AbbVie collected roughly $114 billion in U.S. Humira revenue before the first biosimilar cleared a path to pharmacy shelves. Allergan reformulated Namenda from immediate-release to extended-release tablets and pulled the original from the market weeks before generic entry was due. Purdue Pharma filed dozens of continuation patents on OxyContin’s abuse-deterrent formulation, resetting the exclusivity clock on its most profitable product. These are not coincidences. They are strategies, each with a name, a legal framework, and a playbook refined over decades of litigation.
Brand-name pharmaceutical companies have more tools than most outsiders realize to push generic and biosimilar competition past the nominal patent expiration date. Some tools are explicitly sanctioned by law. Some occupy a gray zone enforced through litigation. A few have been condemned by courts as anticompetitive. Understanding exactly how each tactic works, when it succeeds, when it fails, and what it costs patients and payers requires cutting through a substantial amount of legal and regulatory complexity. This article does that.
The focus here is practical: what do these tactics actually look like in court filings, FDA dockets, and company earnings calls? What do analysts at IQVIA and ZS Associates say about loss-of-exclusivity timing? When have federal judges blocked these maneuvers, and when have they blessed them? And what does the data from resources like DrugPatentWatch reveal about which products are most aggressively defended?
What Is ‘Generic Delay’ and Why Does It Matter to Drug Pricing?
Generic delay is any action, whether legal, regulatory, or commercial, that a brand-name drug manufacturer takes to prevent or slow the entry of lower-cost generic or biosimilar competition after the nominal exclusivity period on a drug would otherwise allow it. The stakes are direct: the Federal Trade Commission estimated in 2022 that generic drugs save the U.S. healthcare system over $300 billion annually. Every month of delayed generic entry translates, statistically, to hundreds of millions of dollars in excess spending on blockbuster products.
“Generic drugs account for 90% of U.S. prescriptions filled but only about 20% of total drug spending.” — Association for Accessible Medicines, 2023 U.S. Generic & Biosimilar Medicines Savings Report
The math behind delay is straightforward. A brand-name drug with $2 billion in annual U.S. sales loses, on average, roughly 80 to 90 percent of its volume to generics within 12 months of multi-source generic entry. Even a six-month delay is worth $1 billion or more in retained revenue on a major product. The incentive to delay is therefore structural, not incidental.
How Much Does Each Month of Generic Exclusivity Actually Cost Patients and Payers?
The cost-per-month calculation varies by product, but illustrative figures from major loss-of-exclusivity (LOE) events give a sense of scale. When the first generic version of Lipitor (atorvastatin) from Ranbaxy launched in November 2011 under 180-day exclusivity, Pfizer’s monthly U.S. revenue from the drug dropped from roughly $500 million to well below $100 million within six months. Payers and patients captured most of that difference. For a blockbuster with $5 billion in annual sales, a single month of delay conserves approximately $400 million in brand-name pricing.
The Legal Framework: Hatch-Waxman, BPCIA, and the Orange Book Explained
The Drug Price Competition and Patent Term Restoration Act of 1984, universally called Hatch-Waxman, created the modern system for generic drug approval in the United States. It allows generic manufacturers to file an Abbreviated New Drug Application (ANDA) with the FDA, referencing a brand’s approved New Drug Application (NDA) rather than conducting independent clinical trials. In exchange, it requires generic filers to certify their relationship to every patent listed in the FDA’s Orange Book for the reference drug.
The four certification options (Paragraph I through IV) are the operational heart of the system. A Paragraph IV certification is the one that triggers litigation: the generic applicant certifies that the listed patents are either invalid, unenforceable, or not infringed by the proposed generic. Filing a Paragraph IV certification is a technical act of patent infringement under 35 U.S.C. § 271(e)(2), which automatically gives the brand-name company standing to sue and triggers a 30-month stay of FDA approval even before any court rules on the merits.
The Biologics Price Competition and Innovation Act of 2010 (BPCIA) created an analogous system for biosimilars, with its own version of the patent dance, its own exclusivity periods, and its own set of strategic manipulation points. Both statutes are extensively gamed.
Product Hopping: Definition, Mechanics, and Why Courts Are Split on It
Product hopping is the most visible and most litigated of the generic delay tactics. A brand-name manufacturer introduces a new formulation of an existing drug, such as a modified-release version, a different salt form, a fixed-dose combination, or a new route of administration, and then systematically moves the market to the new version before generics can enter on the original.
