AbbVie held Humira’s U.S. market alone for twenty years. Not because it held one brilliant patent, but because it built a fortress of 136 of them. When the dust settled, biosimilar manufacturers who had been preparing launches for years found themselves staring at a wall of overlapping intellectual property that made litigation a near-certainty before a single vial shipped.
That is a patent thicket in its mature, industrial form. It is not accidental. It is designed, staffed, budgeted, and executed by IP departments that treat exclusivity extension as a core business function, not a legal afterthought. The strategy has a common name in academic and regulatory circles: evergreening. The name is almost too pleasant for what it describes, which is the systematic layering of secondary and tertiary patents onto a mature drug to push generic and biosimilar entry years, sometimes decades, beyond what a single 20-year compound patent would allow.
This article dissects how that system works, which legal tools fuel it, which drugs have been its most visible beneficiaries, and what the realistic competitive landscape looks like for any company trying to enter a market controlled by a well-constructed thicket.
What Is a Patent Thicket and Why Does It Exist in Pharma?
A patent thicket is a dense web of overlapping patents covering a single product, such that any competitor seeking to manufacture, sell, or import a generic equivalent must either design around every patent in the cluster, license rights from the originator, or litigate its way through. The concept was first formally described in semiconductor contexts, but pharmaceutical companies adopted and refined it into a strategic discipline.
The incentive structure is straightforward. A small-molecule drug patent granted in, say, 1995 expires in 2015 under the standard 20-year rule. But if the originator files additional patents covering a specific salt form, a particular polymorphic crystal structure, a new dosing regimen, a pediatric formulation, a sustained-release delivery mechanism, and a method of treating a newly identified patient subgroup, the effective market exclusivity extends well past 2015, sometimes to 2030 or beyond. Each of those secondary patents can individually be challenged, but a challenger must win every one of them, while the originator only needs to win one to keep the generic off the market, or at least tied up in litigation for additional years.
The asymmetry is deliberate. Originator companies invest heavily in building portfolios where the filing dates, expiry dates, and claim scopes are staggered so that no single court victory by a generic company clears the path. Tools like DrugPatentWatch make these thickets visible, allowing generic manufacturers and payer analysts to map the patent landscape before committing to an ANDA or biosimilar development program. Without that mapping, a generic company can invest hundreds of millions in manufacturing scale-up only to discover that a cluster of late-filed formulation patents blocks launch regardless of whether the core compound patent has fallen.
How the 20-Year Patent Term Interacts With FDA Exclusivity Provisions
The 20-year patent term runs from the filing date, not the grant date. For a drug that spends a decade in development and clinical trials, the effective market exclusivity from the patent is often only eight to twelve years after approval. Congress recognized this erosion in 1984 through the Drug Price Competition and Patent Term Restoration Act, commonly called Hatch-Waxman. That law created two remedies: the ability to restore up to five years of patent life lost during FDA review (through patent term extension, or PTE), and a framework allowing generic companies to file Abbreviated New Drug Applications (ANDAs) challenging those patents before they expire.
Separate from patents, the FDA also grants data exclusivity periods that block generic filings regardless of patent status. New Chemical Entities get five years of exclusivity. New clinical investigations supporting a new indication add three years. Orphan Drug designations provide seven years for rare disease indications. Pediatric studies triggered by FDA Written Request add six months to existing patent terms. These exclusivities stack, and sophisticated originator companies sequence their clinical programs to maximize them.
The result is that the effective exclusivity period for a blockbuster drug rarely reflects the nominal patent expiry date. It reflects the last-expiring patent, the last-expiring exclusivity period, and the resolution date of any outstanding litigation, whichever comes last.
The Orange Book: How Originator Companies Control the Patent Listing Process
The FDA’s Orange Book, formally called Approved Drug Products with Therapeutic Equivalence Evaluations, is the central registry that makes Hatch-Waxman work. Originator companies self-certify patents for listing in the Orange Book, subject to statutory requirements that listed patents must claim the approved drug or a method of use. The FDA does not independently verify the legal validity of listed patents before adding them; it performs a ministerial review of whether the submission is facially complete.
This self-certification mechanism creates an obvious strategic opportunity. Companies have listed patents of questionable scope or relevance, triggering automatic 30-month stays on ANDA approval when generics file Paragraph IV certifications challenging those patents. The 30-month stay runs from the date the originator receives notice of the Paragraph IV filing, regardless of whether the patent at issue would ultimately survive litigation. For a brand company with a drug generating a billion dollars annually, a 30-month stay bought through litigation on even a weak secondary patent is worth $2.5 billion in protected revenues.
The America Invents Act of 2011 and subsequent FDA rulemakings have tried to tighten listing standards, but the Orange Book still contains many patents of uncertain relevance. The FTC has brought actions against companies for improper Orange Book listings, most recently targeting insulin and inhaler manufacturers in 2023 for listing device patents that the agency argued did not legitimately claim the approved drug product itself.
The Core Legal Tools of Evergreening: A Taxonomy
Evergreening is not a single strategy. It is a toolkit. Different companies emphasize different tools depending on the drug’s chemistry, the competitive landscape, and how much remaining time exists on the core compound patent. Here is how the major mechanisms work in practice.
Polymorph and Salt Form Patents: Blocking the Cheapest Manufacturing Route
Most active pharmaceutical ingredients (APIs) can exist in multiple solid-state forms: different crystal structures called polymorphs, or different salt forms created by pairing the API with a different counterion. These variants can differ in solubility, stability, and bioavailability. A compound patent covers the molecule itself, but a polymorph patent covers a specific crystalline arrangement of that molecule, and a salt patent covers a specific ionic pairing.
The strategic logic is simple: even if the core compound patent expires, a generic manufacturer who tries to use the commercially viable salt form may infringe the salt patent, which can expire years later. Clopidogrel bisulfate, marketed as Plavix, illustrates this precisely. The original clopidogrel compound patent expired years before the bisulfate salt patent. Sanofi-Aventis successfully defended the salt patent against Apotex through multiple rounds of litigation, preserving exclusivity well past the compound patent’s expiry. The eventual settlement allowed generic entry in 2012, but only after years of litigation and a disputed launch by Apotex that generated massive damages claims.
Polymorph patents are particularly difficult to design around because a generic company seeking to use a different crystal form must demonstrate that its form does not convert to the patented form under storage or manufacturing conditions, a regulatory and scientific burden that few generics are willing to absorb on top of standard bioequivalence requirements.
Formulation Patents: How Modified-Release Technology Extends Brand Exclusivity
Drug delivery technology, particularly modified-release formulations, is one of the most common and effective evergreening tools. An originator company whose immediate-release formulation faces generic entry can develop an extended-release version with clinical differentiation, file new patents covering the release mechanism, file a new NDA for the reformulated product, and actively promote conversion of its patient base to the new product before generic entry on the old formulation.
AstraZeneca deployed this strategy with omeprazole (Prilosec) and esomeprazole (Nexium). Omeprazole is a racemic mixture; esomeprazole is the S-enantiomer. AstraZeneca filed new patents on esomeprazole and launched Nexium as Prilosec’s generic entry approached. Critics argued the clinical differentiation was marginal. Regulators did not block the strategy. Generic omeprazole launched on schedule, but by that point AstraZeneca had shifted prescriptions and formulary placements to Nexium, which had its own patent wall intact. This is sometimes called the ‘product hop,’ and it is a recognizable pattern across therapeutic categories.
Method-of-Use Patents: Protecting Indications, Not Molecules
A method-of-use patent covers the act of using a drug to treat a specific condition, rather than the drug’s chemical structure. These patents can be filed long after the original compound patent, when clinical data supports a new indication. They extend the Orange Book listing for the new indication and can generate a Paragraph IV certification dispute even when the compound itself is generic.
The challenge for generic companies is that method-of-use patents do not directly block manufacture of the drug; they block use for the patented indication. A generic company can carve out the patented indication from its label, a practice called ‘skinny labeling,’ to launch on the remaining unpatented indications. But if physicians continue to prescribe the drug for the carved-out indication because it is the primary clinical use, the originator may sue for induced infringement, arguing that the generic’s labeling foreseeably leads physicians to use the drug in an infringing manner.
