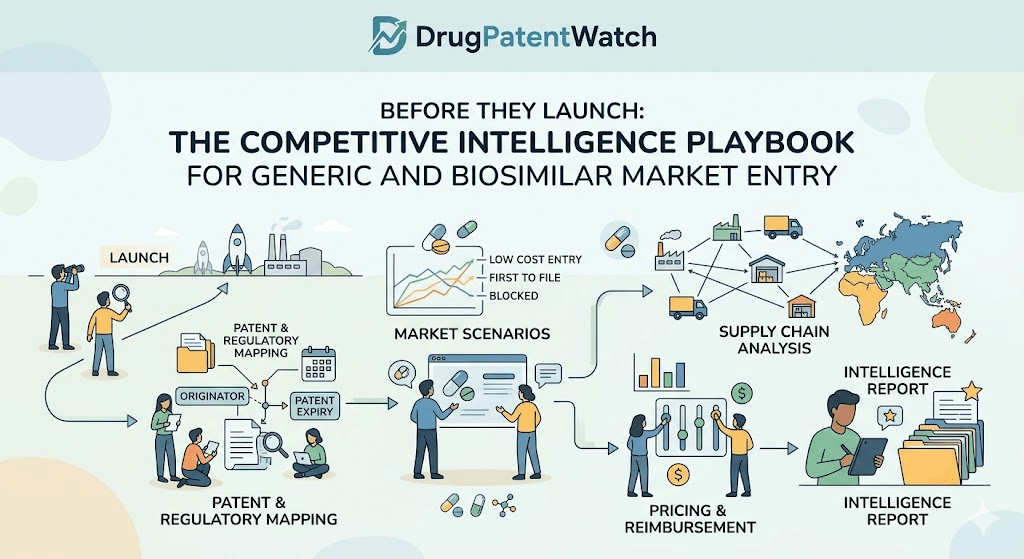
The day a competitor files a Paragraph IV certification against your lead molecule, you have 45 days to file suit or lose your automatic 30-month stay. Most commercial teams learn about that filing from a press release. That is too late.
This guide is built for the analysts, IP strategists, business development leads, and commercial directors who need to know what is coming before it arrives. It covers generic entry tracking, biosimilar launch timing, Hatch-Waxman litigation signals, loss-of-exclusivity (LOE) forecasting, authorized generic strategy, and the manufacturing intelligence that most patent analyses ignore entirely. Every section uses real drugs, real companies, and real docket numbers. No hypotheticals.
The pharmaceutical competitive intelligence function has matured considerably since the Hatch-Waxman Act created the modern generic approval pathway in 1984. But the tools available to analysts have not kept pace with the complexity of today’s patent estates, the multi-wave biosimilar landscape, or the settlement structures that determine actual commercial entry dates better than any legal expiry date does. The gap between what most organizations track and what they need to track is wide, and the cost of that gap is measurable in revenue, capital allocation, and missed launch windows.
This article closes that gap.
What Is Generic and Biosimilar Market Entry Intelligence?
Market entry intelligence, in the pharmaceutical context, means knowing who is filing to compete with a drug, when they plan to enter, on what legal theory, with what manufacturing infrastructure, and at what price point, before they do any of it publicly. It draws on public regulatory filings, patent litigation dockets, FDA databases, manufacturing facility inspection records, settlement agreement disclosures, and commercial pricing data. Done well, it is not a legal function or a commercial function. It sits at the intersection of both and requires both skill sets to execute.
For brand manufacturers, this intelligence determines how to deploy capital into life-cycle management, when to launch an authorized generic, and whether a litigation settlement makes financial sense. For generic and biosimilar developers, it determines which molecules are worth pursuing, which competitors will crowd a market before a return on development investment is possible, and whether first-to-file status is achievable.
Why Traditional Competitive Tracking Fails in Pharma
Standard competitive intelligence practice, the kind borrowed from technology or consumer goods sectors, relies heavily on press releases, earnings calls, SEC filings, and trade publication coverage. In pharma IP, each of those sources carries a structural lag that makes it nearly useless for decision-making.
A generic company files an Abbreviated New Drug Application (ANDA) with the FDA months or years before public disclosure. The FDA notifies the brand only through the Paragraph IV certification mechanism, and even that notification is triggered only if the ANDA includes a patent challenge. For applications filed under Paragraph III (accepting existing patent term), the brand may receive no notification at all until the drug is commercially available.
Biosimilar developers file 351(k) Biologics License Applications under a separate FDA pathway, with their own set of notice requirements under the Biologics Price Competition and Innovation Act (BPCIA). The so-called ‘patent dance’ under the BPCIA requires the biosimilar applicant to share its application with the reference product sponsor, but this process has been subject to litigation, waiver, and strategic manipulation since the law’s passage in 2010.
Conference presentations, LinkedIn announcements, and analyst reports all describe what has already happened. Competitive intelligence requires tracking what is about to happen, which means working directly from primary sources: the FDA’s Paragraph IV certification notice database, the Purple Book, the Orange Book, PACER court dockets, the USPTO assignment database, and facility inspection records from the FDA’s FOIA reading room.
The Data Sources That Actually Matter
Before building any intelligence workflow, you need to know which data sources are authoritative and which are derivative. Here is the tier structure that experienced analysts use:
Tier 1 (Primary, authoritative):
FDA Orange Book (Approved Drug Products with Therapeutic Equivalence Evaluations) for small-molecule patent listings and exclusivity data
FDA Purple Book (Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations)
FDA’s Paragraph IV Certification List (updated monthly)
PACER (Public Access to Court Electronic Records) for ANDA litigation dockets
USPTO Patent Center and Patent Full-Text and Image Database for prosecution history and assignment records
FDA Establishment Inspection Reports (EIRs) via FOIA
FDA Warning Letters and Import Alert database
Tier 2 (Aggregated, high-value):
DrugPatentWatch, which aggregates Orange Book listings, Paragraph IV history, litigation outcomes, and patent expiry data into a searchable commercial platform used by IP counsel, business development teams, and investment analysts across the industry
Drugs@FDA for approval history and labeling
IQVIA MIDAS and IQVIA NSP for commercial sales and volume benchmarks
Citeline Pharma Intelligence and Evaluate Pharma for pipeline and deal data
Tier 3 (Contextual, interpretive):
FTC filings and enforcement actions on pharmaceutical settlements
SEC 10-K and 10-Q filings for litigation disclosures from both brand and generic filers
State pharmacy board substitution law trackers
The fundamental error most organizations make is relying on Tier 3 sources to understand events that are already documented in Tier 1. By the time a settlement appears in an SEC filing or a generic launch appears in a press release, every commercially relevant decision it should have triggered is already overdue.
How to Read the FDA’s Paragraph IV Certification Pipeline
The Paragraph IV certification process is the single most important public signal of competitive intent in small-molecule pharmaceuticals. When a generic manufacturer files an ANDA and certifies under Paragraph IV of 21 U.S.C. 355(j)(2)(A)(vii)(IV) that a listed patent is invalid, unenforceable, or will not be infringed by the generic, the FDA notifies the brand within 20 days. The brand then has 45 days to file an infringement suit in federal district court to trigger an automatic 30-month stay of FDA approval.
The Paragraph IV certification list on the FDA’s website identifies the drug product, the ANDA applicant (by name since the FDA Reauthorization Act of 2017 required disclosure), and the patent number challenged. It does not tell you the legal theory of the challenge (invalidity vs. non-infringement), the proposed launch date, the settlement terms, or whether the filer has first-to-file status. You need the court docket for that.
What a Paragraph IV Filing Tells You About a Competitor’s Strategy
A Paragraph IV filing is a statement of commercial intent. It means the filer has completed or nearly completed an ANDA, believes it can manufacture the product to FDA standards, and has assessed the patent landscape enough to believe the listed patents are challengeable. It does not mean the filer will win in litigation or will even proceed to launch. But it does mean the competitive clock has started.
The specific patents challenged tell you where the generic manufacturer sees weakness. A challenge to a formulation patent but not a composition-of-matter patent signals a non-infringement theory based on a different delivery mechanism. A challenge to a method-of-use patent signals that the generic intends to carve out the patented indication from its label, what the industry calls a ‘skinny label’ or ‘section viii carveout’ under 21 C.F.R. 314.94(a)(12)(iii).
When multiple filers challenge different patents on the same drug, you can often map each filer’s product differentiation strategy from the challenge pattern alone. Sandoz challenging a formulation patent while Teva challenges both formulation and method-of-use suggests Sandoz has a genuinely different formulation and Teva is using a skinny label approach. These are different competitive threats requiring different brand responses.
First-to-File Exclusivity: Who Gets the 180-Day Window and Why It Matters
The first generic manufacturer to file a substantially complete ANDA with a Paragraph IV certification receives 180 days of market exclusivity before any other ANDA can receive final approval. During those 180 days, the first-to-file generic and the brand are the only commercial sources of the drug (unless the brand launches an authorized generic, which it can do freely unless restricted by a settlement agreement).
The commercial value of 180-day exclusivity is enormous in large markets. For a drug with annual US net sales of $3 billion, a single generic entrant at typical day-one pricing of 20% below brand can generate $300-400 million in revenue during the exclusivity window, assuming volume conversion runs at 80-85%, which is the historical norm for primary-care oral solid dosage forms.
Exclusivity is forfeited under six conditions defined in 21 U.S.C. 355(j)(5)(D)(i), most commonly failure to market within 75 days of the earlier of FDA approval or a court decision finding the patent invalid or not infringed. The forfeiture mechanism exists because Congress recognized that first-to-file applicants were warehousing their exclusivity rights to prevent second applicants from launching while collecting settlements from brands. Monitoring forfeiture triggers is a material competitive intelligence task for second and third ANDA filers.
Case Study: Revlimid (Lenalidomide) and the Managed Entry Settlement
Bristol-Myers Squibb’s settlement of Revlimid (lenalidomide, marketed by Celgene prior to BMS’s 2019 acquisition) patent litigation with generic manufacturers became one of the most scrutinized managed entry arrangements in US pharmaceutical history. The settlements, reached between 2015 and 2020 with Natco Pharma, Lotus Pharmaceutical, Hetero Labs, Amneal Pharmaceuticals, and others, allowed generic entry beginning in March 2022, with volume caps that limited each generic to a specified percentage of total lenalidomide demand for the first several years post-entry.