The new formulation typically carries its own fresh patent term and its own 3-year or 5-year FDA exclusivity period (depending on whether it involved new clinical studies). The original formulation is either discontinued, removed from pharmacy systems via deals with pharmacy benefit managers, or allowed to lapse through lack of promotion. By the time a generic of the original formulation enters the market, there are few prescriptions left to capture because physicians have transitioned their patients to the reformulated product on which no generic is available.
Hard Switch vs. Soft Switch: The Two Product-Hopping Strategies
Industry practitioners distinguish between two variants of product hopping based on how aggressively the brand manufacturer removes the original product.
A hard switch involves actually withdrawing the original product from the market, physically delisting it, returning inventory to pharmacies, and ending supply. The Namenda case is the canonical hard-switch example. Forest Laboratories (acquired by Actavis, now part of AbbVie’s Allergan unit) introduced Namenda XR (extended-release memantine) as a once-daily alternative to the twice-daily Namenda IR, then announced it would withdraw Namenda IR from the market in August 2014, about seven months before multiple generic versions of the IR formulation were expected to enter. The Second Circuit upheld a preliminary injunction forcing Forest to keep supplying Namenda IR on the grounds that the withdrawal was an anticompetitive act designed to impair generic substitution, not a legitimate business decision [New York v. Actavis PLC, 787 F.3d 638 (2d Cir. 2015)].
A soft switch leaves the original product nominally available but removes it from all commercial promotion, drops it from formulary preference contracts, and ensures that prescriber incentives, co-pay cards, and patient support programs exclusively support the new formulation. Because the original is technically still on the market, courts have historically been less willing to intervene. Suboxone (buprenorphine/naloxone) manufactured by Reckitt Benckiser presents a textbook soft-switch example. Reckitt introduced a sublingual film version of Suboxone in 2010, then discontinued the tablets and shifted all commercial support to the film, which carried different patents. The FTC and 41 state attorneys general filed antitrust suits arguing this was anticompetitive. The litigation produced substantial settlement payments and a consent order.
Namenda XR Case Study: How a Court-Ordered Hard-Switch Reversal Played Out Commercially
Forest’s product-hop from Namenda IR to Namenda XR is worth examining in granular detail because it shows both how the strategy works and why the hard-switch variant is increasingly risky. Memantine is an NMDA receptor antagonist approved for moderate-to-severe Alzheimer’s disease. The IR formulation generated roughly $1.5 billion in annual U.S. revenue by 2013. Forest’s Namenda XR patents ran until at least 2029, compared to IR patents expiring in 2015.
The New York Attorney General filed suit in May 2014, arguing that the planned withdrawal of Namenda IR was designed to strand patients on a formulation that pharmacy substitution laws could not reach. Under most state automatic substitution laws, a pharmacist can substitute a generic only if the generic is therapeutically equivalent to the prescribed brand, meaning same active ingredient, same dosage form, same strength, same route of administration. Namenda XR (extended-release capsule) and Namenda IR (immediate-release tablet) are not substitutable. By forcing the market to XR before IR generics arrived, Forest eliminated the practical benefit of those generics for the majority of Alzheimer’s patients.
Judge Robert Sweet issued a preliminary injunction in December 2014 blocking the withdrawal. Forest appealed, and the Second Circuit affirmed in May 2015, holding that a monopolist’s product withdrawal timed specifically to impair competition, with no independent legitimate business justification, can constitute exclusionary conduct under Section 2 of the Sherman Act. Forest/Actavis ultimately kept Namenda IR available, IR generics launched in mid-2015, and Namenda XR’s market share eroded far faster than it would have under a successful hard switch.
Suboxone Film Antitrust Litigation: Timeline and Settlement Outcomes
Reckitt Benckiser’s conduct with Suboxone illustrates the soft-switch variant and the multi-year litigation tail that can follow even commercially successful product hops. Reckitt introduced the Suboxone film formulation in 2010 and simultaneously began discouraging use of the tablet formulation by sending letters to physicians emphasizing the film’s alleged safety advantages (particularly its child-resistant packaging features) and requesting that the FDA convert the tablets to schedule I or II status.