The GSK v. Teva case involving carvedilol (Coreg) played out exactly this tension. GSK held method-of-use patents covering carvedilol for heart failure. Teva carved that indication from its label and launched. A jury found induced infringement. The Federal Circuit reversed, then the Supreme Court vacated and remanded. The case produced years of litigation and legal uncertainty that chilled skinny-label launches industry-wide during the proceedings. As of 2025, the rules around induced infringement through skinny labeling remain contested enough that most generic companies approach method-of-use patent situations with significant caution.
Pediatric Exclusivity: How a Six-Month Extension Can Be Worth Billions
The Best Pharmaceuticals for Children Act authorizes the FDA to request that originator companies conduct pediatric studies of their drugs. If the company completes the studies as requested, it receives six months of additional exclusivity tacked onto the end of each existing patent and exclusivity period for the drug, regardless of whether the pediatric indication is actually approved or commercially significant.
Six months sounds modest. On a drug with $5 billion in annual U.S. sales, six months of protected revenue is $2.5 billion. Several originator companies have been documented conducting minimal-cost pediatric studies, primarily to capture this exclusivity bonus, rather than out of genuine pediatric medical need. Abbott’s pediatric study on Depakote and the timing of subsequent exclusivity claims drew scrutiny from the Government Accountability Office in its 2007 report on pediatric exclusivity, which found significant commercial gains accruing from programs with uncertain pediatric benefit [1].
The six-month add-on attaches to every listed patent and every existing exclusivity period. A drug with three Orange Book patents and a five-year NCE exclusivity gets six extra months on each. The cumulative effect can push the last date of exclusivity protection substantially further than any single component would suggest.
Citizen Petitions as Delay Mechanisms: The FDA’s Procedural Vulnerability
The FDA’s citizen petition process allows any party to petition the agency on regulatory matters before final action. Originator companies sometimes file citizen petitions near the anticipated approval date of a competing generic, raising scientific or regulatory concerns that require the FDA to respond before approving the generic. The FDA must respond within 150 days under FDAAA 2007 reforms, but the response can come at any time within that window, and the petition process has historically allowed additional delays that coincide, not coincidentally, with brand sales periods.
The FTC documented this pattern extensively in its 2012 report ‘Navigating the Citizen Petition Process,’ finding that many petitions were filed strategically near ANDA approval dates and that the FDA denied a high proportion of them as unsupported. The FDA subsequently adopted a policy of expediting review of petitions that appear to be filed for delay rather than legitimate regulatory purposes, but the tool remains available and is still used.
Humira’s 136-Patent Fortress: The Most Studied Thicket in Drug History
AbbVie’s adalimumab (Humira) is the textbook case of a patent thicket applied to a biologic drug. The drug launched in 2003. Its core composition-of-matter patent expired in 2016. But through aggressive secondary patent filing covering manufacturing processes, antibody formulations, dosing regimens, specific patient populations, and device designs for the auto-injector, AbbVie assembled a portfolio that the Initiative for Medicines, Access and Knowledge (I-MAK) counted at 136 U.S. patents as of its 2021 analysis [2].
The consequence was that despite biosimilar manufacturers receiving FDA approval as early as 2016, no biosimilar launched in the U.S. until 2023. Every approved biosimilar manufacturer, including Amgen, Samsung Bioepis, Sandoz, and Pfizer, ultimately reached settlement agreements with AbbVie that granted U.S. launch rights beginning January 2023, while European biosimilars launched years earlier due to a different patent landscape and litigation environment.
‘Humira’s U.S. biosimilar delay cost the American healthcare system an estimated $19 billion in excess drug spending between 2016 and 2023, according to analysis published in JAMA Internal Medicine in 2023, comparing U.S. prices to those in markets where biosimilar competition had taken hold.’ [3]
The Humira situation also illustrates how patent thickets interact with biosimilar-specific law. The Biologics Price Competition and Innovation Act (BPCIA) of 2010 established a ‘patent dance’ disclosure process separate from Hatch-Waxman, requiring biosimilar applicants and originators to exchange lists of relevant patents and negotiate which will be litigated before and after launch. AbbVie used this process to control litigation timing, ensure that the most threatening patents were litigated in jurisdictions and sequences favorable to the brand, and negotiate settlements that included launch date restrictions rather than royalties.
How AbbVie’s Settlement Strategy Replaced Litigation Risk With Licensing Control
AbbVie’s approach to the Humira biosimilar landscape was not to win every patent case at trial. It was to reach settlements on terms favorable enough to make litigation unattractive for biosimilar manufacturers, while ensuring those settlement terms still preserved U.S. exclusivity for years beyond European launch dates.
Each settlement granted the biosimilar manufacturer a license to launch in the U.S. at an agreed date, typically January 2023, while also granting earlier launch rights in Europe, where AbbVie’s patent position was weaker. In exchange, the biosimilar manufacturers dropped patent challenges. AbbVie thus avoided the risk of adverse court rulings that might have invalidated key patents, and the biosimilar manufacturers got certainty over their launch timelines rather than years of unpredictable litigation.
From a pure IP strategy perspective, this is settlement as portfolio insurance. AbbVie did not need its entire 136-patent portfolio to withstand legal scrutiny. It needed enough patents to make litigation expensive enough that settlement on AbbVie’s preferred timeline was the rational choice for every biosimilar manufacturer independently. The portfolio’s value was not primarily its courtroom invincibility but its deterrent mass.
Comparable Biologic Thickets: Enbrel, Remicade, and Stelara
Humira is the most visible example, but the pattern repeats across the TNF inhibitor class and beyond. Amgen’s etanercept (Enbrel) faced biosimilar competition that was delayed through a combination of patent portfolios and manufacturing complexity that made biosimilar development technically difficult. Sandoz received FDA approval for an etanercept biosimilar (Erelzi) in 2016 but did not launch until 2023, blocked by patent litigation. Pfizer’s biosimilar (Eticovo) followed a similar trajectory.
Johnson & Johnson’s infliximab (Remicade) saw earlier biosimilar entry than Humira, with Pfizer’s Inflectra launching in 2016 at a discount. But J&J’s contracting strategies with hospital systems and pharmacy benefit managers created non-patent barriers to biosimilar adoption that proved nearly as effective as litigation. The FTC and state attorneys general investigated these contracting practices, and J&J settled related antitrust claims in 2023.
Janssen’s ustekinumab (Stelara) faces patent expiry in 2023 on its core patents, with multiple biosimilar manufacturers including Amgen, Samsung Bioepis, and Fresenius Kabi having received FDA approvals. The launch landscape for Stelara biosimilars in 2024 and 2025 will be the next major test of whether the industry has learned from Humira in terms of settlement structures and launch sequencing.
Small-Molecule Thickets: Gleevec, Revlimid, and the Cancer Drug Playbook
Patent thickets are not exclusive to biologics. Small-molecule oncology drugs have their own documented history of aggressive secondary patent filing and litigation-driven exclusivity extension.
Gleevec/Imatinib: The Indian Supreme Court Ruling That Redrew Global IP Norms
Novartis filed for a patent on a specific beta-crystalline polymorph of imatinib mesylate in India after the compound patent had already been filed. The Indian Patent Office rejected the application under Section 3(d) of the Indian Patents Act, which bars patents on new forms of known substances unless they show significantly enhanced efficacy. Novartis appealed through the Indian court system, ultimately reaching the Supreme Court of India in Novartis AG v. Union of India.
In 2013, the Indian Supreme Court upheld the rejection, ruling that the beta-crystalline polymorph did not demonstrate sufficient therapeutic advantage over the known imatinib compound to warrant patent protection under Indian law [4]. The ruling had no direct legal effect in the U.S., where the polymorph patents were granted and enforced, but it established a legal principle that developing countries could invoke Section 3(d)-type provisions to reject evergreening patents, and it catalyzed significant policy discussion in international IP forums about whether TRIPS flexibilities allowed similar restrictions.
In the U.S., imatinib generics launched in 2016 after the relevant U.S. patents expired or were settled. The drug had cost approximately $92,000 per year at list price. Generic imatinib launched at prices that fell to under $400 per year within two years of generic entry [5], illustrating the scale of price compression that patent thickets delay.