The FTC investigated the settlement structure, and in 2023, the agency filed an administrative complaint alleging the volume restrictions constituted an unlawful reverse payment. The case turned on whether the volume caps, rather than an explicit cash payment, constituted compensation to the generic filers in exchange for delayed or limited entry. As of mid-2025, the litigation continues to define the outer boundaries of permissible settlement terms post-FTC v. Actavis (2013).
For competitive intelligence purposes, the Revlimid situation illustrates two points. First, watching which generic companies settle rather than litigate to judgment tells you about their capital constraints and risk tolerance. Companies that settle quickly are typically operating on thinner margins or have less patent litigation infrastructure than those willing to litigate to judgment. Second, the volume cap structure itself was commercially legible before the FTC complaint: anyone reviewing the settlement terms in BMS’s 10-K could see that generic penetration in the first years post-entry would be far below the historical norm, which should have fed directly into market share models for lenalidomide’s generic competitors.
How Bristol-Myers Squibb Used Paragraph IV Settlements to Protect $10 Billion in Revenue
Revlimid generated approximately $12.8 billion in global net sales in 2021, the year before generic entry. BMS’s settlement strategy deferred meaningful volume erosion by roughly three years beyond the first generic launch date, through the volume cap mechanism. IQVIA data from 2022 and 2023 showed generic lenalidomide capturing share substantially below the typical first-year generic penetration curve, confirming that the settlement terms were functioning as intended from the brand’s perspective.
This type of managed entry strategy is detectable in advance by tracking the gap between the announced settlement entry date and the actual volume share curve post-launch. When a drug’s generic penetration runs 30-40 percentage points below historical norms in the first 12 months, the most likely explanation is a volume-restricted settlement, and the competitive intelligence task is to identify the specific restriction terms before they become public through enforcement proceedings.
Orange Book Patent Listings: The Intelligence Layer Most Analysts Miss
The FDA’s Orange Book lists every patent that the drug manufacturer has certified covers the approved drug product, its active ingredient, or a method of use. These listings are self-reported by the brand, which creates both the principal mechanism for patent protection and the primary target for generic challenges.
The Orange Book currently lists more than 200,000 patents across more than 10,000 approved drug products. Not all of those patents are created equal, and understanding the difference between patent types is the foundation of any serious LOE analysis.
Drug Substance vs. Drug Product vs. Method-of-Use Patents: What Each Tells You
Orange Book patents fall into three functional categories, each with different commercial implications for generic entry timing.
Drug substance patents (also called composition-of-matter patents) cover the active pharmaceutical ingredient itself. These are typically the strongest patents, hardest to design around, and the ones whose expiry date sets the outer boundary of brand exclusivity. When a composition-of-matter patent expires, the generic pathway opens regardless of what other patents remain listed. Examples include the original sildenafil compound patent (US5250534) that Pfizer used to protect Viagra, which expired in 2012, and the atorvastatin compound patent (US4681893) that Pfizer held for Lipitor.
Drug product patents cover formulations, delivery systems, coatings, salt forms, or specific pharmaceutical preparations rather than the molecule itself. Generic manufacturers frequently challenge these as non-infringed if they develop an alternative formulation, or as obvious if the formulation represents an incremental modification of prior art. The lenalidomide case involved compound, polymorph, and formulation patents, which is typical of a heavily protected small molecule.
Method-of-use patents cover specific therapeutic applications of the drug. These are the basis for skinny label strategies. If a drug is approved for three indications and only two are covered by method-of-use patents, a generic manufacturer can file a Paragraph III or IV certification for the unpatented indication and market its product with a label that carves out the patented uses. This creates real-world substitution risk, particularly for physician-dispensed or hospital-administered drugs where the prescribing context is poorly controlled.
How to Use DrugPatentWatch to Map a Brand’s Patent Fortress
DrugPatentWatch provides a structured view of every Orange Book-listed patent for a given drug, organized by expiry date, patent type, and litigation history. For competitive intelligence purposes, the platform’s most useful function is displaying the timeline of patent expirations alongside the history of Paragraph IV challenges to each patent, allowing analysts to see which patents have already been tested in litigation and which remain unchallenged.
An unchallenged late-expiring patent is a red flag, not reassurance. It may mean generic filers see it as a strong patent not worth challenging, or it may mean no one has yet examined it carefully. The correct analytical response is to determine which, by reviewing the prosecution history, the prior art landscape, and whether the patent has been addressed in any inter partes review (IPR) petition before the Patent Trial and Appeal Board (PTAB).
DrugPatentWatch also tracks pediatric exclusivity grants, which add six months of exclusivity to every patent and exclusivity period listed in the Orange Book when the brand conducts FDA-requested pediatric studies under the Best Pharmaceuticals for Children Act. This six-month extension applies even to composition-of-matter patents and can represent hundreds of millions of dollars in brand revenue. Tracking whether a brand has requested or received pediatric exclusivity is a standard step in any LOE model, yet it is missed in a surprising number of analyst forecasts.
Orange Book Delisting Tactics and What They Signal
A brand manufacturer can voluntarily delist a patent from the Orange Book at any time. Under the 2021 CREATES Act amendments to Hatch-Waxman, generic manufacturers can now sue to force delisting of patents they believe are improperly listed. Involuntary delisting has become an active litigation tactic: Mylan (now Viatris) sued to delist AstraZeneca’s quetiapine fumarate formulation patent from the Orange Book in the context of Seroquel XR ANDA litigation, and that case established precedents that later filers have used against brand manufacturers with weaker patent listings.
Voluntary delisting by a brand is a competitive signal worth attention. If a brand delists a patent during active ANDA litigation, it can remove the 30-month stay for that patent and accelerate the generic’s path to approval. Brands typically delist when they have calculated that litigation over a particular patent is unlikely to succeed and that maintaining the patent in litigation creates settlement leverage problems with subsequent filers. The commercial implication is that the brand has internally concluded that particular line of defense is not worth holding.
Evergreening Strategies: Pediatric Exclusivity, NCE, and NME Extensions
Brand manufacturers deploy several FDA-granted exclusivity designations that extend market protection beyond compound patent expiry. Understanding the mechanics of each is essential for accurate LOE forecasting.
New Chemical Entity (NCE) exclusivity grants five years of protection from the date of first approval for drugs containing an active moiety not previously approved. During this period, the FDA cannot accept an ANDA for the same drug. This means no Paragraph IV certifications and no 30-month stays. Generic manufacturers can file their ANDAs one year before the NCE exclusivity expires, but cannot receive final approval until the exclusivity runs out. For drugs with NCE exclusivity, the earliest possible generic launch is five years from approval plus ANDA review time, typically six to seven years from initial approval.
New Molecular Entity (NME) designation, a related but distinct category, determines whether a drug receives a full 10-month FDA review target or a priority review. It is not itself an exclusivity period.
Three-year exclusivity applies to new clinical investigations that support supplemental approvals, such as new indications, new dosage forms, or new dosing regimens. Generic manufacturers can still file ANDAs against the original drug product during this period but cannot reference the new clinical data. The practical effect is that generics can enter the original indication market but are blocked from the newer indication until the three-year period expires.
Pediatric exclusivity, as noted above, adds six months to all existing exclusivity periods and patents for drugs that complete FDA-requested pediatric studies. Because the extension attaches to every listed patent, a drug with a late-expiring formulation patent can receive pediatric exclusivity through a relatively low-cost pediatric trial, extending total protection by six months regardless of what the primary patent situation looks like. Abbott’s Humira received pediatric exclusivity that became commercially relevant during the biosimilar entry period, illustrating that even biologics face evergreening dynamics.
Biosimilar Market Entry: A Different Intelligence Framework
Biosimilar competitive intelligence requires a different analytical framework than generic drug tracking. The regulatory pathway, patent landscape, pricing dynamics, and market access mechanisms all work differently for large-molecule biologics than for small-molecule drugs. Treating them as analogous produces systematically wrong forecasts.
FDA Biosimilar Approval Pathway vs. Generic ANDA: Key Differences
Generic drugs receive FDA approval through the ANDA pathway under Section 505(j) of the Federal Food, Drug, and Cosmetic Act. The core requirement is bioequivalence to the reference listed drug (RLD), established through pharmacokinetic studies. If a generic is bioequivalent, it receives an AB therapeutic equivalence rating and can be substituted at the pharmacy level without prescriber approval in most states.
Biosimilars are approved through the 351(k) pathway under the BPCIA, which requires demonstrating that the product is highly similar to the reference product with no clinically meaningful differences in safety, purity, or potency. The standard of proof is higher than bioequivalence, requiring analytical characterization, preclinical testing, and in most cases clinical pharmacology studies and at least one comparative clinical trial. Full interchangeability designation requires additional switching studies and, once achieved, allows pharmacy-level substitution in states that permit it.
The patent dance under the BPCIA adds another layer of complexity absent from the ANDA process. The biosimilar applicant must notify the reference product sponsor at least 60 days before commercial marketing and disclose its application. The sponsor can then initiate a patent list exchange and negotiation process that determines which patents are litigated and in what sequence. The biosimilar applicant can waive participation in the patent dance, opting instead to provide the 60-day pre-launch notice and accept the risk of a preliminary injunction. Several biosimilar developers have waived the patent dance in recent years, concluding that the litigation-sequencing benefits were outweighed by the delay it introduced.
Interchangeability Designation: What It Means for Market Share Forecasting
Interchangeability is the FDA designation that allows automatic pharmacy substitution of a biosimilar for the reference product, subject to state law. Without interchangeability, substitution requires the prescriber to actively write the biosimilar by name or indicate substitution is acceptable. With it, the pharmacist can substitute without a new prescription, which dramatically lowers the friction for conversion.
For market share forecasting, interchangeability matters most for self-administered biologics with large retail pharmacy volume, particularly subcutaneous injections. For hospital-administered or infusion-center biologics like infliximab (Remicade) and rituximab, the substitution question is managed by formulary committees at the payer and health system level, making interchangeability less operationally relevant at the dispensing point.
The FDA has granted interchangeability to several biosimilars as of 2025, including Cyltezo (adalimumab-adbm, Boehringer Ingelheim) for adalimumab, Hadlima (adalimumab-bwwd, Samsung Bioepis/Organon) for adalimumab, and Semglee (insulin glargine-yfgn, Viatris) for insulin glargine. Each of these designations changed the commercial forecast for the drug’s biosimilar market by increasing the projected volume capture through pharmacy channels.