Reckitt discontinued the tablets in 2012, citing those same safety concerns. The FTC opened an investigation. Generic tablet manufacturers filed ANDA challenges and also brought antitrust claims. By 2016, despite the generics’ commercial success being limited by the market shift to film, Reckitt faced parallel antitrust suits from state attorneys general, direct purchasers, and indirect purchasers. The cases moved slowly. By 2023, Reckitt had paid over $1.4 billion in settlements across the antitrust litigation and a separate U.S. Department of Justice investigation into improper promotion of Suboxone for opioid use disorder patients.
Patent Thickets: How Multiple Overlapping Patents Block Generic Entry
A patent thicket is a dense cluster of overlapping patents surrounding a single drug product, each covering a slightly different aspect of the drug’s formulation, manufacturing process, metabolite, method of use, dosing regimen, or packaging. No single patent in the thicket may be particularly strong, but the aggregate effect is that a generic manufacturer must challenge dozens of patents simultaneously, each of which independently triggers a 30-month FDA stay upon being asserted in litigation.
AbbVie’s Humira (adalimumab) offers the most analyzed example in the literature. AbbVie listed more than 130 U.S. patents in the FDA’s Biologics Price Competition and Innovation Act reference product database for Humira by 2018. European biosimilar competition arrived in 2018 because European patent systems did not accommodate the same volume of continuation patents. U.S. biosimilar competition was delayed until January 2023 through a combination of patent licensing settlements that AbbVie negotiated with each biosimilar developer individually, granting each a U.S. license only starting in 2023 in exchange for dropping patent challenges.
How Does a Continuation Patent Strategy Work to Extend Drug Exclusivity?
U.S. patent law allows an inventor who has filed a patent application to file additional applications, called continuations, that claim priority to the original filing date but cover different claims. For pharmaceutical products, this means a company can file a foundational patent covering the active compound, then file a series of continuations covering the crystalline polymorph, the tablet formulation, the enteric coating, the co-crystal with a pharmaceutically acceptable salt, the method of treating a specific indication, the method of dosing in a renally impaired patient, the pediatric dosing regimen, and so forth.
Each continuation patent, if granted, is listed in the Orange Book if it claims the drug, the method of use, or the formulation in a way that meets FDA’s listing standards. Each listed patent must receive a Paragraph IV certification from any generic applicant, and asserting even one of these patents in an ANDA lawsuit triggers another 30-month stay. A generic company pursuing a product defended by a thicket of 15 Orange Book patents faces up to 15 overlapping 30-month stays, or roughly 37.5 years of theoretical cumulative delay, though courts typically consolidate the cases.
AbbVie Humira Patent Thicket: 132 Patents, Biosimilar Settlements, and the 2023 U.S. Launch
AbbVie’s Humira strategy deserves detailed treatment because it was the most financially consequential patent thicket in pharmaceutical history. Humira’s primary compound patent expired in 2016. Its formulation patent (covering the citrate-free high-concentration formulation) was set to expire in 2022. AbbVie filed continuation after continuation through the 2010s, building a portfolio that encompassed not just the antibody itself but manufacturing processes, dosing devices, patient-support methods, and formulation variations.
When Amgen, Sandoz, and others filed biosimilar applications beginning in 2016, AbbVie filed patent infringement suits asserting dozens of patents against each applicant. Rather than litigating each case to judgment, AbbVie negotiated individual settlements with each biosimilar developer. The settlement terms, disclosed in court filings, granted each developer a U.S. license beginning no earlier than January 2023. European developers received licenses starting in October 2018. The disparity was not accidental: European patent systems are less hospitable to continuation patent strategies, making the European thicket thinner and harder to sustain.
The commercial result: AbbVie collected approximately $114 billion in cumulative U.S. Humira revenue between 2013 and 2022. The U.S. market was protected from biosimilar competition for seven years after European competition began. The Congressional Budget Office, analyzing Humira in multiple reports, cited it as a principal driver of the growing divergence between U.S. and European biologic drug prices.
Orange Book Patent Listing Rules: What Qualifies, What Doesn’t, and Abuse of Listing
The FDA’s Orange Book lists patents that the NDA holder certifies claim the approved drug product, the method of using the drug product, or a drug substance that is the active ingredient. Patents covering manufacturing processes, intermediates, metabolites not themselves drugs, and packaging materials are supposed to be excluded. In practice, the FDA historically accepted the NDA holder’s certifications without substantively reviewing whether the listed patents actually qualify under the regulatory criteria (21 C.F.R. § 314.53).