Revlimid (Lenalidomide): How Bristol Myers Squibb Built a Post-2019 Settlement Wall
Lenalidomide (Revlimid), marketed by Celgene and later Bristol Myers Squibb after its 2019 acquisition, is the most commercially significant example of a small-molecule patent thicket in recent memory. Revlimid generated approximately $12.8 billion in global sales in 2021 [6]. Its core compound patent expired in 2019, but through a combination of formulation patents, method-of-use patents covering specific hematologic malignancies, and REMS (Risk Evaluation and Mitigation Strategy) program restrictions on distribution, Celgene constructed barriers that delayed full generic competition.
The REMS program for lenalidomide, called RevAssist, required enrollment for all prescribers, pharmacies, and patients due to the drug’s teratogenicity. Celgene restricted distribution to authorized specialty pharmacies and, critics argued, used REMS requirements to prevent generic manufacturers from obtaining sufficient drug samples for bioequivalence testing, a practice that triggered FTC scrutiny and eventual antitrust litigation.
Celgene settled patent litigation with multiple generic manufacturers, granting volume-capped generic entry beginning in 2022, with full competition only in 2026. The volume-cap structure allowed Celgene and then BMS to continue generating substantial branded revenues while technically allowing generic competition, a settlement design that critics argued constituted an anticompetitive market division rather than a genuine patent license.
The FTC filed a complaint against BMS in 2023 challenging the settlement terms as anticompetitive under FTC Act Section 5, arguing that the volume-cap provisions were not the product of genuine patent dispute resolution but were designed to maintain above-competitive prices by restricting generic volume below what full competition would produce [7].
What the Revlimid Settlement Structure Means for Generic Entry Timelines
The Revlimid case introduces a category of evergreening that operates through settlement design rather than patent filing. Even when a generic company successfully challenges enough of a thicket to reach a settlement, the settlement’s commercial terms can function as a patent extension by restricting the volume, timing, or geography of generic sales.
Regulatory economists at the University of Chicago and the Congressional Budget Office have both noted that volume-capped settlements create outcomes economically indistinguishable from extended exclusivity, even when the settlements nominally permit generic entry [8]. The policy implication is that Hatch-Waxman’s exclusivity framework cannot be evaluated purely by looking at patent expiry dates; the commercial terms of any litigation settlements that precede those dates matter equally.
Paragraph IV Litigation Strategy: How Originators Pick Their Battles
When a generic company files an ANDA with a Paragraph IV certification, it certifies that the listed Orange Book patents are either invalid or not infringed by the generic product. This certification is a legal act of patent challenge, and it triggers a 45-day window during which the originator can file an infringement suit that automatically activates a 30-month stay on FDA approval of the generic.
The strategic question for originators is not whether to sue, but which patents to sue on, in which district, and in which sequence. The choice of patents for the initial complaint matters because the 30-month stay runs from notice regardless of which patents are in suit, and originators generally want to use the 30 months to litigate patents where they have a strong position, while reserving weaker patents for later rounds or allowing them to be settled with favorable terms.
Which District Courts Hear the Most ANDA Patent Litigation and Why It Matters
For most of the 2000s and 2010s, the District of New Jersey and the District of Delaware dominated ANDA patent litigation, together accounting for over 70% of Hatch-Waxman cases. This concentration was not random. Most major pharmaceutical originators are incorporated in Delaware, making it a proper venue. New Jersey hosts many pharmaceutical headquarters and research facilities, creating venue connections. Both courts developed specialized local rules and experienced judges for patent matters.
Venue concentration matters for thicket litigation because district courts develop local culture around patent issues. Delaware’s specialized patent docket has historically moved at a pace that brand companies found manageable for a 30-month stay, while plaintiff-friendly discovery rules in some jurisdictions were more favorable to generic challengers. Patent reform advocacy groups have argued that venue shopping by both originators and generics shapes case outcomes in ways that have nothing to do with patent merits.
The TC Heartland Supreme Court decision in 2017 tightened venue rules for patent cases generally, requiring that corporate defendants be sued in their state of incorporation or where they have a regular place of business and have committed acts of infringement. For ANDA cases, where the generic manufacturer typically has its principal operations, this concentrated cases in states like New York, New Jersey, Indiana, and West Virginia, redistributing case loads from Delaware and creating new judicial learning curves at receiving courts [9].
First-Filer Exclusivity: Why Generic Companies Race to Challenge Patent Thickets
Hatch-Waxman awards 180 days of generic market exclusivity to the first ANDA filer to submit a Paragraph IV certification for a given drug. During that 180-day period, no other generic can launch, effectively giving the first filer a duopoly with the brand for six months before the broader generic market opens. On a large drug, 180 days of first-filer exclusivity can generate hundreds of millions in profits, creating a strong incentive to challenge patent thickets early.
This creates the races to challenge that Hatch-Waxman intended. Multiple generics often file substantially simultaneously on the first day a Paragraph IV filing is legally possible, triggering what is called ‘shared first-filer’ status when two or more ANDAs are filed on the same day. The FDA treats shared first-filers as jointly holding the 180-day exclusivity, reducing each company’s share of the prize but still creating a meaningful duopoly period.
Originators have responded to first-filer races by building thickets with staggered patent expiries, so that successfully challenging the earliest-expiring patents only clears the way to the next layer, reducing the first-filer prize for early challengers who crack the thicket’s outer ring without addressing the inner layers.
Reverse Payment Settlements: When Originators Pay Generics to Stay Out
A reverse payment settlement, also called a pay-for-delay agreement, occurs when an originator pharmaceutical company pays a generic company to drop a patent challenge and delay entering the market. The payment runs from the entity with the stronger commercial position (the originator) to the entity doing the challenging (the generic), which is the reverse of the normal damages-payment direction in patent litigation.
The Supreme Court addressed reverse payment settlements in FTC v. Actavis, Inc. in 2013, ruling that they are not automatically illegal but must be evaluated under a rule-of-reason antitrust standard [10]. The FTC argued that large reverse payments are evidence of an underlying weak patent, because an originator with a strong patent would simply win at trial rather than pay the challenger to go away. The Court agreed that payment size is relevant evidence but declined to adopt a presumption of illegality.
Post-Actavis, cash payments in Paragraph IV settlements became less common, replaced by non-cash consideration such as authorized generic licenses (allowing the generic company to sell a branded-manufacturer generic under the brand company’s NDA), co-promotion rights, supply agreements, and other business arrangements. These non-cash transfers are harder to value and therefore harder to challenge as anticompetitive, even though they serve the same commercial function as cash payments.
Biosimilar-Specific Legal Complexity: The BPCIA’s ‘Patent Dance’ and Its Exploitation
The BPCIA created a distinct patent litigation pathway for biologic drugs, different from Hatch-Waxman. The biosimilar applicant must provide the reference product sponsor (the originator) with its complete biologics license application after filing, initiating a multi-step information exchange and negotiation process about which patents will be litigated. This process is the ‘patent dance.’
The dance has multiple stages: the originator provides a list of patents it believes could be infringed; the biosimilar applicant provides a statement of why it disagrees; the parties negotiate a litigation list; and, if negotiation fails, both parties file lists and the originator sues within 30 days on its chosen patents. After a second notice 180 days before commercial marketing, a second round of litigation on remaining patents can begin.
Originator companies have used the dance to control litigation sequencing in ways that maximize the deterrent effect of large portfolios. By choosing which patents to litigate in the pre-launch phase and reserving others for the post-launch phase, they can force biosimilar manufacturers to calculate the risk of at-risk launches against a second wave of patent litigation even after prevailing in the first wave. AbbVie employed this strategy in multiple Humira biosimilar litigations, as documented in court records from the Northern District of Illinois [11].
Amgen v. Sandoz: The Supreme Court Ruling That Shapes Biosimilar Notice Requirements
The first major Supreme Court interpretation of the BPCIA came in Amgen Inc. v. Sandoz Inc. in 2017. The dispute centered on whether participation in the patent dance is mandatory and what the 180-day commercial marketing notice requirement means for biosimilar launch timing.
The Court held that the patent dance is not mandatory, that biosimilar applicants may opt out, and that in such cases the originator can immediately seek injunctive relief for patent infringement after FDA approval. The Court also held that the 180-day marketing notice cannot be given before FDA approval, meaning the earliest possible commercial launch is 180 days after approval, not 180 days after the BLA filing [12].
The practical effect of Amgen v. Sandoz was to confirm that originators cannot use mandatory dance participation to extract information about biosimilar manufacturing before FDA approval, but also that biosimilar manufacturers cannot use pre-approval notice to compress the post-approval launch timeline. The ruling clarified the litigation calendar but did not reduce the total patent count or narrow the scope of secondary patents that originators can list and enforce.