The Humira Biosimilar Wave: Eight Launches and What Happened
Humira (adalimumab), AbbVie’s blockbuster immunology biologic, generated approximately $21.2 billion in global net sales in 2022, making it the best-selling prescription drug in history at the time. Seven biosimilar developers reached settlement agreements with AbbVie granting US launch rights beginning January 2023. Two more followed with later dates. The commercial result was the largest single LOE event in biosimilar history.
The January 2023 wave included Amgen’s Amjevita, Sandoz’s Hyrimoz, Samsung Bioepis/Organon’s Hadlima, Boehringer Ingelheim’s Cyltezo, Coherus BioSciences’ Yusimry, Pfizer’s Abrilada, and Fresenius Kabi’s Idacio. Each launched with discounts of approximately 5% to 55% below AbbVie’s then-list price of roughly $6,900 for a 40mg/0.8mL pre-filled syringe.
By mid-2024, however, AbbVie maintained over 85% of US adalimumab market share by volume. The explanation lies in the rebate wall AbbVie had constructed in the years before biosimilar launch. AbbVie offered pharmacy benefit managers (PBMs) rebates conditioned on exclusive or preferred formulary placement for Humira, with rebate rates reportedly in the range of 40-60% off list price for top-tier placement. Biosimilar entrants, priced at list discounts of 5-20%, could not generate net prices competitive with post-rebate Humira without cutting their own prices far enough to undermine return on investment.
The lesson for competitive intelligence analysts: for biologics with established large-account PBM contracts, list price discounts are not a reliable proxy for commercial access. Tracking rebate contract renewal timelines, payer formulary tier placement decisions, and PBM contract expiry dates provides more actionable launch timing intelligence than patent litigation outcomes alone.
Biosimilar Launch Timing vs. Settlement Dates: Remicade and Enbrel as Case Studies
Infliximab (Remicade, Janssen/Johnson & Johnson) and etanercept (Enbrel, Amgen/Pfizer) provide contrasting case studies in biosimilar launch dynamics.
For Remicade, Pfizer’s Inflectra (infliximab-dyyb) launched in November 2016 after winning patent litigation against Janssen and after Pfizer agreed to launch without waiting for the BPCIA patent dance to complete. Celltrion’s Inflectra had already launched in Europe. Despite FDA approval and a competitive price, Inflectra achieved only approximately 20-25% US market share in infliximab by 2020, reflecting J&J’s aggressive contracting with hospital systems and group purchasing organizations (GPOs) that created switching costs and volume incentives for staying on Remicade.
Enbrel’s biosimilar situation played out differently. Amgen held composition-of-matter patent protection in the US that kept biosimilar competitors off the market until 2029, later than most analysts had modeled, because Amgen had been granted patent term extensions and had obtained late-expiring manufacturing process patents not originally appreciated in early LOE models. Samsung Bioepis’s etanercept biosimilar SB4 (Benepali) launched in Europe in 2016, where patent protection was different, but Amgen’s US position remained intact. The gap between European and US biosimilar availability for Enbrel illustrates why US-specific Orange Book analysis, rather than global patent mapping, is essential for US commercial forecasts.
Tracking ANDA Filers: How to Build a Competitor Filing Database
Monitoring who has filed ANDAs against a drug of interest requires combining the FDA’s Paragraph IV certification list with the Orange Book ANDA approval history and, where available, the ANDA applicant information published in FDA approval letters. For drugs not yet subject to Paragraph IV filings, the absence of ANDA activity is informative in its own right: it may mean no one has tried, no one has found a viable challenge, or ANDAs have been filed under Paragraph III or Paragraph II and are simply waiting for patent expiry.
How Many Generic Applicants Are Too Many? The Economics of Crowded Markets
A market with more than six generic entrants at the time of first launch will typically see price erosion exceeding 80% within 12-18 months. The FDA’s 2019 Drug Competition Action Plan data showed that markets with 10 or more generic competitors see price declines exceeding 90% relative to brand price, with average generic prices under $0.10 per unit for oral solid dosages in some cases.
For generic manufacturers, this means that pursuing an ANDA in a market already targeted by seven or eight other filers, without first-to-file exclusivity, is frequently not economically rational. The expected return on ANDA development, plus litigation costs if Paragraph IV certification is filed, plus product launch costs, may not clear the hurdle rate when the forecast price environment is 90% below brand within 24 months of first entry.
Tracking the number of ANDA filers for a given molecule is therefore a market sizing input as well as a competitive threat assessment tool. DrugPatentWatch provides historical ANDA applicant data that allows analysts to benchmark the number of filers per molecule against similar drugs, giving a rough probability distribution for the long-run price floor.
“Markets with two to five generic competitors retain an average retail price of approximately 39% of the original brand price. Markets with more than six competitors see prices fall to an average of 13% of original brand price.” (FDA, Drug Competition Action Plan, 2019)
Authorized Generics: When the Brand Fights Back
An authorized generic (AG) is a branded product sold under the generic drug regulatory approval with a label identical to the brand but marketed under a different name, typically by the brand itself or a partner. The brand can launch an AG at any time, including during the first-to-file generic’s 180-day exclusivity window. This was confirmed by the US Supreme Court’s 2006 ruling in Solvay Pharmaceuticals Inc. v. Brotech Corp. and codified in subsequent case law.
AG launches during the 180-day window are among the most commercially devastating tactics available to a brand manufacturer facing generic entry. With the AG sharing the first-to-file generic’s exclusivity window, the first-to-file generic loses the pricing power that makes the 180 days valuable. Instead of a duopoly with the brand, the first generic faces a three-way market with the brand, its own product, and the brand’s AG, all of which are bioequivalent. Prices drop to the 40-50% range almost immediately instead of the 80% or above that generic manufacturers typically model into first-year revenues.
The commercial intelligence signal here is whether a brand has historically launched AGs against first-to-file generics. Companies with AG strategies include Pfizer (which frequently partners with Greenstone, its AG subsidiary), Novartis (which partners with Sandoz, its generic division, in specific circumstances), and Sanofi (through its generics partnerships). Tracking a brand’s historical AG behavior is a key input to first-to-file economics modeling for any new target.
The Role of the First-to-File Generic in Pricing Strategy
First-to-file generics typically enter at 10-25% below brand list price. The pricing strategy is calibrated to: maintain a price premium over anticipated second-wave generics, capture volume-conversion from payers seeking mandatory generic substitution, and maximize revenue during the 180-day window before the price collapse that follows multi-source entry.
Modeling the optimal entry price for a first-to-file generic requires knowing three things that are knowable from competitive intelligence: who else has filed an ANDA, what their manufacturing cost structure likely is based on their API sourcing and facility infrastructure, and when they are likely to receive FDA approval after the 180-day exclusivity period begins. Generic companies with higher-cost manufacturing, typically US-based or European manufacturers, will have less room to cut price than Indian or Chinese generic manufacturers with lower labor and facility costs. This cost structure differential affects the competitive price floor and should factor into first-to-file pricing models.
When Does a Second Generic Applicant Actually Win? The 180-Day Forfeiture Rules
The 180-day forfeiture provisions in the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (MMA) created six circumstances under which a first-to-file applicant can lose its exclusivity period before using it, allowing second applicants to advance. The most commercially relevant forfeiture triggers are: failure to market within 75 days of the earlier of FDA approval or court decision, withdrawal of the ANDA, and failure to obtain timely tentative approval.
Competitive intelligence around forfeiture potential requires tracking whether the first-to-file generic has received tentative approval, whether it has filed marketing applications in other markets (a signal of commercial readiness), and whether it is in active litigation that could delay final approval. If a first-to-file applicant’s exclusivity is at risk of forfeiture, second applicants may rationally accelerate their own approval timelines to be positioned to launch as soon as the forfeiture triggers.
The Patent Cliff Forecast: How to Build a Loss-of-Exclusivity Timeline
An LOE timeline is not simply a list of patent expiry dates. The most common mistake in LOE modeling is equating legal patent expiry with commercial generic entry. These dates rarely align, and the gap between them, which can run from zero to several years, is entirely determined by factors that patent expiry dates do not capture.
What Data Points Go Into a Reliable LOE Model
A complete LOE model incorporates at least seven data layers:
Patent expiry dates for every Orange Book-listed patent, including patent term extensions (PTEs) and pediatric exclusivity additions
Non-patent exclusivity periods (NCE, orphan drug exclusivity, NME exclusivity, three-year new clinical investigation exclusivity)
ANDA filing status: how many filers, their Paragraph certification status, and tentative approval dates
Litigation status for each Paragraph IV challenge: trial dates, judgment history, and settlement terms
Manufacturing readiness signals for leading ANDA filers: facility inspection history, FDA warning letter history, import alert status
AG strategy probability based on brand’s historical behavior and any disclosed contractual commitments in settlement agreements
Payer contract and formulary dynamics for high-rebate drugs where list-price-based generic entry does not translate to formulary conversion
Excluding any one of these layers produces a systematically biased forecast. The most common omission in publicly available LOE models is layer 7, the formulary and rebate dynamics that have determined actual biosimilar and specialty generic adoption rates in post-2015 markets.
The Gap Between Legal Expiry and Commercial Entry: Why They’re Never the Same
Patent expiry tells you when entry is legally possible, not when it will happen. The gap runs in both directions. Generic entry can occur before final patent expiry if: a court rules a patent invalid or not infringed, a settlement grants an entry date before expiry, or the FDA grants approval after all exclusivities have run but the brand challenges the approval in court without seeking a preliminary injunction.
Entry can occur after patent expiry if: no ANDA has been filed, ANDA applicants are blocked by manufacturing deficiencies, or the market is too small to attract generic development at all. For branded drugs with less than $100 million in US annual sales, generic entry frequently does not occur for years after patent expiry because the development economics do not support it.
The average gap between final patent expiry and first generic launch, for drugs with annual US sales above $500 million, is approximately six months, based on IQVIA analysis of LOE events from 2015-2023. For drugs with sales above $2 billion, the average gap is closer to 12-18 months because the commercial stakes drive more aggressive brand litigation and more complex settlement negotiations. For drugs with sales below $100 million, the average gap is over three years, and roughly 30% of such drugs see no generic entry within five years of patent expiry at all.