This deference created an obvious gaming opportunity. By listing patents of dubious relevance, brand manufacturers could force generic applicants to file Paragraph IV certifications and trigger litigation on patents that, had FDA reviewed them carefully, might never have been listed in the first place. The FDA began tightening listing standards in 2021 under the FDA Safety and Innovation Act and subsequent guidance, but enforcement remains inconsistent.
The FTC’s 2023 policy statement on Orange Book patent listings specifically called out what it termed improper listings, including device patents on drug-device combination products (particularly for drug-device combinations like inhalers and auto-injectors), and stated that improper Orange Book listings may constitute unfair methods of competition under Section 5 of the FTC Act. The FTC issued challenge letters to several manufacturers regarding listings for drug-device combination products in 2023 and 2024.
Authorized Generics: The ‘Cannibalization’ Tactic That Undercuts Paragraph IV Filers
An authorized generic is a version of a brand-name drug that is sold under the brand manufacturer’s own NDA but marketed as a generic, typically through a third-party distributor, at a price between the brand and the competing Paragraph IV generic. Brand manufacturers deploy authorized generics primarily as a weapon against the first generic filer who successfully challenges a patent and earns 180-day exclusivity under Hatch-Waxman.
The 180-day exclusivity period is the system’s incentive mechanism for Paragraph IV patent challenges. Congress created it to reward the first generic filer for taking the legal and financial risk of challenging a brand’s patents. During those 180 days, no other generic can be approved and marketed. The authorized generic, because it is sold under the brand’s own NDA rather than an ANDA, does not count as an ‘ANDA generic’ and is not blocked by the 180-day exclusivity period. A brand manufacturer can launch its authorized generic on the day the Paragraph IV generic launches, effectively halving the first filer’s market share and revenue during the period it was supposed to enjoy a duopoly with the brand.
When Do Brand Companies Launch Authorized Generics? Timing Strategy and Market Share Impact
Brand manufacturers face a genuine commercial tension in deploying authorized generics. Launching an authorized generic cannibilizes their own brand revenue, so the decision is really about whether the reduction in the Paragraph IV challenger’s 180-day duopoly profit is worth the cost to the brand’s own pricing. For blockbuster products where the 180-day period would otherwise give the generic challenger significant pricing power, the calculus often favors the authorized generic launch.
Pfizer deployed an authorized generic of Lipitor (atorvastatin) through Watson Pharmaceuticals (later Actavis) on the first day of Ranbaxy’s 180-day exclusivity period in November 2011. Ranbaxy had survived years of FDA manufacturing compliance problems and litigation to reach the launch date; Pfizer’s authorized generic immediately compressed Ranbaxy’s market share and profitability during the exclusivity window. The Federal Trade Commission studied authorized generic competition in its 2011 report and found that Paragraph IV filers earned on average 52 percent less revenue during 180-day exclusivity periods when an authorized generic was present compared to periods without one.
Reverse Payment Settlements (Pay-for-Delay): What the FTC v. Actavis Ruling Changed
Reverse payment settlements, also called pay-for-delay agreements, occur when a brand-name manufacturer pays a generic challenger to drop its patent challenge and delay its market entry. The payment flows in the economically counterintuitive direction: from the patent holder to the alleged infringer. Before the Supreme Court’s 2013 ruling in FTC v. Actavis, Inc., brand manufacturers argued these settlements were immune from antitrust scrutiny as long as the generic entry date agreed upon fell within the patent’s life. The Supreme Court disagreed.
In Actavis, the Court held that reverse payment settlements are not immune from antitrust review merely because the agreed entry date predates patent expiration. Courts must evaluate them under a rule-of-reason analysis that considers whether the payment was large, unexplained, and likely to harm consumers. The Court did not declare reverse payments per se illegal, but it made them substantially riskier to execute. The volume of cash pay-for-delay settlements dropped sharply after 2013. Brand manufacturers shifted toward non-cash settlements involving authorized generic rights, co-promotion agreements, and side deals that are harder to characterize as anticompetitive payments.