Interchangeability Designation: The Regulatory Hurdle That Delays Pharmacist Substitution
FDA approval of a biosimilar as ‘biosimilar’ to the reference product does not automatically allow pharmacists to substitute it for the brand without physician authorization, unlike with small-molecule generics, which receive automatic substitution rights through state pharmacy laws when rated therapeutically equivalent in the Orange Book. A biosimilar must separately obtain ‘interchangeable’ designation from the FDA, requiring additional clinical data demonstrating that switching back and forth between the biosimilar and the reference product does not produce clinically meaningful differences.
The interchangeability requirement creates a commercial disadvantage for biosimilars that does not exist for small-molecule generics. Physicians and pharmacists who might automatically substitute a generic in the small-molecule context must actively choose the biosimilar in the biologic context, absent interchangeability designation. Originator companies have used this regulatory distinction in contracting with pharmacy benefit managers, arguing that non-interchangeable biosimilars should not receive automatic formulary preference over the brand.
The FDA granted its first interchangeable biosimilar designation to Viatris’s insulin glargine (Semglee) in July 2021 [13]. As of 2025, interchangeability designations have been granted for a small but growing number of biosimilars, with the FDA’s updated guidance making the pathway somewhat clearer. But the requirement remains a structural advantage for originator biologics that has no equivalent in the small-molecule space.
Regulatory Strategy and Data Exclusivity: Beyond Patent Protection
Patent thickets operate in conjunction with FDA exclusivity periods, and sophisticated originators treat both as integrated tools in their exclusivity management program. Understanding the interaction between patent terms and regulatory exclusivity periods is essential to accurately forecasting loss-of-exclusivity (LOE) dates.
How New Chemical Entity Exclusivity Blocks ANDA Filings, Not Just Approvals
The five-year NCE exclusivity prevents the FDA from accepting an ANDA filing until four years have passed (five years if no Paragraph IV challenge is contemplated). This is a filing bar, not just an approval bar. The four-year filing window is specifically designed to allow an 18-month litigation period plus a 30-month stay to resolve before NCE exclusivity ends, so that generics can conceivably launch at the five-year mark if they prevail quickly.
In practice, NCE exclusivity combined with Orange Book patents produces a scenario where no ANDA can even be filed for four years after approval, litigation cannot begin until filing, the 30-month stay begins at filing notice, and by the time the stay expires, Orange Book patents may still have years of life remaining. The sequence ensures that rapid generic entry is structurally impossible for NCEs regardless of how weak secondary patents might be, because the four-year filing bar means the litigation clock cannot start early enough to matter.
Orphan Drug Exclusivity: When Rare Disease Designations Become Commercial Tools
Orphan Drug designation, available for drugs treating conditions affecting fewer than 200,000 Americans, provides seven years of marketing exclusivity after approval. The program was designed to incentivize development of drugs for diseases where market size is insufficient to recoup R&D investment at normal commercial pricing. It has also been used for drugs where the orphan indication is a subset of a much larger patient population, effectively granting premium exclusivity for a drug with broad commercial potential.
Celgene’s thalidomide (Thalomid) and lenalidomide (Revlimid) both received Orphan Drug designations for multiple myeloma, a condition affecting approximately 34,000 Americans annually. But lenalidomide expanded in clinical use to myelodysplastic syndromes and other hematologic malignancies, all of which could receive separate Orphan Drug applications. Each approved orphan indication generated its own seven-year exclusivity period. Combined with compound patents, formulation patents, and method-of-use patents covering each hematologic indication, the result was a layered exclusivity structure that multiple generic companies found commercially forbidding even after core patent challenges.
REMS Programs as Market Access Barriers: When Safety Becomes Strategy
Risk Evaluation and Mitigation Strategy programs are FDA-mandated safety programs for drugs with serious risks that require management beyond standard prescribing information. REMS programs can require prescriber certification, pharmacy registration, patient enrollment, and laboratory testing. For drugs with REMS requirements, generic manufacturers must demonstrate that their REMS program is the same as or comparable to the brand’s REMS, requiring cooperation between the generic company and the brand company during development.
The FTC has documented cases where originator companies refused to provide generic manufacturers with drug samples needed for bioequivalence testing, citing REMS restrictions, and where originator companies structured their REMS programs in ways that made generic development more difficult. The Creating and Restoring Equal Access to Equivalent Samples (CREATES) Act of 2019 addressed this by giving generic manufacturers a private right of action against originators who refuse to provide access to drugs for testing purposes, but only after lengthy negotiations and demonstrated refusals [14].
The CREATES Act has been invoked in litigation. Amneal Pharmaceuticals sued Noven Therapeutics in 2021 over alleged REMS-related sample denial for a transdermal patch product. The litigation generated discovery about how originator companies document and communicate their REMS access policies, creating a paper trail that subsequent litigation may use. Whether the Act has materially improved generic sample access across the industry remains debated among generic industry trade groups as of 2025.
The Financial Arithmetic of Evergreening: LOE Dates, Revenue Cliffs, and Wall Street’s Reaction
Originator pharmaceutical companies report patent expiry dates and exclusivity end dates in their SEC filings, but those disclosed dates are often optimistic from a competition perspective and can differ substantially from the dates on which generic competition will actually begin to erode revenues. Analysts at investment banks and specialized services like DrugPatentWatch track the full patent landscape, including secondary and tertiary patents not always prominently disclosed, to build more realistic LOE models.
How Analysts Model Revenue Cliffs When Patent Thickets Are Involved
A standard LOE model for a small-molecule drug assumes revenue declines of 70-90% within two years of generic entry, based on historical data across dozens of drug markets. But this model applies to markets with robust generic competition, typically six or more generic entrants. When a patent thicket produces settlement-based entry with volume caps, or single-generic entry under a first-filer exclusivity that the brand company manages through authorized generic competition, the revenue decline is slower and more controlled.
Authorized generics, where the brand company itself markets a generic version of its drug under the originator NDA, can compete directly with the Paragraph IV first-filer during the 180-day exclusivity period, splitting the generic market and reducing the first-filer’s profits. This reduces the first-filer’s incentive to challenge the thicket, since the authorized generic undercuts the very prize that makes the challenge worthwhile. Several originator companies have formal authorized generic programs that are deployed as strategic tools rather than commercial afterthoughts.
The revenue profile of a thicketed drug therefore looks different from a clean-LOE drug: a smaller, more gradual revenue decline, possibly with a prolonged tail of partial generic competition, followed by a steeper decline when the thicket’s last patent expires or is invalidated and full market competition opens. Wall Street models the two scenarios very differently, and analysts who correctly identify the last-patent-expiry date of a thicket rather than the first-patent-expiry date can significantly improve their revenue forecasts for originator companies.
What Loss-of-Exclusivity Means for Drug Pricing: The Speed of Price Erosion
Price erosion after generic entry follows a well-documented pattern. In the first month after generic entry with multiple filers, generic prices are typically 70-80% below brand list price. Within six months, generic prices fall to 20-30% of brand price. Brand price often increases slightly after generic entry, targeting remaining brand-loyal prescribers who are less price-sensitive, while the volume shifts rapidly to generics. For Medicaid programs with mandatory generic substitution, brand volume can fall to single-digit percentages within weeks of generic availability.
For specialty drugs with patient assistance programs, copay cards, and formulary protections, the brand volume decline is slower. Originator companies with sophisticated commercial teams use these tools to preserve brand revenues during what they call ‘competitive entry periods,’ recognizing that the revenue cliff is not a cliff at all for managed brands in managed care environments.
The most commercially damaging LOE scenario for an originator is uncontrolled, rapid generic entry without settlement constraints, which happens when the originator loses key patent trials rather than settling. Pfizer’s experience with atorvastatin (Lipitor) in 2011 illustrates this: following patent expiry, multiple generics entered simultaneously (after the first-filer exclusivity resolved), and Lipitor revenues fell from approximately $10 billion to approximately $3 billion within two quarters [15].
Congressional and FTC Responses to Evergreening: Legislation, Enforcement, and Limitations
The policy response to pharmaceutical patent thickets has been slow, incremental, and frequently outpaced by industry adaptation. Major legislative and enforcement actions have addressed specific tactics, but no comprehensive legislative solution has restructured the incentive framework that makes thicket-building rational for originator companies.