Top 10 Drugs Facing LOE Between 2025 and 2030
The following molecules face meaningful US LOE risk in the 2025-2030 window, representing combined annual US revenues exceeding $80 billion based on 2023-2024 reported sales:
Keytruda (pembrolizumab, Merck): compound patent expiry expected 2028; biosimilar development active across multiple companies
Eliquis (apixaban, Bristol-Myers Squibb/Pfizer): generic entry settlement agreements with multiple ANDA filers allow launch from April 2028
Stelara (ustekinumab, Johnson & Johnson): biosimilar approvals received by Amgen, Samsung Bioepis, and others; J&J settlements allow US launch from late 2023, with ongoing commercial rollout
Ozempic/Wegovy (semaglutide, Novo Nordisk): composition-of-matter patent expires 2032 in US; no generic entry expected before then absent successful patent challenge
Xarelto (rivaroxaban, Bayer/J&J): US patent expires 2024; multiple generics already in market
Biktarvy (bictegravir/emtricitabine/tenofovir alafenamide, Gilead): complex co-formulation patent estate; generic entry not before 2032 based on Orange Book analysis
Dupixent (dupilumab, Sanofi/Regeneron): biologic with exclusivity into the 2030s; no biosimilar approvals yet; patent estate includes multiple formulation and manufacturing process patents
Jardiance (empagliflozin, Boehringer Ingelheim/Eli Lilly): compound patent expires 2025; generic entry imminent with multiple Paragraph IV filers
Entresto (sacubitril/valsartan, Novartis): key patents litigated extensively; settlement allows generic entry from July 2025
Skyrizi (risankizumab, AbbVie): biologic with compound patent expiry 2033; biosimilar development in early stages
Keytruda (Pembrolizumab) Patent Strategy and What Competitors Are Doing Now
Keytruda, Merck’s anti-PD-1 antibody, generated approximately $25 billion in global net sales in 2023, making it the best-selling prescription drug in the world. Its US compound patent, US9908948, is expected to expire in 2028. Merck has built a secondary patent estate covering formulations, delivery devices, and manufacturing processes that extends protection into the early 2030s.
Biosimilar development activity is extensive. Samsung Bioepis filed for FDA approval of SB27, its pembrolizumab biosimilar, in early 2025. Alvotech, Celltrion, and Amgen are all in various stages of development or regulatory submission. The biosimilar pathway for antibodies requires demonstrating analytical similarity against the reference product, which for pembrolizumab means demonstrating comparable binding characteristics against PD-1 and comparable Fc-mediated effector functions, both of which are analytically tractable.
The commercial question for Keytruda is not whether biosimilars will enter but whether they will gain formulary access in the oncology setting. PD-1 inhibitors are administered primarily in oncology clinics and hospital infusion centers, where formulary decisions are made at the pharmacy and therapeutics (P&T) committee level. The willingness of oncologists and institutions to substitute pembrolizumab biosimilars will depend heavily on clinical evidence, institutional contract terms, and whether Merck constructs a rebate wall comparable to AbbVie’s Humira strategy. Given AbbVie’s experience, Merck is almost certain to attempt aggressive contracting, and any pembrolizumab biosimilar launch strategy must account for that.
Litigation Intelligence: Reading Court Dockets for Commercial Signals
ANDA litigation under Hatch-Waxman is the primary mechanism through which brands delay generic entry beyond patent expiry. For competitive intelligence purposes, the litigation docket is the most information-dense public record available, containing not just legal arguments but scheduling orders, settlement conferences, discovery disputes, and expert witness designations that collectively reveal each party’s assessment of their legal position and commercial priorities.
ANDA Litigation Under Hatch-Waxman: Timeline, Costs, and Outcomes
When a brand files an infringement action within 45 days of receiving a Paragraph IV notice, the 30-month stay clock starts. The FDA cannot grant final approval to the ANDA during the stay period unless a court enters judgment before the 30 months expire. Given the typical timeline for ANDA litigation, including Markman hearings, fact discovery, expert discovery, and trial, many cases settle or reach judgment within the 30-month window, but a significant fraction require full trial and appellate resolution.
The average cost of ANDA litigation through trial runs $10-30 million per case on each side, based on law firm billing data cited in pharmaceutical litigation surveys. For a drug with $2 billion in annual US sales, the cost-benefit calculation strongly favors litigating to delay even by six to 12 months, because one additional month of protected sales at full brand price typically generates $100-200 million in revenue. The asymmetry drives aggressive brand litigation regardless of the underlying merits of the patent claim.
Historical win rates in Hatch-Waxman litigation favor generic challengers. A 2019 study by the Lex Machina legal analytics platform found that generic manufacturers prevailed in approximately 57% of ANDA trials where a final judgment was entered. The finding is consistent with earlier studies dating to the early 2000s and reflects the systematic nature of Orange Book listing, where brands often list patents that represent incremental over prior-art improvements and are therefore more vulnerable to obviousness challenges than primary composition-of-matter patents.
Inter Partes Review vs. District Court: Which Path Do Generic Challengers Prefer?
Since the America Invents Act created the inter partes review (IPR) pathway at the PTAB in 2012, generic manufacturers have used IPR petitions as a parallel or primary vehicle for challenging Orange Book patents. IPR offers several advantages over district court litigation: a lower burden of proof (preponderance of evidence rather than clear and convincing evidence), a faster decision timeline (typically 18 months from institution), lower cost, and the ability to challenge patents before an ANDA is even filed, allowing manufacturers to test a patent’s vulnerability before committing to full ANDA development.
The limitation of IPR for generic purposes is that an IPR decision, even a favorable one finding the patent invalid, does not automatically result in ANDA approval. The FDA still requires an ANDA to be filed and reviewed. IPR-driven invalidity, however, collapses the litigation risk that would otherwise accompany a Paragraph IV filing and eliminates the 30-month stay if the patent is cancelled before the brand can file suit.
Coalitions of generic manufacturers have increasingly coordinated IPR petitions, with multiple filers challenging the same patent in coordinated IPRs to share costs and prevent estoppel under 35 U.S.C. 315(e). The 2017 Supreme Court ruling in SAS Institute Inc. v. Iancu and subsequent PTAB procedural guidance changed the economics of coordinated IPR somewhat, but the strategy remains common for high-value pharma patent targets.
The Delaware District Court and the SDNY: Why They Attract Most ANDA Cases
The US District Court for the District of Delaware and the US District Court for the Southern District of New York (SDNY) handle the majority of ANDA litigation filings, reflecting the fact that most major pharmaceutical companies are incorporated in Delaware and have principal places of business in the New York metropolitan area.
Delaware is particularly dominant for ANDA cases. Chief Judge Colm Connolly’s 2021 standing order requiring disclosure of litigation funding arrangements in Delaware cases, and the court’s historically efficient Markman-to-trial scheduling, have made it the preferred venue for both brands and generic challengers. The court’s institutional familiarity with Hatch-Waxman mechanics also means fewer procedural delays driven by judicial unfamiliarity with pharmaceutical patent concepts.
For competitive intelligence purposes, the choice of venue by a brand in its ANDA infringement complaint tells you something about its assessment of how the case will proceed. Filing in Delaware typically signals an expectation of efficient, predictable litigation. Filing in New Jersey (District of New Jersey, the other major ANDA venue given the large pharma concentration in that state) or in the home district of the generic manufacturer suggests either convenience-based choices or an attempt to secure what the brand perceives as a favorable judge assignment.
How Litigation Settlements Reveal Approved Entry Dates Before the FDA Does
Hatch-Waxman settlements are disclosed in SEC filings and, in cases where the FTC reviews them under its consent agreement authority, in FTC enforcement actions. But the most consistent and detailed disclosure source is actually the court’s docket itself. When a case settles, the parties typically file a stipulation of dismissal and, in some jurisdictions, are required to file the settlement agreement under seal with a public redacted version.
The critical commercial information in a settlement agreement is the agreed entry date. This date is almost always earlier than any publicly disclosed patent expiry timeline, because the brand is granting early entry in exchange for the generic dropping its patent challenge. Reading that date before it becomes public through press releases or SEC filings gives a competitive intelligence team weeks or months of advance notice for commercial planning.
A structured docket monitoring workflow, covering all active ANDA cases in Delaware, New Jersey, and the SDNY, and triggering alerts on stipulation filings, can capture this signal reliably. PACER charges by page for downloads, but at $0.10 per page, the cost of monitoring a universe of 200-300 active ANDA cases is under $10,000 per year and generates intelligence that cannot be purchased from any commercial database at any price.
Settlement Structures: What Reverse Payments Tell You About Competitive Timing
The concept of a reverse payment, sometimes called a ‘pay-for-delay’ settlement, refers to a Hatch-Waxman litigation settlement in which the brand manufacturer pays the generic challenger to accept a delayed entry date rather than proceeding to trial. The payment can take the form of cash, a royalty-free license to another product, a co-promotion agreement, an agreement not to launch an authorized generic, or, as in the lenalidomide case, volume restrictions.
FTC Scrutiny of Pay-for-Delay Settlements Post-Actavis
The Supreme Court’s 2013 decision in FTC v. Actavis, Inc. held that reverse payment settlements can violate antitrust law and directed lower courts to evaluate them under the rule of reason. The decision overturned the Eleventh Circuit’s holding that such settlements were presumptively lawful as long as the generic entry date fell within the patent term and resolved more than a decade of lower court disagreement about the applicable standard.
Post-Actavis, the FTC has investigated and challenged a range of settlement structures beyond cash payments. The Solvay pharmaceutical settlement itself, on which Actavis was decided, involved a payment of $42 million to Actavis (then Watson Pharmaceuticals). Subsequent FTC actions have targeted non-cash compensation including authorized generic exclusions, royalty arrangements, and business side deals that effectively compensate generic challengers for delaying entry.
For competitive intelligence purposes, the FTC’s enforcement docket is a real-time map of what settlement structures brands are attempting and which have attracted regulatory scrutiny. Reading FTC complaints and consent decrees in pharmaceutical matters tells you the boundaries of what is legally risky versus what is currently tolerated, which directly informs your assessment of whether a given settlement includes implicit compensation.
Permitted vs. Impermissible Settlement Terms: Reading the FTC’s Enforcement Signals
The current state of enforcement suggests the following rough framework for settlement term permissibility:
Likely permissible: agreed entry date within patent term, no-AG provisions limited to the 180-day exclusivity period, royalty licenses at fair market value, co-promotion agreements with commercial justification independent of patent litigation.
High risk/under scrutiny: no-AG provisions extending beyond the 180-day period, above-market royalties on unrelated products, volume caps on generic sales post-entry (as in the lenalidomide matter), bundled deal payments that lack arms-length commercial justification.