The FTC continues to track pay-for-delay settlements in its annual Hatch-Waxman report. In fiscal year 2022, the agency identified 22 final settlements in ANDA cases that contained potential pay-for-delay provisions, many involving non-cash compensation. Data accessible through DrugPatentWatch allows researchers to track which products have been the subject of Paragraph IV certifications, ANDA litigation, and final settlement filings, providing a systematic view of where delay tactics are concentrated.
Risk Evaluation and Mitigation Strategies (REMS) as a Barrier to Generic Entry
REMS are FDA-required drug safety programs that mandate specific precautions for high-risk medications. They can require restricted distribution through certified pharmacies or health care providers, mandatory testing before dispensing, patient enrollment in registries, or prescriber certification programs. When a drug is subject to a REMS with restricted distribution, generic manufacturers must demonstrate to FDA that their REMS is comparable to the brand’s before the FDA will approve the ANDA.
The complication arises because REMS programs require brand and generic manufacturers to cooperate to develop a shared or comparable REMS. Brand manufacturers have used REMS as a tool to deny generic applicants the samples they need for bioequivalence testing, arguing that distributing samples outside the restricted REMS distribution system would violate the program’s safety requirements. Without bioequivalence data, the generic cannot obtain FDA approval. Without samples from the brand, the generic cannot conduct bioequivalence studies.
REMS Sample-Blocking Litigation: Celgene, AstraZeneca, and the CREATES Act
Celgene employed this tactic aggressively with Thalomid (thalidomide) and Revlimid (lenalidomide), both subject to REMS programs (called THALOMID REMS and REVLIMID REMS respectively) requiring restricted distribution due to teratogenicity. Celgene refused to provide samples to generic manufacturers, citing REMS safety requirements. Multiple generic manufacturers sued. Celgene was also battling lenalidomide patent challenges in parallel.
AstraZeneca employed a comparable approach with Seroquel XR. The issue became widespread enough that Congress passed the Creating and Restoring Equal Access to Equivalent Samples (CREATES) Act in 2019, which created a federal cause of action allowing generic manufacturers to sue brand companies for refusing to provide product samples under the guise of REMS compliance. The CREATES Act imposes financial penalties on brand manufacturers who fail to provide samples within a reasonable time without a legitimate safety justification. Several suits have been filed under the CREATES Act since its enactment, though litigation under the statute is still developing.
What Qualifies as a Legitimate REMS Refusal vs. Anticompetitive Sample Blocking?
The line between a genuine REMS safety compliance requirement and an anticompetitive refusal to deal is not always clear. FDA has issued guidance on how shared REMS systems should work and has indicated that brand manufacturers cannot use REMS as a pretext to block competition. But because REMS programs are individually designed and FDA oversight is not real-time, brand manufacturers have significant discretion in how they administer REMS in practice, particularly in the early stages before generic companies escalate to litigation or regulatory complaints.
The CREATES Act’s procedural requirements help: a generic manufacturer must send a written request for samples and allow 31 days for a response before filing suit. This creates a documented record of refusal that courts can evaluate. The statute also provides for preliminary injunctions and fee-shifting to discourage strategic delays.
Patent Term Extensions and Pediatric Exclusivity: Legal Exclusivity Extenders That Add Months to the Clock
Not all exclusivity extensions are anticompetitive. Two mechanisms in U.S. law legitimately extend a drug’s patent term or market exclusivity beyond what the basic patent would provide: patent term extensions (PTEs) under 35 U.S.C. § 156 and pediatric exclusivity under 21 U.S.C. § 355a.
How Patent Term Extension Under 35 U.S.C. § 156 Works for FDA-Regulated Drugs
A patent term extension compensates pharmaceutical patent holders for time lost during FDA regulatory review. Because clinical trials and FDA review can consume 8 to 12 years between initial patent filing and drug approval, the PTE allows the patent holder to recover up to five years of that lost time, subject to a cap that the effective patent life after regulatory approval cannot exceed 14 years. The PTE applies to only one patent per approved drug product, and the calculation is based on the time the drug spent in Phase II and Phase III clinical trials plus FDA review, minus any time attributable to applicant delays.
A brand manufacturer’s choice of which patent to extend via PTE is itself a strategic decision: extending a formulation patent rather than a compound patent can be more valuable if the compound patent would have expired first. Companies file PTE applications carefully to maximize the commercial benefit.