The Inflation Reduction Act’s Drug Pricing Provisions: Impact on Evergreening Incentives
The Inflation Reduction Act of 2022 included several provisions relevant to pharmaceutical pricing, including Medicare drug price negotiation for a defined set of high-cost, single-source drugs, penalties for drug price increases that exceed inflation, and changes to Medicare Part D cost-sharing. The price negotiation provisions apply to drugs that have been on the market for a defined number of years without generic or biosimilar competition.
The IRA’s negotiation provisions create a marginal disincentive to evergreening by reducing the value of extended exclusivity for Medicare-eligible drugs. If a drug faces negotiated Medicare pricing after nine years (for small molecules) regardless of its patent status, the revenue value of each additional year of exclusivity after year nine is reduced because the negotiated price will be lower than the list price. Critics of the law argue that the nine-year threshold was set too high to materially alter originator behavior on thicket-building, while supporters argue the provision will accumulate in impact as the first set of negotiated drugs reaches their price-setting dates.
The first ten drugs subject to IRA negotiation were announced in August 2023, including Eliquis, Jardiance, Xarelto, and Januvia. The negotiated prices, effective in 2026, represent the first direct price controls on Medicare Part D drugs in the program’s history [16].
FTC Enforcement Actions Against Improper Orange Book Listings: The 2023 Campaign
In 2023, the FTC launched a coordinated challenge to dozens of Orange Book patent listings, formally disputing listings it argued did not meet statutory requirements that listed patents must claim the approved drug or method of use. The challenged listings included device patents on insulin auto-injectors and inhaler systems that the FTC argued claimed the device rather than the drug itself, such that generics would not need to infringe the device patent to market a generic drug in any auto-injector device.
The FTC’s position, if sustained by the FDA and ultimately by courts, would remove a category of secondary patents from Orange Book listing and the associated Paragraph IV challenge framework, reducing the automatic 30-month stay availability for these patents. Originator companies challenged the FDA’s authority to act on FTC disputes and argued that device-drug combinations are properly listed when the device is part of the approved combination product.
As of 2025, the dispute has generated significant legal briefing and FDA administrative proceedings but no definitive court ruling on the scope of Orange Book listing requirements. Several drug companies have voluntarily removed disputed device patents from the Orange Book in response to FTC pressure, suggesting that the campaign has already achieved partial deterrent effect even without final judicial resolution.
PREVAIL Act and Patent Challenge Reform Proposals: What Might Change
Congress has repeatedly considered legislation that would modify the inter partes review (IPR) process at the Patent Trial and Appeal Board, which has become a major vehicle for generic and biosimilar companies to challenge patent thickets outside of district court litigation. The IPR process, created by the America Invents Act, allows any party to petition the PTAB to review an issued patent’s validity on prior art grounds. Successful IPR petitions have invalidated numerous secondary pharmaceutical patents, including polymorph patents, formulation patents, and process patents.
Originator industry trade groups, primarily PhRMA and BIO, have advocated for limiting IPR standing, increasing the evidentiary standard for institution of IPR reviews, and restricting the ability of hedge funds and other non-practicing entities to file IPR petitions for financial purposes. Generic industry groups and patient advocates have argued the opposite, seeking to expand IPR accessibility. The legislative balance remains contested, and no comprehensive IPR reform has passed as of 2025.
Generic and Biosimilar Entry Strategies: How Challengers Navigate Thickets
Generic and biosimilar companies facing a patent thicket have four realistic options: design around patents, challenge patents through litigation or IPR, negotiate a settlement that includes a launch date, or wait for the thicket to expire naturally. Each option has distinct cost, risk, and timeline profiles.
IPR Petitions as Thicket-Clearing Tools: Success Rates and Strategic Limitations
The PTAB has instituted and decided IPR reviews on pharmaceutical patents at significant scale since the America Invents Act’s implementation in 2012. Institution rates for petitions seeking review of pharmaceutical patents have historically been around 60-65%, and petitions that are instituted result in final written decisions canceling all challenged claims approximately 75% of the time, with partial invalidations adding to that total.
For thicket-clearing purposes, IPR petitions are most useful for secondary patents that are clearly vulnerable on prior art grounds, such as formulation patents that a generic company can demonstrate were obvious in light of published prior art. They are less useful for method-of-use patents, which often face different legal standards, and for process patents where the prior art may be less accessible in the public record.
The strategic limitation of IPR as a thicket-clearing tool is that it addresses individual patents, not portfolios. A generic company that successfully IPRs five formulation patents may still face litigation on ten additional patents covering delivery systems, patient subpopulations, and co-administration with companion drugs. The PTAB’s estoppel provisions also limit the grounds that can be raised in IPR to prior art that the petitioner reasonably could have raised at the time of the IPR petition, potentially closing off grounds for later litigation challenges if the IPR fails.
Designing Around: When Chemical Modification Avoids Infringement
For small-molecule drugs, a sophisticated generic company may be able to design around specific patents by manufacturing the API using a different synthetic route (avoiding process patents), using a different polymorphic form (avoiding polymorph patents), or formulating the product differently while maintaining bioequivalence (partially avoiding formulation patents). Each design-around must be validated through regulatory testing and patent analysis, and the work must be documented in a way that supports a non-infringement argument if litigation follows.
Design-around strategies require significant investment in medicinal chemistry and pharmaceutical sciences, costs that smaller generic companies cannot absorb and that larger companies must weigh against the probability that the design-around will survive litigation scrutiny. Design-around documents become discovery targets in subsequent patent litigation, and an unsuccessful design-around, or one that was implemented after awareness of the patent, can create willful infringement exposure with treble damages.
Authorized Generic Deals vs. Patent Licenses: Understanding the Commercial Distinction
When an originator company and a generic company settle Paragraph IV litigation, the settlement can take several commercial forms. A patent license grants the generic company rights to make and sell the drug under the originator’s patents from a specified date. An authorized generic arrangement grants the generic company the right to market the originator’s NDA-approved product, typically at a royalty, without requiring the generic to manufacture independently.
The authorized generic arrangement avoids the need for the generic company to independently scale manufacturing for the drug, reducing its capital investment. It also ensures the originator maintains quality control over the generic product and receives ongoing royalties. Critics argue that authorized generic deals reduce the incentive for genuine competition because the originator participates commercially in the generic market rather than facing it as a true competitive threat.
From a patient cost perspective, authorized generic arrangements generate price competition but typically less than full generic competition with independent manufacturers. The authorized generic is typically priced at 10-20% below brand list price in exchange for a formulary position, while independent generics compete more aggressively when they have their own manufacturing economics to optimize.
International Comparisons: Why the Same Drug Has Different Competition Timelines in Different Markets
The Humira example highlighted that biosimilar competition began in Europe years before the U.S., not because the science was different, but because the patent landscape differed and European courts applied different legal standards to secondary patents. This pattern recurs across numerous drugs.
European Patent Opposition: A Faster Path to Thicket Clearance
The European Patent Office (EPO) allows third parties to oppose granted European patents within nine months of grant. The opposition process is faster and less expensive than U.S. district court patent litigation, and the EPO applies an inventive step standard that is generally considered stricter on secondary pharmaceutical patents than U.S. obviousness law. As a result, many polymorph, formulation, and salt form patents that survive in U.S. courts are revoked or narrowed through EPO opposition.
European biosimilar manufacturers used EPO oppositions to clear much of AbbVie’s secondary patent portfolio for Humira before the core composition-of-matter patent expired in 2018 in Europe. This created the conditions for biosimilar launch in Europe in 2018, while the U.S. market remained locked until 2023. The five-year gap in competition onset between the two markets produced substantially lower European prices for adalimumab treatment relative to U.S. prices during that period.
Canada, Australia, and the UK: How Patent Linkage Regimes Affect Generic Entry
Patent linkage, the connection between drug regulatory approvals and patent challenge, exists in different forms in different countries. Canada has a linkage system under the Patented Medicine (Notice of Compliance) Regulations that broadly parallels Hatch-Waxman but applies to a narrower set of patents than the Orange Book. Australia has a linkage system with yet different characteristics. The UK’s system does not link regulatory approval and patent challenges in the same way, meaning generic companies can obtain regulatory approval and then launch at commercial risk pending patent litigation outcomes.