Clearly impermissible post-Actavis: large reverse cash payments with no independent commercial justification, agreements to restrict price competition after generic launch, agreements not to challenge related patents on other drugs.
Case Study: AbbVie’s Humira Biosimilar Settlements with AstraZeneca and Samsung Bioepis
AbbVie’s settlement strategy for Humira biosimilar entry is the most commercially instructive recent example of brand-side settlement intelligence. AbbVie held approximately 130 patents covering adalimumab, its formulations, delivery devices, and manufacturing processes as of 2020. The breadth of this patent fortress, sometimes called a ‘patent thicket’ in academic and advocacy literature, was itself a source of litigation: Mylan filed suit in 2019 alleging that AbbVie’s patent listings were improperly listed in the Orange Book (note: biologics are listed in the Purple Book, not the Orange Book, but the delisting claim involved related small-molecule components).
AbbVie’s settlement with AstraZeneca for Amgevita (adalimumab-atto, Amgen’s product licensed to AstraZeneca for non-US markets) and separately with Samsung Bioepis for Hadlima each granted US launch rights in January 2023, approximately two years before AbbVie’s final US patents for Humira would expire. In exchange, the biosimilar developers dropped their patent challenges, and AbbVie retained its late-expiring patent estate without having to defend it at trial.
The commercial rationale for AbbVie was to avoid the risk of adverse judgments that could have opened the market to all biosimilar entrants simultaneously while preserving the appearance of a defended patent estate for future biologics. For biosimilar developers, the January 2023 entry date represented early access at a defined date, which enabled capital planning and commercial launch preparation that would not have been possible without a settled entry date.
Reading a Settlement Agreement for Implicit Commercial Intelligence
Settlement agreements in Hatch-Waxman and BPCIA litigation contain several provisions beyond the entry date that carry commercial intelligence value:
No-AG clauses specify whether the brand commits not to launch an authorized generic during the first-to-file exclusivity period. These clauses protect the first generic’s pricing power and are commercially very valuable. A settlement without a no-AG clause signals that the brand intends to compete aggressively from day one of generic entry.
Sublicense provisions specify whether the settling generic can license its ANDA to other manufacturers. Some settlements prohibit sublicensing, effectively limiting post-settlement entry to one generic source. This reduces competition and maintains prices above the multi-source erosion floor.
Sales milestone triggers sometimes restart or extend restriction periods if the generic exceeds a certain market share. These provisions, less common after the FTC’s focus on volume caps, can dramatically alter penetration curves in ways not visible from the headline entry date.
Biosimilar Naming, Interchangeability, and Pharmacy-Level Substitution Dynamics
The FDA’s biosimilar naming convention, state substitution laws, and interchangeability designation together determine whether a biosimilar’s commercial performance resembles a generic drug or a branded competitor. Getting this layer of intelligence right is essential for forecasting biosimilar market share.
FDA’s Four-Letter Suffix Rule and What It Means for Market Intelligence
All biosimilars approved in the US receive a nonproprietary name consisting of the reference product’s INN (international nonproprietary name) followed by a four-letter suffix, for example adalimumab-atto, adalimumab-adaz, adalimumab-bwwd. The suffix requirement was finalized in FDA guidance in 2017 and was designed to enable pharmacovigilance tracking of adverse events to specific biosimilar products.
The intelligence implication is that prescription data, dispensing data, and adverse event reports are all separable by product at the suffix level. IQVIA’s prescription tracking distinguishes adalimumab-atto (Amjevita) from adalimumab-adbm (Cyltezo) from adalimumab-bwwd (Hadlima), enabling market share analysis by individual biosimilar rather than at the class level. This granularity allows competitive intelligence teams to compare the commercial performance of interchangeable vs. non-interchangeable biosimilars, different launch timing strategies, and different pricing approaches, all within the same reference product market.
State Substitution Laws: The Battleground Below the FDA
FDA approval of a biosimilar, even with interchangeability designation, does not automatically authorize pharmacy-level substitution everywhere in the US. Pharmacy substitution is governed by state law. As of 2025, 49 states and the District of Columbia have enacted biosimilar substitution laws, but the specific requirements vary. Most require pharmacist notification to the prescriber within 48-72 hours of substitution, recordkeeping, and, in some states, patient notification.
The commercial effect of these notification requirements has been debated. The original concern from brand manufacturers was that notification requirements would discourage pharmacists from substituting, reducing biosimilar uptake. The empirical evidence from insulin glargine biosimilar substitution, after Semglee received interchangeability designation in 2021, suggests that notification requirements do not substantially reduce substitution rates when payer formularies support biosimilar use, but they do slow the initial months of substitution as pharmacy systems are updated.
Which States Have Automatic Substitution for Biosimilars?
No state has implemented completely automatic substitution equivalent to the AB-rated generic substitution model for small molecules. All state biosimilar substitution laws include at least a prescriber notification requirement or a prescriber opt-out mechanism. California’s biosimilar substitution statute (California Business and Professions Code Section 4073.5) is among the most permissive, requiring pharmacist notification to the prescriber within five business days and patient notification, but not requiring prescriber pre-authorization. Florida, Texas, and New York have similar models.
The states with more restrictive substitution laws, requiring prescriber authorization before substitution rather than just post-substitution notification, effectively require biosimilar developers to drive demand through prescriber education and sales force effort rather than relying on pharmacy-level switching. This changes the commercial model substantially: more sales force investment, longer conversion timelines, and higher customer acquisition costs per unit than in states with notification-only regimes.
How Interchangeability Changes the Commercial Forecast for Biologics Like Adalimumab
For adalimumab specifically, the interchangeability designations granted to Cyltezo (Boehringer Ingelheim) and Hadlima (Samsung Bioepis/Organon) as of 2023 should theoretically enable pharmacy-level switching in states with permissive substitution laws. The empirical reality, as noted in the earlier Humira section, is that payer formulary placement driven by rebate contracts has dominated over interchangeability designation as the primary determinant of actual switching.
The commercial forecast implication: for biosimilars of high-rebate brands, interchangeability adds approximately 5-10 percentage points of additional market share over non-interchangeable biosimilars in the 24 months post-launch, based on IQVIA biosimilar performance data. This is a materially smaller effect than the theoretical substitution model would predict. The correct competitive forecast adjusts for formulary dynamics first, then adds the interchangeability premium on top of the formulary-adjusted base case.
Manufacturing and Supply Chain Intelligence for Generic Entry
A competitor can have FDA approval for an ANDA and still be months or years away from commercial launch if their manufacturing infrastructure is not ready. FDA warning letters, import alerts, consent decrees, and facility inspection observations are public documents that track manufacturing quality issues in real time and provide advance warning of launch delays that would not otherwise be visible from regulatory filing databases alone.
API Sourcing Concentration: Why India and China Dominate and What the Risks Are
Approximately 80% of the active pharmaceutical ingredient (API) supply for US generic drugs is manufactured in India or China, with India alone accounting for roughly 40% of finished generic drug imports by volume. This concentration creates two types of competitive intelligence signals: supply chain risk signals for manufacturers dependent on concentrated API sources, and competitive entry timing signals when API suppliers face regulatory issues.
The FDA maintains import alerts that restrict entry of products from specific facilities. Import Alert 66-40, which restricts entry of drugs manufactured at facilities in India and elsewhere that have not met FDA current good manufacturing practice (cGMP) standards, affects dozens of active generic manufacturers. When a leading ANDA filer’s primary API supplier appears on Import Alert 66-40, that filer’s commercial launch timeline extends by at minimum several months, more typically 12-18 months, creating an opening for competitors with alternative API sources.
Tracking API supplier relationships for generic ANDA filers requires combining drug master file (DMF) citations in ANDA applications (available in FDA’s DMF database with supplier identity redacted but cross-referenceable to public information) with import alert and warning letter databases. This synthesis reveals which generic manufacturers are exposed to specific supply chain risks, allowing competitive positioning adjustments when those risks materialize.
How to Track FDA Warning Letters and Import Alerts as Entry Delay Signals
FDA warning letters for pharmaceutical manufacturers are public documents published on the FDA’s website typically within 30 days of issuance. Warning letters cite 21 CFR Part 211 (cGMP for finished pharmaceuticals) or 21 CFR Part 210 (minimum cGMP requirements) violations and require a response from the manufacturer within 15 business days.
The commercial significance of a warning letter depends on its specific observations. Data integrity violations, which cite fabrication or manipulation of manufacturing records, typically result in the longest remediation timelines, often 18-36 months before the FDA will re-inspect and approve pending applications from that facility. Equipment and cleaning validation failures are typically remediable in six to 12 months. Process control failures fall somewhere in between.
For a competitive intelligence analyst tracking a specific ANDA filer, the receipt of a warning letter by that company’s primary dosage form manufacturing site means that any ANDA citing that facility will not receive final approval until the warning letter is resolved. This is a definitive launch delay signal. In markets where the warning-letter recipient is the first-to-file generic holding 180-day exclusivity, the delay extends to all other ANDA applicants as well, because the FDA cannot grant approval to non-first-filers while the first-filer’s exclusivity clock has not started running.
The Role of Facility Inspections in Generic Launch Timing
Beyond warning letters, routine FDA pre-approval inspections (PAIs) are a less publicized but equally important launch timing variable. The FDA conducts PAIs at the manufacturing facility for all generic drug applications before granting final approval. PAI scheduling can add three to 12 months to the approval timeline depending on FDA workload, facility location, and whether the application is high-priority under the Drug Competition Action Plan’s complex drug designation.
Post-COVID, the FDA’s international inspection program experienced significant backlogs. FDA inspection data published in its annual reports showed that the backlog of overseas facility inspections reached historic highs in 2021 and 2022, with some facilities in India and China awaiting inspection for more than 24 months past their scheduled dates. While the FDA has made progress in clearing this backlog, manufacturers heavily dependent on Indian and Chinese API suppliers face inspection-driven delays that US-based or European manufacturers with more accessible facilities do not.
Post-COVID API Shortages and How They Shifted Generic Launch Strategies
The COVID-19 pandemic’s disruption of global API supply chains had lasting effects on generic launch strategy that continue to shape competitive intelligence in 2025. Several medium-volume generic manufacturers shifted their sourcing strategies to include US or European-manufactured APIs for select products, accepting higher COGS in exchange for supply chain resilience and better FDA inspection access.