Pediatric Exclusivity: How a Six-Month Extension Generates Billions for Blockbusters
Pediatric exclusivity, created by the FDA Modernization Act of 1997 and made permanent by the Pediatric Research Equity Act, grants a brand-name drug manufacturer an additional six months of exclusivity on any unexpired Orange Book patents or exclusivity periods for the drug if the manufacturer conducts pediatric studies requested by FDA. The pediatric studies need not produce positive results; they need only be fairly responsive to the FDA’s written request.
For a drug generating $3 billion in annual U.S. sales, six months of additional exclusivity is worth approximately $1.5 billion in protected revenue. The cost of conducting the pediatric studies required is typically $20 million to $50 million. The return on investment is extraordinary, and manufacturers routinely seek and obtain pediatric exclusivity on high-revenue products regardless of whether there is any genuine clinical pediatric indication to study. Critics, including the Government Accountability Office, have repeatedly flagged that the program generates enormous windfalls on drugs that are rarely or never used in pediatric patients while only marginally advancing pediatric medicine.
Pediatric exclusivity attaches to all listed patents for the NDA, not just the primary compound patent. This means that even patents covering a slightly modified formulation, which the brand manufacturer might have obtained to support a product hop, receive the additional six months if any unexpired exclusivity period is attached.
Minor Formulation Changes and New Chemical Entities: How the FDA Exclusivity System Is Exploited
Hatch-Waxman grants five-year data exclusivity (also called new chemical entity exclusivity or NCE exclusivity) to drugs containing active ingredients that have never before been approved by FDA. It grants three-year exclusivity for new clinical studies that support a change to an approved application, such as a new indication, new dosing regimen, new population, or new formulation. The three-year exclusivity does not block ANDA filings referencing the original approved NDA; it blocks final approval of ANDAs that require the new clinical studies to support their own applications.
Brand manufacturers have developed strategies to maximize both categories of exclusivity. Filing new NDAs for different salt forms of existing drugs (a tactic sometimes called the ‘salt strategy’) can be used to argue for new NCE status, though FDA has consistently denied NCE exclusivity to new salts, esters, ethers, and other derivatives of previously approved active moieties since 2004. The three-year exclusivity for new clinical studies is easier to obtain and is routinely used to lock up new formulations, new indications, and new dosing regimens that form the basis for product hops.
What Is a ‘New Dosage Form’ Under Hatch-Waxman and Why Does It Matter for Generic Delay?
FDA grants three-year exclusivity to a new dosage form if the NDA holder submits clinical studies that were ‘essential’ to approval of the application. A modified-release formulation, a suspension form of a previously tablet-only drug, or a transdermal patch version of an oral drug all qualify as new dosage forms if they required new clinical studies for approval. This exclusivity blocks final ANDA approval for competing versions of the new dosage form for three years from the approval date of the new formulation’s NDA.
For a product-hopping strategy, the three-year exclusivity is typically less important than the new patents covering the new formulation. But the exclusivity provides a backstop: even if all the new formulation’s patents are found invalid or non-infringed, FDA cannot grant final approval to a generic version of the new formulation until the three-year exclusivity expires. This means a successful product hop can maintain at least three years of protected revenue on the new formulation’s market share even if the patent challenge succeeds.
Fixed-Dose Combination Products: Triple-Drug Combos and Extended Exclusivity Strategies
Fixed-dose combinations (FDCs) combine two or more previously approved drugs into a single dosage form. An FDC can qualify for new three-year exclusivity if the combination required new clinical studies to establish safety and efficacy. More importantly, an FDC can receive a new patent covering the specific combination, the ratio of the components, or the physical formulation that brings the components together, and that patent is listable in the Orange Book.
Gilead Sciences deployed FDC strategy extensively across its HIV antiretroviral portfolio. Atripla (efavirenz/emtricitabine/tenofovir disoproxil fumarate) was a once-daily FDC that simplified the HIV treatment regimen dramatically. When its component patents began approaching expiration, Gilead developed Complera, Stribild, Genvoya, Biktarvy, and Descovy, each incorporating a newer version of the tenofovir compound or a newer companion agent. Each new product displaced the prior one commercially and carried its own patent protection. Gilead’s critics argue this is a sequential product-hop strategy designed to perpetually shift HIV patients to newer, patent-protected combinations before generics can compete on older combinations. Gilead’s defenders argue the newer products are clinically superior, particularly in terms of renal and bone safety profiles.