The UK’s at-risk launch environment, where a generic can launch and face damages if it later loses a patent case, creates different strategic incentives than the U.S. system’s automatic 30-month stay. UK generic companies and their litigation teams must make launch-or-wait decisions based on their assessment of patent validity, rather than relying on a regulatory stay to preserve the status quo during litigation. This produces faster generic entry in many cases but also occasional significant damages awards when generic companies lose litigation after launching.
The Role of Specialized Patent Intelligence Tools in Competitive Strategy
Navigating a patent thicket requires systematic patent intelligence. Individual lawyers and scientists reading patent files can miss relevant claims, overlook continuation applications, or fail to identify international patent family members that might inform U.S. claim construction arguments. Tools like DrugPatentWatch provide structured databases of pharmaceutical patent information, including Orange Book listings, patent expiry dates, ANDA filing histories, Paragraph IV challenge histories, and litigation outcomes, in a format accessible to business development professionals, generic company strategists, and payer analysts.
Generic companies use patent intelligence at multiple stages: during target selection, when evaluating whether a product is worth developing against its patent exposure; during formulation development, when identifying which formulation approaches are covered by existing patents; during ANDA preparation, when constructing Paragraph IV certifications; and during settlement negotiations, when assessing the strength of remaining patents relative to the commercial value of a negotiated launch date.
Payer organizations, including pharmacy benefit managers and hospital formulary committees, use patent intelligence to forecast LOE dates and build contracting strategies that time benefit design changes around expected generic availability. When a PBM incorrectly estimates LOE by relying only on the core compound patent expiry date rather than the full thicket landscape, it may make formulary commitments to brand products that prove difficult to exit when expected generic entry fails to materialize on schedule.
Investment banks and hedge funds trading pharmaceutical equity use patent intelligence to identify discrepancies between market-consensus LOE dates and realistic competitive entry timelines. When a thicket extends effective exclusivity beyond what analysts are modeling, the originator company’s stock may be underpriced. When a thicket is weaker than disclosed LOE dates suggest, the originator’s revenue projections may be overstated. Detailed patent mapping of secondary and tertiary patents, the kind available through DrugPatentWatch, drives some of these analytical edges.
How Generic Companies Use Patent Landscape Analysis Before Filing ANDAs
A generic company evaluating whether to develop a product for ANDA filing typically conducts a freedom-to-operate (FTO) analysis covering all Orange Book-listed patents plus any non-listed patents that might be asserted against manufacture, use, or sale of the generic. The FTO analysis involves claim charting, where the generic company’s proposed product and process are compared against the claims of each relevant patent to assess infringement risk.
For a heavily thicketed drug, the FTO analysis may identify twenty or more patents requiring individual analysis, some of which will require opinions from outside patent counsel. The legal fees for this analysis can reach $500,000 to $2 million before a single ANDA is filed, creating a barrier to entry for smaller generic companies and favoring large generic manufacturers who can spread this cost across multiple products. This is one mechanism through which patent thickets reduce generic competition even for patents that would not withstand litigation scrutiny, because the cost of analyzing them deters companies who might otherwise challenge.
Drug-Specific Case Studies: Nexium, Abilify, and Sovaldi
Nexium (Esomeprazole): The Product Hop That Defined a Generation of PPI Competition
AstraZeneca’s move from omeprazole to esomeprazole is the defining case study of the product hop strategy. Omeprazole, the original proton pump inhibitor, was a massive commercial success. As its U.S. patent protection eroded in the early 2000s, AstraZeneca launched esomeprazole under the Nexium brand, patenting the single-enantiomer formulation and claiming clinical advantages in acid suppression that were medically modest but commercially sufficient.
The critical question in product hops is whether the new product represents genuine therapeutic advancement or primarily patent extension. For esomeprazole versus omeprazole, studies showed that esomeprazole provided slightly better acid suppression at equivalent doses in some patient populations, but that the clinical difference was small and arguably not worth the price premium of the new brand. The FDA approved esomeprazole on its clinical merits regardless of the competitive strategy underlying its development.
Generic omeprazole captured the cost-sensitive market segment rapidly after its launch, but Nexium held significant market share through physician brand loyalty, managed care formulary positioning, and AstraZeneca’s direct-to-consumer advertising, which was particularly prominent for Nexium. AstraZeneca spent over $500 million annually on Nexium DTC advertising at the drug’s commercial peak, a level of marketing investment that generic omeprazole competitors could not match [17].
Abilify (Aripiprazole): How Bristol-Myers Squibb Extended Exclusivity Through Multiple Secondary Patents
Aripiprazole (Abilify), an atypical antipsychotic marketed jointly by Bristol-Myers Squibb and Otsuka, faced patent expiry on its core compound patent in October 2014. Multiple generic manufacturers had filed ANDAs and resolved their Paragraph IV certifications through settlements that allowed generic entry in April 2015, after BMS’s contractual protections with Otsuka expired. Within months of generic entry, aripiprazole became one of the fastest generic switches on record, with generic aripiprazole capturing over 80% of prescriptions within six months [18].
BMS and Otsuka filed secondary patents covering depot formulations (long-acting injectables) of aripiprazole, marketed as Abilify Maintena. The depot formulation represents a genuine pharmaceutical advancement for patients who benefit from monthly rather than daily dosing, but it also effectively extended brand revenues through a next-generation product with its own patent protection running well past 2015. As of 2025, Abilify Maintena continues to generate branded revenues while oral aripiprazole is generically available at commodity prices.
Sovaldi and Harvoni: Short-Exclusivity Windows in a Disease Category Solved Too Fast
Gilead’s sofosbuvir (Sovaldi) and sofosbuvir/ledipasvir (Harvoni) for hepatitis C represent an unusual evergreening failure, not because Gilead did not try to build a secondary patent portfolio, but because the clinical landscape for hepatitis C therapy changed so rapidly that the commercial window for first-generation direct-acting antivirals was compressed by the arrival of second-generation competitors from AbbVie and Merck rather than by generics.
Gilead filed numerous secondary patents covering sofosbuvir formulations, combination products, treatment regimens, and patient subpopulations. But when AbbVie launched Viekira Pak (ombitasvir/paritaprevir/ritonavir/dasabuvir) in 2014 and later Mavyret (glecaprevir/pibrentasvir) in 2017, they competed as brand-to-brand innovator competition rather than generic entry, undermining Gilead’s revenues through formulary competition rather than patent challenge. The patent thicket Gilead built was most relevant for blocking generic entry, but the competitive threat came from therapeutic alternatives, which patents cannot block.
Sofosbuvir faces generic entry in some international markets and eventual U.S. generic entry when core patents expire in the late 2020s. The secondary patent portfolio will matter then. But it illustrates a limit of evergreening as a business strategy: it protects against generic substitution but not against innovator competition within the therapeutic class.
Manufacturing Patents and Process Claims: The Hidden Layer of Pharmaceutical Thickets
Most public discussion of pharmaceutical patent thickets focuses on product patents (compound, salt, polymorph, formulation) and method-of-use patents. Process patents, which cover the method of manufacturing an API or formulation, receive less attention but can be equally effective barriers to generic entry.
Why Process Patents Are Hard to Challenge Without Manufacturing Data
A process patent infringement claim requires showing that the generic manufacturer uses the patented process. Generic manufacturers do not generally disclose their synthesis routes in their ANDAs. When an originator company asserts a process patent, it may obtain discovery into the generic’s manufacturing process, but this information is commercially sensitive and vigorously protected. Courts have developed protocols for handling confidential manufacturing process information in litigation, but the protective order regime for these disclosures adds procedural complexity and cost to process patent cases.
The 35 U.S.C. Section 295 burden-shifting provision allows a court to presume that the generic uses a patented process if the product produced by the process is new and the patentee has made a reasonable effort to determine the process used but has been unable to do so. This provision is rarely invoked but is available when a patentee can establish sufficient factual predicates.
For biologic drugs, manufacturing process patents are particularly significant because the specific process used to produce a biologic directly affects the product’s safety and efficacy profile. Biosimilar manufacturers must demonstrate that their product is highly similar to the reference biologic despite potentially using different manufacturing processes, a requirement that effectively forces disclosure of process information to the FDA even when it is kept confidential from competitors. Process patents in the biologic space can cover specific cell culture conditions, purification techniques, glycosylation patterns, and aggregation prevention methods, all of which are manufacturing know-how that the originator invested heavily to develop.