The FDA’s domestic drug manufacturing initiative, including incentives under the National Defense Authorization Act and the CHIPS and Science Act-adjacent provisions for pharmaceutical supply chain security, created a modest but measurable increase in US-based API investment between 2022 and 2024. Companies including Lannett, Hikma’s US operations, and Pfizer’s CentreOne API manufacturing business have invested in US API capacity for specific molecules. For the competitive intelligence analyst, a competitor’s shift to domestic API sourcing signals both improved supply chain resilience and potentially higher manufacturing costs, affecting the competitor’s sustainable price floor in the post-launch pricing battle.
Pricing Intelligence: What Happens to Market Price After Generic Entry
Generic price erosion follows a well-documented curve that varies by number of entrants, dosage form complexity, API sourcing structure, and whether an authorized generic is present. Understanding the shape of this curve for a specific drug is the commercial intelligence task that links all the upstream patent and regulatory analysis to its financial consequences.
The Generic Price Erosion Curve: 30%, 80%, and 90% Scenarios
The standard generic price erosion model used in pharma commercial planning uses three scenarios based on entrant count:
Two entrants (first generic plus brand AG): Price settles at approximately 60-70% of brand price within 90 days. The presence of only two generics, both bioequivalent to the same product, produces limited price competition because payer mandatory substitution drives volume but does not force aggressive price competition when there are only two sources.
Three to five entrants: Price erodes to 20-40% of brand price within 12-18 months. This range reflects the typical outcome for drugs with annual US sales of $300 million to $1 billion, which attract enough generic development interest to produce a modestly competitive generic market without complete commoditization.
Six or more entrants: Price erodes to 5-15% of brand price within 24-36 months. This is the commodity scenario for primary care oral solids with uncomplicated IP, high-volume prescribing, and low API manufacturing barriers. Lisinopril, metformin, atorvastatin (post-2012), and omeprazole all followed this curve.
Complex dosage forms, injectables, and modified-release formulations erode more slowly because manufacturing barriers limit the number of qualified entrants. Inhalation products, for example, require specialized manufacturing and device compatibility, and markets like fluticasone propionate/salmeterol (Advair’s generic market) took years longer to commoditize than oral solid dosage markets with equivalent patent landscapes.
Biosimilar Price Discounts vs. Generic Price Erosion: A Structural Comparison
Biosimilar price competition does not follow the generic erosion curve. The structural reasons for this divergence are well-documented but frequently understated in commercial forecasts.
Generic drugs achieve rapid and deep price erosion because: bioequivalence guarantees that all AB-rated generics are interchangeable at the pharmacy level, payers mandate substitution through formulary tiering, and manufacturing costs for oral solid dosages are low relative to product revenue, enabling price competition to the manufacturing cost floor.
Biosimilars achieve slower and shallower discounts because: biologic manufacturing costs are 10-50x higher per gram than small-molecule API costs, therapeutic equivalence (interchangeability) is harder to establish and rarely carries automatic substitution rights, payer formulary placement is determined by rebate negotiations rather than automatic substitution mandates, and physician and patient comfort with switching is lower for complex biologic therapies than for established small-molecule generics.
The empirical result: biosimilars launched in the US between 2016 and 2024 achieved average launch price discounts of 15-30% below reference product list price, compared to 20-40% average erosion in small-molecule generic launches at the same stage. The spread widens dramatically in the second and third year post-launch. Generic markets are typically 80-90% eroded two years after first entry. Biosimilar markets, except in infliximab where hospital formulary management drove aggressive conversion, are typically 20-40% eroded at the same two-year mark.
How Rebate Walls and Formulary Access Slow Biosimilar Uptake
A rebate wall is an informal term for the contractual structure by which a brand biologic manufacturer offers payer rebates conditioned on preferred formulary placement, with rebate rates calibrated to make generic or biosimilar competition economically unattractive to the payer even at significant biosimilar list price discounts.
The mechanics work as follows. A brand offers a PBM a 40% rebate on list price in exchange for exclusive formulary tier-1 placement with no non-preferred or excluded tier for biosimilars. The effective net price to the payer is 60% of list. A biosimilar entering at 20% list discount offers a net price of 80% of brand list without additional rebates, which is 33% more expensive than the post-rebate brand. The biosimilar would need to offer rebates exceeding 25-30% of its own list price to match the brand’s post-rebate net price, which at 20% list discount is impossible without pricing below manufacturing cost.
This rebate wall dynamic is why the adalimumab biosimilar market in the US looked so different from the European market. In Europe, where biosimilar tendering models and formulary exclusion mechanisms differ, adalimumab biosimilars captured 70-80% market share within 18 months of first launch in Germany, France, and the Nordic countries. In the US, AbbVie’s rebate structure maintained over 85% share two years after biosimilar entry despite eight competing products.
What Humira Biosimilar Pricing in 2024 Tells You About the Adalimumab Market
By 2024, two distinct adalimumab pricing tiers had emerged in the US market. Manufacturers including Amgen (Amjevita), Boehringer Ingelheim (Cyltezo), and Organon (Hadlima) had adopted ‘high-list’ versions of their biosimilars, priced at only 5% below Humira’s list price, alongside ‘low-list’ versions priced at 55-80% below list. The dual-pricing strategy was designed to match the payer rebate model: high-list products generate room for PBM rebate negotiations; low-list products compete on net price in self-pay, Medicare Part D low-income subsidy, and value-based formulary segments.
The existence of dual-list-price biosimilars tells analysts something specific about the payer landscape: a meaningful segment of payer volume is accessible only at high net prices through rebate-matching, while another segment is price-sensitive and accessible at low net prices. The proportions of each segment are themselves intelligence data, trackable through IQVIA national sales data and payer formulary audits by specialty pharmacy tracking services.
Building a Real-Time Competitive Intelligence System
The components described above, patent databases, ANDA filing monitors, litigation docket alerts, manufacturing inspection trackers, and pricing databases, must be integrated into a workflow that delivers actionable intelligence before commercial decisions are due, not after. Most organizations have access to some of these data sources but lack the workflow infrastructure to convert raw data into timely commercial decisions.
Tools, Databases, and Monitoring Workflows That Work
A functional pharmaceutical competitive intelligence system requires four operational components:
First, a patent and exclusivity tracker that monitors the Orange Book and Purple Book for changes to patent listings, new exclusivity grants, and pediatric exclusivity notifications on all drugs of commercial interest. DrugPatentWatch provides this function with automated alerts and historical patent litigation data that would take weeks to compile from primary sources. For biosimilars, the Purple Book equivalent requires separate monitoring workflows because its data structure differs from the Orange Book.
Second, an ANDA and BLA filing monitor that tracks the FDA’s Paragraph IV certification list for new filings, the FDA’s ANDA approval database for new tentative and final approvals, and the PACER docket system for new case filings and significant case events in active ANDA litigation. This workflow requires daily or near-daily monitoring, not weekly, because a 45-day response window runs from a Paragraph IV notification that may itself arrive with delay.
Third, a manufacturing quality tracker that monitors the FDA’s warning letter database, import alert database, and Form 483 observation disclosures for facilities associated with key ANDA filers. This monitoring should be triggered by any facility associated with a competitor in a drug of commercial interest and should generate an automatic impact assessment when a quality event occurs.
Fourth, a pricing and formulary tracker that monitors payer formulary tier changes, wholesale acquisition cost (WAC) changes for biosimilar and generic products, and specialty pharmacy contract announcements. IQVIA’s suite of pricing databases provides the most comprehensive US pricing data, supplemented by CMS drug spending data for Medicare and Medicaid-significant markets.
How to Structure Internal Patent Watch Alerts
Patent watch alerts should be tiered by urgency and by the type of decision they inform. Three tiers work for most commercial organizations:
Tier A (immediate escalation, same-day response required): new Paragraph IV certifications against any product with annual US sales above $200 million; any court filing indicating settlement or judgment in active ANDA litigation on these products; FDA approval of a competing generic or biosimilar product.
Tier B (48-hour response): new IPR petitions against Orange Book or Purple Book patents for products of interest; FDA warning letters issued to facilities of the top five ANDA or biosimilar BLA filers for key products; new Paragraph IV filings in the broader drug class (competitor molecules that affect the market even if they do not directly target a specific product).
Tier C (weekly review): new ANDA tentative approvals for products three or more years from expected LOE; new drug master file (DMF) submissions suggesting API development activity; payer formulary tier changes for competitor biosimilars in related therapeutic areas.
Integrating DrugPatentWatch, FDA Databases, and Court Dockets Into One Feed
No single commercial platform aggregates all four components of the competitive intelligence system described above. Building a unified feed requires either custom API integration or a workflow platform that ingests multiple data sources. Several approaches are in active use at major pharma companies:
DrugPatentWatch’s alert API can be configured to trigger on specific drugs, patent numbers, or ANDA applicant names and feeds into internal analytics platforms via webhook or email. Its patent expiry and litigation history data is particularly useful for the LOE modeling layer.
PACER’s Case Locator and RSS feeds for specific courts provide docket-level alerts for case filings and document entries. These require manual filtering to extract commercially relevant documents but are the only real-time source for ANDA litigation events.
The FDA’s Paragraph IV certification list is updated monthly and is downloadable as a machine-readable file, enabling automated comparison against the prior month’s list to identify new certifications. The FDA’s warning letter database is similarly structured for automated monitoring.
Integration platforms including Palantir Foundry, Veeva Vault, and custom Python or R workflows built on public API access are all in active use at pharmaceutical companies of different sizes. The technology choice matters less than the process discipline: who receives the alert, what decision it should trigger, and what the escalation path looks like when a Tier A alert arrives on a Friday afternoon.
When to Trigger a Commercial Response vs. a Legal Response
Not every competitive intelligence signal requires the same response type. Distinguishing between signals that require commercial action (repricing, formulary negotiation, AG launch planning, life-cycle management investment) and signals that require legal action (filing an infringement suit, filing an IPR petition, asserting an AG launch right) is a governance question that most organizations handle through a cross-functional patent committee with standing commercial, legal, and regulatory representation.
The decision framework should be pre-built, not ad hoc. When a Paragraph IV certification arrives, the organization should already have documented answers to: what is the commercial value of the 30-month stay, what is the probability of prevailing in litigation on each challenged patent, what is the cost of litigation versus the benefit of the stay, and what commercial responses (AG, authorized pricing, contracting adjustments) are available if litigation is not pursued or if it is lost. Building this framework before a Paragraph IV certification arrives reduces response time and improves decision quality.