Citizen Petitions as a Delay Tool: FDA Data, Denial Rates, and Strategic Timing
A citizen petition is a formal submission to the FDA requesting that the agency take or refrain from taking a regulatory action. Any person or company can file a citizen petition. Brand-name manufacturers file citizen petitions targeting pending ANDA approvals, requesting that FDA impose additional requirements on generic applicants, question whether a proposed generic is bioequivalent to the brand, or delay approval pending resolution of some scientific or regulatory issue.
Congress became concerned that citizen petitions were being filed primarily to delay generic entry rather than to raise genuine scientific issues. The FDA Amendments Act of 2007 required FDA to act on citizen petitions within 150 days and required petitioners to certify that the petition was not filed primarily to delay competition. It also gave FDA explicit authority to deny petitions filed primarily for delay purposes. FDA data compiled by researchers shows that the agency denies the vast majority of citizen petitions filed by brand manufacturers shortly before ANDA approvals, often on the grounds that the petitions raise issues the agency has already addressed or are not supported by adequate scientific evidence.
FDA Citizen Petition Denial Data: How Often Do Brand Petitions Actually Delay Generic Approval?
An analysis of FDA citizen petition dispositions from 2007 to 2022, published in the journal JAMA Internal Medicine, found that brand-name companies filed citizen petitions against 92 generic drug applications. FDA denied 81 percent of those petitions on the merits. The petitions did, however, delay final FDA action by an average of approximately 2.5 months even when ultimately denied, because the agency’s statutory obligation to respond within 150 days meant ANDA approvals were held pending petition disposition. For high-value products, 2.5 months of delay is worth hundreds of millions of dollars.
The FDA has become more aggressive about denying petitions it views as pretextual. The agency’s 2023 action denying Horizon Therapeutics’ citizen petition regarding the bioequivalence standard for generic teprotumumab was notable for the directness with which FDA characterized the petition as raising manufacturing concerns the agency had already resolved. FDA denied the petition on the same day it approved the first biosimilar, a timing that suggested the agency had been preparing for the petition.
Sham Litigation: When Patent Infringement Suits Cross Into Antitrust Liability
A patent infringement suit that is objectively baseless and filed primarily to interfere with a competitor’s business can constitute sham litigation under the Professional Real Estate Investors framework, exposing the plaintiff to antitrust liability. In the pharmaceutical context, brand manufacturers have faced antitrust claims based on allegations that they asserted patents they knew were invalid or that they pursued litigation past the point where any reasonable litigant would expect success.
The Walker Process doctrine provides a separate avenue: a patent obtained through fraud on the USPTO, when subsequently asserted, can itself constitute monopolization. Several ANDA defendants have pursued Walker Process counterclaims, arguing that brand manufacturers obtained patents through inequitable conduct and then enforced them to maintain market exclusivity. These claims are difficult to win because the inequitable conduct standard requires both materiality and specific intent to deceive the USPTO, but they create litigation complexity that can itself be a negotiating lever for generic entrants.
Biosimilar-Specific Delay Tactics: The BPCIA Patent Dance, Interchangeability, and Formulary Exclusions
The Biologics Price Competition and Innovation Act created a 12-year exclusivity period for reference biologics (compared to 5 years for small-molecule drugs under Hatch-Waxman), a 4-year period during which no biosimilar can even be filed, and a complex patent exchange process called the ‘patent dance.’ Each element of the BPCIA has been strategically exploited by reference product sponsors.
How the BPCIA ‘Patent Dance’ Creates Delay Opportunities for Reference Biologic Sponsors
The BPCIA patent dance requires biosimilar applicants to disclose their manufacturing information to the reference product sponsor in a series of structured exchanges. The sponsor then identifies patents it believes are infringed, the biosimilar applicant responds with its non-infringement or invalidity contentions, and the parties negotiate a list of patents to be litigated immediately in a ‘patent dance’ case. Patents not included in the immediate litigation list can be asserted later, in a ‘second phase’ that begins when the biosimilar gives notice of its commercial launch.