The Future of Patent Thickets: Regulatory Pressure, Litigation Trends, and Policy Options
The pharmaceutical patent thicket landscape in 2025 is evolving under pressure from multiple directions: FTC enforcement campaigns, IRA pricing provisions, FTC-focused antitrust scrutiny of settlement terms, European pricing pressures that remove revenue justification for some secondary patent filings, and the growing sophistication of generic and biosimilar company legal teams that have decades of experience challenging secondary patents.
Will the Inflation Reduction Act’s Negotiation Provisions Reduce Thicket-Building?
The IRA’s negotiation provisions cap Medicare revenues for selected drugs after nine years for small molecules and thirteen years for biologics. If the negotiated price is substantially below list price, the marginal value of each additional year of exclusivity beyond year nine or thirteen is reduced to the negotiated price rather than the list price. This reduction in marginal exclusivity value theoretically reduces the return on investment from secondary patent filing and thicket-building for drugs that will be subject to negotiation.
The magnitude of this effect depends on which drugs are selected for negotiation and what the negotiated prices are. CMS negotiated 2026 prices for the first ten drugs at discounts ranging from 38% to 79% below list price [19]. If the negotiated prices are approximately 50% of list price, the value of a year of exclusivity is halved for negotiated drugs, which reduces but does not eliminate the return on secondary patent investment. Whether this reduction is sufficient to alter originator IP filing behavior in practice remains an empirical question that will take years of data to answer.
Forecast: Which Drugs Face the Largest Thicket-Related Exclusivity Extensions Through 2030?
Based on published patent data and ANDA filing histories available through the FDA’s Orange Book and patent intelligence sources including DrugPatentWatch, several high-revenue drugs face patent landscapes where secondary patent portfolios could produce effective exclusivity extending well past core compound patent expiry through 2030.
Dupixent (dupilumab, Regeneron/Sanofi) has a biologic compound patent expiry in the late 2020s but has accumulated secondary patents covering formulation, dosing, and manufacturing. Given the lessons of Humira, biosimilar developers will be watching Regeneron’s secondary patent filings closely to assess whether a comparable settlement period is emerging.
Keytruda (pembrolizumab, Merck) faces compound patent expiry in the early 2030s, but with multiple approved indications across oncology and a substantial secondary patent filing program, the biosimilar entry landscape is complex. Merck has aggressively filed patents on manufacturing processes and combination regimens. As of 2025, no pembrolizumab biosimilar has received FDA approval, and the regulatory timeline for biosimilar development means the first biosimilar clinical data would be maturing now for an approval in the late 2020s.
Ozempic and Wegovy (semaglutide, Novo Nordisk) have attracted intense generic and biosimilar attention given their weight-loss indication’s commercial scale. Novo Nordisk’s semaglutide patent portfolio includes compound, formulation, delivery device, and method-of-use patents. The FDA approved the first GLP-1 receptor agonist biosimilar pathway under the BPCIA in early 2025, and the semaglutide competitive landscape will develop rapidly through the late 2020s. The patent thicket for semaglutide will be a defining test of whether the Humira precedent shapes biosimilar settlement structures in the next major therapeutic category.
What Systematic Patent Challenge Programs Mean for Thicket Deterrence
The Medicines Patent Pool, I-MAK, and academic groups including researchers at Yale and Johns Hopkins have published systematic analyses of secondary pharmaceutical patent portfolios, identifying patents they argue are legally weak and commercially exploitative. These analyses create public records that generic and biosimilar companies can use in IPR petitions and litigation, and they influence regulatory and legislative discussions about patent reform.
Whether public patent challenge programs materially accelerate generic entry depends on whether they identify and attack patents that are actually blocking competition, rather than patents that the industry would litigate around or settle regardless. For drugs with thickets of fifty or more patents, eliminating ten through advocacy may have limited effect if the remaining forty continue to block entry. The strategic value of systematic patent challenges may be more in deterrence, by making patent thicket-building more expensive to defend, than in clearing individual patents.
Key Takeaways
A pharmaceutical patent thicket is a deliberate strategic construction, not a coincidental accumulation of patents. Companies build them with specific intent to extend exclusivity beyond the original compound patent’s 20-year term.
The core tools include polymorph and salt form patents, formulation and modified-release patents, method-of-use patents covering new indications, pediatric exclusivity add-ons, and citizen petitions as procedural delays.
Humira’s 136-patent thicket is the industry’s most studied example, producing a five-year biosimilar entry gap between Europe and the U.S. that cost American payers an estimated $19 billion in excess spending.
Reverse payment settlements and volume-capped generic entry arrangements can extend effective exclusivity even after patent challenges succeed, substituting commercial restrictions for legal ones.
The Orange Book’s self-certification mechanism, the 30-month automatic stay, and FDA exclusivity periods all amplify the impact of secondary patents in ways that legislators and courts have only partially addressed.
IPR petitions at the PTAB provide a faster and less expensive route to patent challenge than district court litigation but address individual patents rather than portfolios, limiting their thicket-clearing utility.
International markets, particularly in Europe, see earlier generic competition on many drugs due to EPO opposition procedures that effectively narrow secondary patent coverage and court standards that are less favorable to secondary pharmaceutical patents.
The IRA’s Medicare price negotiation provisions reduce the marginal value of exclusivity for negotiated drugs but at levels unlikely to fully eliminate the return on thicket-building investment.
Patent intelligence tools, including DrugPatentWatch, enable generic companies, payers, and investors to identify realistic competitive entry timelines that differ materially from disclosed core patent expiry dates.
GLP-1 receptor agonists, PD-1/PD-L1 checkpoint inhibitors, and dupilumab are the most commercially significant thicket-building theaters to watch through 2030.
Frequently Asked Questions
1. What is the difference between a patent thicket and normal patent protection in pharma?
Normal patent protection covers the active compound discovered by the originator company, typically granted for 20 years from filing. A patent thicket extends beyond the compound patent to cover every associated attribute of the drug: its salt forms, crystal structures, manufacturing processes, delivery mechanisms, dosing regimens, and approved indications. The compound patent is often the thinnest layer; the thicket is the dense accumulation of secondary and tertiary patents that surrounds it.
2. How long can pharmaceutical companies realistically extend exclusivity through evergreening?
Documented cases show extensions of 10-20 years beyond the core compound patent. Humira’s effective U.S. biosimilar-free period extended from the core composition patent’s 2016 expiry to 2023, a seven-year extension through secondary patents. Revlimid’s full generic competition is delayed to 2026 through settlement structures built on a secondary patent portfolio. Extensions beyond twenty years past compound patent expiry are rare but have been claimed in some orphan drug contexts.
3. What is a Paragraph IV certification and how does it trigger a patent thicket battle?
A Paragraph IV certification is a statement by a generic ANDA filer that the brand’s listed Orange Book patents are either invalid or not infringed by the generic product. Filing a Paragraph IV certification constitutes a legal act of patent challenge. If the originator sues for infringement within 45 days, an automatic 30-month stay on FDA generic approval activates, effectively buying the originator 2.5 years of additional exclusivity regardless of the merits. Patent thickets exploit this mechanism by requiring generic companies to certify against dozens of patents, each potentially triggering litigation.
4. Can a generic company launch before all thicket patents expire?
Yes, by launching at risk: commercially selling the generic while patent litigation remains pending. An at-risk launch exposes the generic company to damages if it loses the remaining patent cases, potentially covering all profits made during the at-risk period. The calculation depends on the generic company’s assessment of the remaining patents’ strength and the commercial opportunity in the drug market. Large generic companies like Teva and Sandoz have launched major at-risk products when their patent counsel assessed remaining thicket patents as weak.
5. How does the BPCIA ‘patent dance’ differ from the Hatch-Waxman Paragraph IV process?
The Hatch-Waxman Paragraph IV process is a unilateral patent challenge by the generic applicant, triggered by the ANDA filing and notification to the originator. The BPCIA patent dance is a bilateral information exchange process where the biosimilar applicant shares its BLA with the originator, both parties exchange patent lists and positions, and they negotiate a litigation list before any suit is filed. The dance is optional (per Amgen v. Sandoz), while the Paragraph IV process is automatically triggered by the ANDA filing. The BPCIA does not include an automatic stay equivalent to Hatch-Waxman’s 30-month stay.