What This Means for Brand Manufacturers Facing Generic or Biosimilar Entry
Brand manufacturers operating in the 24-36 months before LOE have more decision levers than most commercial teams actively deploy. The tendency to view LOE as an inevitable decline, rather than a managed transition with multiple strategic options, understates the commercial opportunity in the pre-LOE period and overestimates the inevitability of post-LOE share loss.
The 12 Months Before LOE: A Tactical Playbook
The 12 months before expected first generic launch are the highest-leverage period for brand commercial strategy. The following actions, all supported by competitive intelligence inputs, should be completed before the LOE event:
Contracting for AG economics: If the brand plans to launch an AG, the contract terms with the AG distributor (often a generic subsidiary or partner) must be negotiated and executed well before launch. AG distribution agreements, volume commitments, and pricing floors all require lead time and internal approval cycles that cannot be compressed into the 45-day window following a Paragraph IV notification.
Payer contract restructuring: For specialty or biologic products, payer rebate contracts often have renewal cycles of 12-24 months. Structuring the next contract cycle to include biosimilar or generic entry provisions, including pre-agreed formulary protection terms for the post-LOE period, requires re-negotiating before the LOE event, not after.
Patient assistance and adherence program adjustments: For complex dosage forms or biologics where patient switching costs are high, enhancing adherence programs in the pre-LOE period retains patients who are less likely to switch during an involuntary formulary-driven transition if they are actively enrolled in brand support programs.
LOE-specific commercial education for the salesforce: The salesforce needs accurate information about the competitive landscape, not generic narratives about ‘fighting back.’ They need to know specifically which generic or biosimilar products are approved or expected, what their clinical differentiation profile looks like (if any), and what the formulary placement situation is, all of which flow from the competitive intelligence system described above.
Authorized Generic vs. Life-Cycle Management: Choosing the Right Defense
The choice between an AG strategy and a life-cycle management strategy is not mutually exclusive, but the resource allocation between them must be made before LOE arrives, not during it. Life-cycle management includes formulation improvements (extended-release, abuse-deterrent, co-formulation), indication expansion, delivery device improvements, and line extensions that qualify for new exclusivity periods.
The value of life-cycle management investments depends on whether the new formulation or indication can maintain clinical preference over the generic product, whether payers will maintain premium formulary tier for the newer version, and whether the patent estate protecting the life-cycle extension is robust enough to withstand challenge. Reformulation patents protecting extended-release versions have been invalidated in multiple ANDA litigations, and the commercial value of a reformulation that payers immediately equate with the generic product is minimal.
The AG strategy is simpler and often more immediately valuable for large oral solid dosage markets without complex dosage form differentiation opportunity. For a $2 billion primary-care oral solid entering generic competition, an AG launched at first generic entry can capture 30-50% of the generic volume share, maintaining equivalent revenue per unit to the generic price while reducing the brand’s own revenue only modestly compared to the scenario where a competitor’s generic captures all of the converted volume. Several major pharma companies, including AstraZeneca (through its Prilosec OTC strategy, a related concept), Pfizer (through Greenstone), and Teva’s brand-protection AG launches, have demonstrated the financial viability of this model.
What This Means for Generic and Biosimilar Developers
Competitive intelligence for generic and biosimilar developers runs in the opposite direction from brand intelligence: instead of monitoring who is targeting you, you are monitoring which targets are worth pursuing and which competitive fields are too crowded. The decision to initiate ANDA or BLA development for a specific drug is a multi-million-dollar commitment that should be preceded by a systematic intelligence assessment of the IP landscape, competitive field, regulatory requirements, and commercial opportunity.
How to Prioritize Targets Using IP Intelligence
Target prioritization for generic development begins with the Orange Book analysis described earlier: identifying the key patents, assessing their challenge-ability based on prosecution history and prior art, and determining whether first-to-file exclusivity is achievable. DrugPatentWatch’s ANDA intelligence data shows the number of existing filers per molecule and their Paragraph certification status, which immediately identifies whether first-to-file exclusivity is still available or already taken.
Beyond the IP layer, target economics require modeling the expected number of competitors at the time of anticipated launch, not the number today. A molecule with two Paragraph IV filers today may have eight by the time a new filer completes ANDA development three years hence. Using today’s competitive count systematically overestimates the commercial opportunity for any target with a development timeline longer than 18 months.
Manufacturing complexity is an underweighted variable in most target prioritization models. Sterile injectables, modified-release oral solids, nasal sprays, topical products with complex vehicle systems, and transdermal patches all require specialized manufacturing infrastructure that limits competitive entry. For generic manufacturers with relevant existing manufacturing capabilities, complex dosage forms are attractive because competitive crowding is constrained by manufacturing barriers, not just IP barriers. For manufacturers without those capabilities, the capital investment required to enter a complex dosage form market may not be recoverable at the expected generic price.
Reading the Competitive Landscape Before Filing an ANDA or BLA
Before committing to an ANDA or BLA, a complete competitive landscape assessment covers six dimensions:
Patent landscape: every Orange Book or Purple Book patent, its expiry, challenge history, and prosecutorial strengths and weaknesses. IPR filings against these patents, whether successful or unsuccessful, reveal what prior art has already been surfaced.
ANDA/BLA filer landscape: number of existing filers, their Paragraph certifications, tentative approval status, manufacturing readiness, and settlement history. A filer with a tentative approval but unresolved warning letter is a different competitive threat than a filer still in FDA review.
Brand defense posture: historical AG launch behavior, life-cycle management investment, litigation aggressiveness, settlement flexibility, and rebate strategy. A brand that has historically settled quickly on favorable terms for generic filers is a different commercial environment than a brand that litigates to judgment on every challenged patent.
Market structure: payer formulary dynamics, therapeutic substitutability, physician prescribing patterns, and retail pharmacy concentration for the relevant drug class. For biosimilars, hospital formulary committee composition and historical substitution behavior for prior biosimilar launches in the same therapeutic area are particularly important inputs.
Regulatory pathway clarity: whether the FDA has issued product-specific guidance for the drug, whether bioequivalence or biosimilarity criteria are established or still in development, and whether the drug is classified as complex under the FDA’s complex drug evaluation framework, which affects both the development investment required and the approval timeline.
Financial modeling: integrating all five dimensions into a net present value model for the development program, with explicit probability weights for first-to-file exclusivity achievement, litigation outcome, manufacturing approval timing, and post-launch price erosion trajectory. For biosimilar programs, development costs typically run $100-250 million, requiring a commercial opportunity well above $500 million in peak annual revenue to generate an adequate return even in optimistic scenarios.
Key Takeaways
Paragraph IV certifications are the primary public signal of generic competitive intent. A monitoring workflow covering the FDA’s monthly certification list, supported by PACER docket alerts for resulting litigation, provides the earliest commercially actionable warning of competitive entry.
Orange Book patent listings reveal not just expiry dates but patent type and challenge history. Drug substance, drug product, and method-of-use patents each carry different generic entry implications, and unchallenged late-expiring patents should be analyzed rather than accepted as strong without examination.
Biosimilar competitive intelligence requires a separate analytical framework from generic drug tracking. The regulatory pathway, interchangeability rules, state substitution laws, and payer rebate dynamics all operate differently, and generic-market assumptions systematically underestimate the resilience of biologics’ brand market share post-biosimilar entry.
Settlement entry dates, disclosed in court stipulations and SEC filings, are more reliable predictors of commercial generic and biosimilar launch timing than legal patent expiry dates. Building a settlement monitoring workflow generates earlier and more accurate LOE timelines than patent-expiry-based models.
Manufacturing quality events, specifically FDA warning letters and import alerts affecting key ANDA filers, are reliable launch delay signals that are tracked in real time through public FDA databases and can shift competitive dynamics by months or years.
Rebate walls have proven more effective than patent protection in delaying biosimilar commercial entry in the US, as demonstrated by the Humira adalimumab market. Formulary and payer contract analysis is a required input to any commercial LOE forecast for high-rebate biologics.
DrugPatentWatch provides a structured aggregation of Orange Book patent data, Paragraph IV history, and ANDA filer intelligence that materially reduces the time required to build an accurate competitive IP assessment for any drug of interest.
The 12 months before LOE are the highest-leverage commercial planning period for brand manufacturers. Pre-LOE contracting, AG launch preparation, and payer agreement restructuring all require lead time that cannot be recovered once a generic or biosimilar enters the market.
Generic and biosimilar target prioritization must account for the expected competitive field at the time of launch, not today’s. Modeling competitive crowding three years forward is more accurate than using current ANDA filer counts.
A functional competitive intelligence system requires integration across patent monitoring, regulatory filing tracking, litigation docket alerts, manufacturing quality surveillance, and commercial pricing data. No single commercial platform provides all four components, and the workflow infrastructure to integrate them is itself a competitive asset.
Frequently Asked Questions
1. What is the difference between a Paragraph III and Paragraph IV ANDA certification?
A Paragraph III certification states that the patents listed in the Orange Book for the reference drug will have expired by the time the generic enters the market. The ANDA applicant is agreeing not to launch before the patent expires. No patent litigation is triggered, and the brand receives no advance notice of entry timing other than the ANDA itself. A Paragraph IV certification states that the listed patents are invalid, unenforceable, or will not be infringed. This triggers a notice requirement and allows the brand to file an infringement suit within 45 days to obtain the 30-month stay. Paragraph IV is the aggressive strategy; Paragraph III is the wait-and-see approach. From a competitive intelligence standpoint, Paragraph IV filings are visible and generate litigation records; Paragraph III filings are far less visible until the generic actually receives approval.
2. How do you find out which companies have filed ANDAs for a specific drug?
The FDA’s Paragraph IV certification list provides ANDA applicant names for challenges filed since the FDA Reauthorization Act of 2017 required disclosure. For pre-2017 filings, applicant names are available in approval letters published on Drugs@FDA after final approval. DrugPatentWatch aggregates ANDA applicant data across multiple sources and is the most efficient starting point. For drugs under active Paragraph IV litigation, the defendant names in the ANDA infringement suits (publicly available on PACER) identify filers by name regardless of approval status. For Paragraph III filers, the best public proxy is ANDA approval letters, which disclose applicant names at the time of approval but provide no advance notice.
