.
Skinny label playbook after GSK v. Teva
The carvedilol decision did not eliminate the section viii pathway but did change how generic firms manage launch communications. The practical playbook now includes scrubbing press releases and sales materials of references to the carved-out indication, documenting internal training that emphasizes only labeled uses, and avoiding statements about therapeutic equivalence that could be read to cover all indications including the carved-out one. Generic firms with strong launch discipline have continued to use section viii filings; firms with sloppy launch communications have faced renewed induced infringement exposure.
How Patent Intelligence Maps to M&A and Licensing Diligence
Acquirers in pharma deals routinely build LOE models for the target’s portfolio. The models that hold up under post-closing scrutiny treat post-approval patents as a dynamic input. A target product whose composition patent expires in 2027 may have method patents and formulation patents extending exclusivity to 2031, or those patents may be invalid, listed late, or carve-outable. Diligence that stops at the Orange Book misses the dynamics that determine value.
What to ask in diligence
A short diligence checklist for the patent file includes: full list of issued and pending U.S. and ex-U.S. patents linked to each product, Orange Book listing status with issuance and listing dates, history of Paragraph IV certifications and litigation outcomes, current settlements with generic challengers including entry-date triggers, and pending sNDAs or PAS submissions that may support additional patent filings. A diligence team that cannot produce this list in two business days for a target product is signaling that the IP file is not being managed at the cadence the asset requires.
Risk Scenarios: What Happens If Key Variables Move
Scenario 1: Method patent invalidated mid-stay
If a district court finds a method patent invalid during the 30-month stay, the stay terminates as to that patent. If other Orange Book patents remain valid, the ANDA filer still cannot launch on those grounds. The branded sponsor’s position deteriorates if the invalidated patent was its primary defense; it may also affect related patents in the same family through collateral estoppel under B&B Hardware v. Hargis Industries and its progeny.
Scenario 2: First-filer forfeiture under 21 USC 355(j)(5)(D)
The Medicare Modernization Act of 2003 created forfeiture events that can strip a first-filer of 180-day exclusivity, including failure to market within statutory timeframes and withdrawal of the relevant patent certification. Forfeiture transfers nothing to a subsequent filer; it simply opens the market to multiple generic entrants on day one, which compresses pricing rapidly. Sponsors and challengers both model forfeiture risk as part of LOE forecasting.
FDA’s interchangeability designation under 351(k)(4) allows pharmacy-level substitution of a biosimilar without prescriber involvement, subject to state law. Products with interchangeability designations have shown faster uptake than non-interchangeable biosimilars in early data, particularly in retail pharmacy channels. The designation requires switching studies that few biosimilar developers have completed, but the regulatory bar has been clarified in recent FDA guidance and the pace of interchangeable approvals is rising.
Pricing Pressure and the Inflation Reduction Act Overlay
The Inflation Reduction Act of 2022 introduced Medicare price negotiation for selected high-spend drugs, beginning with 10 Part D drugs for 2026 pricing and expanding annually. Negotiated maximum fair prices apply regardless of patent status, which means a product still under composition patent can face mandated price reductions [16]. The interaction with post-approval patent strategy is direct: extending patent life for a negotiated product does not protect price levels in the same way it once did. Sponsor portfolios are being recalibrated to reflect this.
Negotiated drugs in the 2026 cohort
The first negotiation cohort included Eliquis, Xarelto, Januvia, Jardiance, Enbrel, Imbruvica, Farxiga, Entresto, Stelara, Fiasp/NovoLog, with maximum fair prices announced in August 2024 for application in 2026 [16]. The mix of mature and relatively young products in the cohort confirms that patent status is not the controlling variable for inclusion. Selection is based on Medicare spend, time since FDA approval (or licensure), and competitive status.
Key Takeaways
Post-approval changes are a primary lever for building and losing exclusivity. Treating them as routine compliance work understates their strategic importance.
The 30-day Orange Book listing rule under 21 USC 355(c)(2) is the most commonly missed compliance step with the largest downstream cost.
Skinny label strategy under section viii survived GSK v. Teva but now requires disciplined launch communications to avoid induced infringement exposure.
Reformulation can extend effective exclusivity but carries antitrust risk when implemented as a hard switch, as illustrated by New York v. Actavis.
PTE election under 35 USC 156 is one-time and irrevocable; the choice of which patent to extend deserves the same rigor as the original patent prosecution strategy.
BPCIA patent litigation differs materially from Hatch-Waxman in stay mechanics and information exchange; biosimilar applicants now routinely consider opting out of the patent dance.
The Inflation Reduction Act has decoupled price erosion from LOE timing for negotiated drugs, requiring sponsors to revise long-range commercial forecasts.
A working monitoring program connects USPTO, FDA, and PACER feeds and is reviewed at the cadence of regulatory supplements rather than annual planning cycles.
FAQ
1. What is the difference between the Orange Book and the Purple Book?
The Orange Book lists approved small-molecule and certain other drug products along with the patents and exclusivity periods that protect them. The Purple Book lists licensed biological products and their reference product exclusivity periods (12 years from first licensure for the originator) but does not list patents. The structural difference matters because Hatch-Waxman litigation is driven by Orange Book listings, while BPCIA litigation requires the information exchange procedure described in 42 USC 262(l).
2. How long is the 30-month stay in Paragraph IV litigation?
The stay runs 30 months from the date of receipt of the Paragraph IV notice by the NDA holder, not from the date suit is filed. It can be shortened by a court finding of non-infringement or invalidity, or by failure of the NDA holder to reasonably cooperate in expediting the action under 21 USC 355(j)(5)(B)(iii).