6. What is an authorized generic and how does it affect thicket-related settlements?
An authorized generic is a version of a branded drug marketed by the originator company (or a company authorized by it) under the existing brand NDA, typically at a lower price. In Hatch-Waxman settlements, originators sometimes grant the settling generic company authorized generic rights as partial consideration, allowing that company to sell the drug without independent manufacturing. Authorized generics compete with first-filer generic companies during the 180-day exclusivity period, reducing the first-filer’s profit and the commercial value of patent challenges, which can reduce generic companies’ incentive to challenge thickets.
7. How do payers and pharmacy benefit managers account for patent thickets in drug cost projections?
Sophisticated PBMs model pharmaceutical LOE dates using full patent portfolio analysis rather than just core patent expiry. They identify the last-expiring Orange Book patent, review ANDA challenge histories, track litigation settlement terms for volume caps and launch date restrictions, and monitor IPR proceedings that might accelerate competitive entry. Databases like DrugPatentWatch support this analysis. PBMs use these models to time formulary changes, negotiate contract terms with brand companies, and advise plan sponsors on drug spend forecasts.
8. What is the significance of the Federal Circuit’s role in pharmaceutical patent thicket cases?
The U.S. Court of Appeals for the Federal Circuit has exclusive appellate jurisdiction over patent cases from all district courts. Its precedents directly shape the legal standards applied to secondary pharmaceutical patents nationwide. The Federal Circuit has interpreted obviousness standards, patent eligibility, and claim construction in ways that affect whether polymorph, formulation, and method-of-use patents survive challenge. Federal Circuit judges have also addressed the skinny-label/induced infringement issue in GSK v. Teva, creating legal uncertainty that functionally operates as a thicket element even without additional patent filings.
9. How does the inter partes review process at the PTAB compare to district court patent litigation for attacking thickets?
IPR at the PTAB is faster (typically 12-18 months from petition to final decision versus 3-5 years in district court), less expensive (petitioner costs often $500,000 to $1 million versus $5-20 million for district court through trial), and limited to prior art grounds. District court litigation allows broader validity challenges including written description, enablement, and inequitable conduct claims, plus full infringement analysis and injunctive relief. For thicket-clearing, IPR is most efficient for patents with clear prior art vulnerability; district court is necessary for patents requiring broader invalidity theories or where infringement is in genuine dispute.
10. What should investors watch to identify drugs where the real LOE date differs from Wall Street consensus?
Investors should examine the full Orange Book patent listing for a drug rather than just the headline patent expiry date, check for continuation patent applications that extend filing dates beyond the parent patent, review ANDA challenge histories to identify which patents have not yet been challenged or litigated, assess whether any settlements include volume caps or launch date restrictions that preserve brand revenues post-generic entry, and monitor IPR filings at the PTAB that signal generic companies’ assessments of thicket patent strength. Discrepancies between consensus LOE dates and detailed patent landscape analysis can identify drugs where brand revenues are under- or over-estimated relative to market expectations.
Sources
U.S. Government Accountability Office. (2007). Pediatric Drug Research: Studies Conducted Under Best Pharmaceuticals for Children Act (GAO-07-557). https://www.gao.gov/products/gao-07-557
Initiative for Medicines, Access & Knowledge (I-MAK). (2021). Humira Has Cost the US Healthcare System $19.6 Billion in Excess Spending Over the Last Five Years. https://www.i-mak.org/humira/
Rome, B. N., Egilman, A. C., & Kesselheim, A. S. (2022). Trends in prescription drug launch prices, 2008–2021. JAMA, 327(21), 2145–2147. https://doi.org/10.1001/jama.2022.5542
Novartis AG v. Union of India & Ors., Civil Appeal Nos. 2706-2716 of 2013 (Supreme Court of India, April 1, 2013).
Mulcahy, A. W., Schwam, D., & Qureshi, N. (2021). Prices for and spending on specialty drugs in the commercial and Medicare Part D sectors. RAND Corporation. https://www.rand.org/pubs/research_reports/RRA788-1.html
Federal Trade Commission. (2023). FTC Files Complaint Against Bristol Myers Squibb for Illegally Blocking Competition to Cancer Drug Revlimid. https://www.ftc.gov/news-events/news/press-releases/2023/01/ftc-files-complaint-against-bristol-myers-squibb
Congressional Budget Office. (2021). Research and Development in the Pharmaceutical Industry. https://www.cbo.gov/publication/57025
TC Heartland LLC v. Kraft Foods Group Brands LLC, 581 U.S. 258 (2017).
FTC v. Actavis, Inc., 570 U.S. 136 (2013).
AbbVie Inc. v. Boehringer Ingelheim International GmbH et al., No. 1:16-cv-01093 (N.D. Ill.).
Amgen Inc. v. Sandoz Inc., 582 U.S. 1 (2017).
U.S. Food and Drug Administration. (2021, July 28). FDA approves first interchangeable biosimilar insulin product for treatment of diabetes. https://www.fda.gov/news-events/press-announcements/fda-approves-first-interchangeable-biosimilar-insulin-product-treatment-diabetes
Creating and Restoring Equal Access to Equivalent Samples (CREATES) Act of 2019, Pub. L. No. 116-94, 133 Stat. 2534 (2019).
Lipitor (atorvastatin calcium) revenue data derived from Pfizer Inc. 10-K filings (2011–2013). https://www.pfizer.com/science/drug-product-pipeline/resources/reports
Centers for Medicare & Medicaid Services. (2023, August 29). Medicare drug price negotiation program: Negotiated prices for initial price applicability year 2026. https://www.cms.gov/newsroom/press-releases/cms-announces-historic-low-prices-first-10-drugs-selected-medicare-drug-price-negotiation-program
Donohue, J. M., Cevasco, M., & Rosenthal, M. B. (2007). A decade of direct-to-consumer advertising of prescription drugs. New England Journal of Medicine, 357(7), 673–681. https://doi.org/10.1056/NEJMsa070502
IQVIA Institute for Human Data Science. (2016). Medicines Use and Spending in the U.S.: A Review of 2015 and Outlook to 2020. https://www.iqvia.com/insights/the-iqvia-institute/reports
Centers for Medicare & Medicaid Services. (2024). Medicare Drug Price Negotiation Program: Final Negotiated Prices for 2026. https://www.cms.gov/inflation-reduction-act
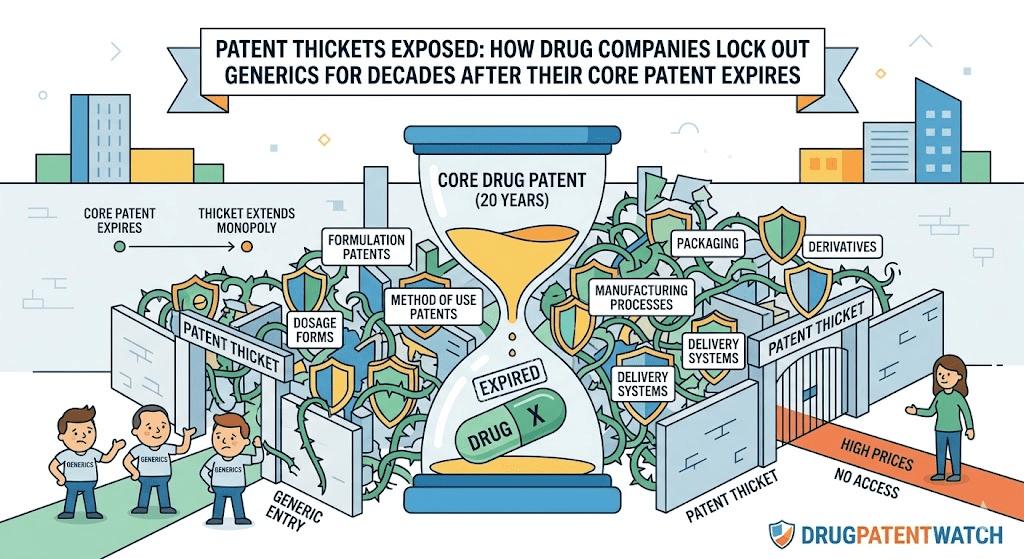



")






