3. What is 180-day generic exclusivity and when does it get forfeited?
The 180-day exclusivity period is the window during which only the first generic manufacturer to file a substantially complete ANDA with a Paragraph IV certification can be commercially active in the generic market. The FDA cannot grant final approval to any other ANDA during this period. Forfeiture occurs when: the first filer fails to market within 75 days of the earlier of FDA approval or a court decision finding the patent invalid/not infringed; the first filer withdraws its ANDA; the first filer fails to obtain tentative approval within 30 months of filing without adequate justification; or the first filer’s 180-day period has been triggered by a court decision or FDA approval but the filer chooses not to market. Forfeiture monitoring is commercially important for any manufacturer holding second or subsequent ANDA position, because forfeiture allows them to advance to a position equivalent to first entry.
4. What is the BPCIA patent dance and can a biosimilar applicant opt out?
The BPCIA patent dance is the information-sharing and negotiation process under 42 U.S.C. 262(l) between a biosimilar applicant and the reference product sponsor. It involves the biosimilar applicant providing its 351(k) application to the reference product sponsor, followed by patent lists from both parties, negotiation over which patents to litigate, and litigation sequencing provisions. Yes, a biosimilar applicant can opt out of the patent dance by electing not to provide the application to the reference product sponsor. If it does so, it must provide 60 days notice before commercial marketing and accepts that the reference product sponsor can then sue for infringement of any patent it believes is relevant. Several biosimilar developers have found opting out strategically advantageous because it removes the patent dance timeline requirements and allows the biosimilar applicant to control the timing of litigation initiation.
5. How does the FDA’s Complex Drug Evaluation framework affect generic entry timing?
The FDA’s complex drug list, maintained under the Drug Competition Action Plan, identifies drugs where the development of approved generics is particularly challenging due to formulation complexity, unique drug-device combination requirements, or difficult bioequivalence standards. Drugs on the complex list receive additional FDA resources for product-specific guidance development, which clarifies the development requirements for generic applicants and is expected to speed development timelines for complex drugs. However, complex drugs also have longer ANDA review timelines and higher development costs, which reduces the number of developers willing to pursue them. The net commercial effect is that complex drugs tend to see fewer generic entrants, slower price erosion post-entry, and longer lag times between patent expiry and first generic launch than comparable-volume simple oral solid dosage drugs.
6. How should a biosimilar developer forecast commercial performance when a rebate wall exists?
The starting point is determining which formulary tier the reference biologic holds across the top five PBMs and the top ten health plans by covered lives. If the reference product holds exclusive or preferred tier-one placement backed by high rebates, the biosimilar forecast must model access at the non-preferred tier rather than assuming the same formulary position as the brand. Non-preferred tier access means patients face higher cost-sharing, reducing demand relative to a co-equal formulary model. The forecast should then model what net price the biosimilar needs to offer PBMs to achieve preferred tier placement, and compare that net price to the biosimilar’s manufacturing cost plus target margin. If the math does not work at preferred tier, the biosimilar developer should model a segmented strategy: winning access in Medicare Part D (where the IRA’s inflation reduction provisions have changed incentives), hospital system formularies (where different contracting dynamics apply), and state Medicaid programs (which have separate formulary processes) rather than assuming parity with the reference product in commercial insurance.
7. What do FDA Form 483 observations mean for a competitor’s ANDA approval timeline?
A Form 483, officially an Inspectional Observations form, is issued to a drug manufacturer at the conclusion of an FDA inspection that identified deviations from cGMP. Form 483s are not warnings; they are observations that require a response. The manufacturer typically has 15 business days to submit a corrective action plan. If the FDA is satisfied with the response, no further action may be taken and ANDA approvals from the facility can proceed. If the FDA is not satisfied, the next steps include a warning letter or, in severe cases, consent decree proceedings. The commercial intelligence implication depends on the severity and nature of the 483 observations. Data integrity observations are high-risk and frequently precede warning letters. Equipment and process observations are lower risk if the corrective action plan is credible. Tracking 483s for competitor facilities requires FOIA requests because Form 483s are not automatically public, unlike warning letters, but they are often released in response to FOIA submissions and are sometimes disclosed voluntarily by companies in earnings calls when the observations are not material.
8. When does a pay-for-delay settlement violate antitrust law after FTC v. Actavis?
Post-Actavis, a settlement can violate antitrust law when the value transferred from the brand to the generic challenger, in whatever form, constitutes an inducement for the generic to delay entry that is not explained by the independent value of the services or rights provided. The analysis focuses on whether the payment is large and unexplained. An above-market royalty on an unrelated drug license, an exclusion of the brand’s own AG from the market for a period that benefits the settling generic, or a volume cap that restricts generic output all qualify as value transfers subject to antitrust scrutiny. The rule of reason test means neither party can predict with certainty whether a specific settlement will survive challenge, but settlements where the brand payment can be credibly justified by independent commercial value are more defensible than settlements where the only explanation for the payment is delayed generic entry.
9. How far in advance can a biosimilar developer predict the US launch date for a reference biologic?
For a reference biologic with disclosed patent expiry dates, no active litigation, and no settlement, the earliest reliable launch date prediction is approximately 24-36 months from the expected composition-of-matter patent expiry, accounting for BLA development and review time. If the brand has already settled with leading biosimilar developers, the settlement entry dates are typically disclosed in public SEC filings or court documents and provide the most reliable launch date signal available, sometimes years in advance. For biologics still in primary patent protection without settlement disclosures, the practical launch date uncertainty window spans three to five years, making financial modeling highly sensitive to patent expiry assumptions. Monitoring IPR petitions at the PTAB against the reference biologic’s patents provides earlier signals about which patents are likely to survive, narrowing this uncertainty range.
10. What is the commercial impact of an orphan drug designation on generic or biosimilar entry timing?
Orphan Drug Designation (ODD) under the Orphan Drug Act grants seven years of market exclusivity for the designated indication from the date of FDA approval. During the orphan exclusivity period, the FDA cannot approve a marketing application for the same drug for the same indication from a competitor, including a generic or biosimilar. For small molecules, the seven-year orphan exclusivity period often runs concurrently with compound patent protection, reducing its independent value. For biologics, where composition-of-matter patent protection may not extend as long relative to approval date as it does for small molecules, orphan exclusivity can be a significant independent barrier to biosimilar entry. Monitoring orphan exclusivity expiry alongside patent expiry is a required step in any LOE analysis for drugs in rare disease indications, where orphan designation is common.
References
Food and Drug Administration. (2019). Drug Competition Action Plan: Progress report 2019. U.S. Department of Health and Human Services. https://www.fda.gov/media/109456/download
Food and Drug Administration. (2017). Nonproprietary naming of biological products: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/93218/download
Federal Trade Commission. (2013). FTC v. Actavis, Inc., 570 U.S. 136. U.S. Supreme Court. https://www.ftc.gov/enforcement/cases-proceedings/ftc-v-actavis
Federal Trade Commission. (2023). In the matter of AbbVie Inc. (Humira biosimilar settlement investigation). FTC Docket. https://www.ftc.gov/legal-library/browse/cases-proceedings
Federal Trade Commission. (2023). In the matter of Bristol-Myers Squibb Company and Celgene Corporation (Revlimid). FTC Administrative Complaint. https://www.ftc.gov/legal-library/browse/cases-proceedings
Hatch-Waxman Act, Drug Price Competition and Patent Term Restoration Act of 1984, Pub. L. No. 98-417, 98 Stat. 1585 (codified at 21 U.S.C. § 355).
Biologics Price Competition and Innovation Act of 2009, Pub. L. No. 111-148, §§ 7001–7003, 124 Stat. 119 (codified at 42 U.S.C. § 262).
Medicare Prescription Drug, Improvement, and Modernization Act of 2003, Pub. L. No. 108-173, 117 Stat. 2066 (180-day exclusivity forfeiture provisions at 21 U.S.C. § 355(j)(5)(D)).
IQVIA Institute for Human Data Science. (2024). Biosimilars in the United States 2019–2023: Competition, savings, and sustainability. IQVIA. https://www.iqvia.com/insights/the-iqvia-institute/reports
AbbVie Inc. (2022). Annual report on Form 10-K for the fiscal year ended December 31, 2022. U.S. Securities and Exchange Commission. https://www.sec.gov/cgi-bin/browse-edgar?action=getcompany&CIK=abbv
Bristol-Myers Squibb Company. (2022). Annual report on Form 10-K for the fiscal year ended December 31, 2022 (Revlimid settlement disclosures). U.S. Securities and Exchange Commission. https://www.sec.gov/cgi-bin/browse-edgar?action=getcompany&CIK=bmy
Merck & Co., Inc. (2024). Annual report on Form 10-K for the fiscal year ended December 31, 2023 (Keytruda patent disclosures). U.S. Securities and Exchange Commission. https://www.sec.gov/cgi-bin/browse-edgar?action=getcompany&CIK=mck
Feldman, R., & Frondorf, E. (2017). Drug wars: A new generation of generic pharmaceutical delay. Harvard Journal on Legislation, 53(2), 499–543.
Hemphill, C. S., & Sampat, B. N. (2012). Evergreening, patent challenges, and effective market life in pharmaceuticals. Journal of Health Economics, 31(2), 327–339. https://doi.org/10.1016/j.jhealeco.2012.01.004
U.S. Patent and Trademark Office. (2023). Inter partes review statistics through Q4 FY2023. USPTO. https://www.uspto.gov/patents/patent-trial-and-appeal-board/statistics
DrugPatentWatch. (2024). Pharmaceutical patent intelligence database. Medicine Equity Partners. https://www.drugpatentwatch.com
Engelberg, A. B., Kesselheim, A. S., & Avorn, J. (2009). Balancing innovation, access, and profits: Market exclusivity for biologics. New England Journal of Medicine, 361(20), 1917–1919. https://doi.org/10.1056/NEJMp0908496
California Business and Professions Code § 4073.5 (2023). (Biosimilar substitution at pharmacy level, State of California).
Food and Drug Administration. (2022). Considerations in demonstrating interchangeability with a reference product: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/124907/download
Grabowski, H., Guha, R., & Salgado, M. (2014). Biosimilar competition: Lessons from Europe. Nature Reviews Drug Discovery, 13(2), 99–100. https://doi.org/10.1038/nrd4230
Sanofi-Aventis U.S. LLC v. Mylan Inc., No. 1:17-cv-09105 (D.N.J.) (Paragraph IV ANDA litigation, illustrative docket reference).
SAS Institute Inc. v. Iancu, 584 U.S. 357 (2018). (PTAB IPR institution scope requirement).