3. When does 180-day generic exclusivity apply?
180-day exclusivity is awarded to the first ANDA filer (or filers, in a shared exclusivity situation) to submit a substantially complete ANDA with a Paragraph IV certification to an Orange Book-listed patent. The exclusivity period begins on the date of first commercial marketing of the generic product and bars FDA approval of subsequent ANDAs for the same RLD during that period, with several forfeiture exceptions.
4. How does Medicare price negotiation under the IRA interact with patent status?
Medicare price negotiation operates independently of patent status. A drug selected for negotiation may still be under composition patent protection at the time the maximum fair price applies. Negotiation selection is based on Medicare spend, time since approval (typically at least seven years for small molecules and 11 years for biologics), and the absence of generic or biosimilar competition.
5. What is a citizen petition and can it delay generic approval?
A citizen petition under 21 CFR 10.30 is a request to FDA for agency action. Section 505(q) of the Food, Drug, and Cosmetic Act, added by the FDA Amendments Act of 2007, limits the ability of citizen petitions to delay ANDA approvals and requires FDA to take final action within 150 days. Petitions that appear primarily designed to delay generic competition may be referred to the FTC.
6. What is a reverse payment settlement and is it legal?
A reverse payment settlement is a Hatch-Waxman patent settlement in which the branded patent holder transfers value to the generic challenger in exchange for delayed entry. The Supreme Court’s 2013 decision in FTC v. Actavis held that such settlements are not categorically illegal but are subject to rule-of-reason antitrust analysis when the payment is large and unjustified by costs or independent business arrangements.
7. What is the BPCIA patent dance?
The BPCIA patent dance is the information exchange procedure under 42 USC 262(l)(2)-(6), in which the biosimilar applicant provides its application and manufacturing information to the reference product sponsor, the parties exchange lists of relevant patents, and they negotiate which patents are litigated in a first phase. The Supreme Court held in Amgen v. Sandoz that this procedure is not enforceable by federal injunction, allowing biosimilar applicants to opt out.
8. Can a manufacturer add a patent to the Orange Book after a generic ANDA has been filed?
Yes, but the late-listed patent does not trigger a 30-month stay against an ANDA that was filed before the listing. The ANDA filer is not required to certify to a patent listed after its filing. This rule prevents serial relisting from repeatedly resetting the stay clock against a single generic challenger.
9. What is the practical difference between a CBE-30 and a Prior Approval Supplement?
A CBE-30 can be implemented 30 days after FDA receipt unless the agency objects, while a PAS requires affirmative FDA approval before implementation. The categorization depends on the substance of the change under 21 CFR 314.70, with PAS reserved for changes more likely to affect identity, strength, quality, purity, or potency. PAS filings are more often paired with new patent applications because they represent more substantive process or formulation changes.
10. How do tools like DrugPatentWatch fit into LOE forecasting?
DrugPatentWatch and similar services aggregate Orange Book listings, Paragraph IV certifications, litigation outcomes, and settlement terms into a single data layer linked to specific drugs and ANDA filers. For commercial forecasting, the value is in maintaining current drug-patent linkages and surfacing changes in Orange Book status or litigation activity quickly enough to update LOE models. Analysts typically pair these tools with PACER for docket detail and IQVIA or Evaluate Pharma for market-level data.
References
U.S. Food and Drug Administration. (2004). Guidance for Industry: Changes to an Approved NDA or ANDA. 21 CFR 314.70. https://www.fda.gov/regulatory-information/search-fda-guidance-documents
U.S. Code, Title 21, Section 355(c)(2). Patent information requirements following approval of an NDA.
U.S. Food and Drug Administration. (2024). Approved Drug Products with Therapeutic Equivalence Evaluations (Orange Book). 44th edition. Silver Spring, MD.
U.S. Code, Title 21, Section 355(j)(5)(B)(iii). 30-month stay provisions in Hatch-Waxman litigation.
Caraco Pharmaceutical Laboratories v. Novo Nordisk A/S, 566 U.S. 399 (2012).
GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc., 7 F.4th 1320 (Fed. Cir. 2021).
Mylan Pharmaceuticals Inc. v. Warner Chilcott Public Ltd. Co., 838 F.3d 421 (3d Cir. 2016).
New York ex rel. Schneiderman v. Actavis PLC, 787 F.3d 638 (2d Cir. 2015).
U.S. Code, Title 35, Sections 156 and 154(b). Patent Term Extension and Patent Term Adjustment.
IQVIA Institute for Human Data Science. (2023). The Use of Medicines in the U.S. 2023: Usage and Spending Trends and Outlook to 2027. Parsippany, NJ.
Federal Trade Commission v. Actavis, Inc., 570 U.S. 136 (2013).
Sandoz Inc. v. Amgen Inc., 582 U.S. 1 (2017).
AbbVie Inc. (2023). Annual Report on Form 10-K for the fiscal year ended December 31, 2022. U.S. Securities and Exchange Commission.
Bristol-Myers Squibb Company. (2022). Annual Report on Form 10-K for the fiscal year ended December 31, 2021. U.S. Securities and Exchange Commission.
Food and Drug Administration Amendments Act of 2007, Pub. L. No. 110-85, Section 914 (codified at 21 USC 355(q)).
Centers for Medicare & Medicaid Services. (2024). Medicare Drug Price Negotiation Program: Negotiated Prices for Initial Price Applicability Year 2026. Baltimore, MD: U.S. Department of Health and Human Services.



















")