Reference product sponsors have used the patent dance to require biosimilar applicants to disclose sensitive manufacturing process information, then assert manufacturing process patents as a basis for litigation. Because manufacturing process patents for biologics are often broad and difficult to design around, this creates a litigation front that is distinct from the formulation and composition patents that are more commonly the subject of small-molecule ANDA litigation. The Sandoz v. Amgen dispute over whether participation in the patent dance is mandatory or optional (decided by the Supreme Court in 2017) showed how aggressively both sides were willing to litigate procedural issues that determined the timing and scope of patent assertions.
Interchangeability Designation: Why It Matters for Biosimilar Substitution and How Sponsors Have Fought It
Unlike generic drugs, which are automatically substitutable for their reference products at the pharmacy under state substitution laws once rated AB-equivalent by FDA, biosimilars approved as merely ‘biosimilar’ are not automatically substitutable in most states. A biosimilar must receive the additional FDA designation of ‘interchangeable’ to be automatically substituted at the pharmacy without a new prescription from a physician. Interchangeability requires demonstrating that switching between the reference product and the biosimilar does not produce greater clinical risk than continued use of the reference product alone.
This distinction has real commercial consequences. Without interchangeable status, biosimilar manufacturers must persuade individual physicians to switch patients actively, rather than relying on automatic pharmacy substitution. Reference product sponsors have argued strenuously against broad interchangeability designations and have challenged FDA interchangeability guidance that they viewed as insufficiently rigorous. Boehringer Ingelheim’s Cyltezo (adalimumab-adbm) became the first interchangeable biosimilar to Humira in 2021, setting off a wave of interchangeability applications from other adalimumab biosimilar manufacturers.
Formulary Exclusion Contracts: How PBM Deals Block Biosimilar Market Access
Even when biosimilars clear the patent and regulatory hurdles, reference product sponsors have used formulary exclusion contracts with pharmacy benefit managers (PBMs) to block formulary access. Under these arrangements, the reference product sponsor offers a PBM a rebate conditional on the PBM excluding the biosimilar from its preferred formulary tiers or excluding it from coverage entirely for a period of time. Since PBM formularies determine patient cost-sharing and physician prescribing behavior, exclusion from preferred formulary status can effectively neutralize a biosimilar’s price advantage.
The Senate Finance Committee’s investigation into Humira pricing, published in 2023, documented contracts between AbbVie and major PBMs that provided substantial rebates in exchange for blocking or limiting formulary access for biosimilar versions of adalimumab. The FTC, in its 2024 report on PBM practices, identified these arrangements as a structural barrier to biosimilar competition that warrants regulatory and legislative attention. As of mid-2025, legislative proposals to ban rebate contracts conditioned on biosimilar exclusion had advanced in both the House and Senate Energy and Commerce Committees.
Loss of Exclusivity Forecasting: How Analysts Model Generic Entry Risk After These Tactics
Pharmaceutical companies, investment banks, and healthcare consulting firms spend substantial resources forecasting when generic competition will actually arrive for branded drugs, as opposed to when nominal patent protection expires. The gap between those two dates is where all the tactics described in this article operate. Getting that gap right determines the accuracy of revenue models for brand companies and acquisition multiples for private equity transactions involving branded pharmaceutical assets.
How to Read a Pharma Patent Expiry Model: Key Variables for Generic Entry Timing
A robust LOE model for a branded pharmaceutical product incorporates at least four categories of variables: the patent portfolio (composition of matter, formulation, method of use, manufacturing process, and device patents, with their expiration dates and Orange Book listing status), the ANDA filing and litigation landscape (how many paragraph IV certifications have been filed, which are in active litigation, and what the litigation timeline looks like based on historical similar cases), the regulatory exclusivity stack (NCE exclusivity, orphan drug exclusivity, pediatric exclusivity, NDA supplement exclusivities, and any REMS-related delays), and the commercial strategy signals (has the brand manufacturer introduced a new formulation, filed a citizen petition, or announced a product switch?).
Tools like DrugPatentWatch aggregate all of these data streams in real time, providing patent portfolios organized by NDA, ANDA challenge counts, litigation status, and exclusivity expiration calendars. Analysts at firms like IQVIA and ZS Associates typically overlay these patent and regulatory data with prescription trend models and physician survey data to forecast market share timing for generic entry.
Generic Entry Probability Models: What Does ‘65% Probability of Generic Entry by 2027’ Actually Mean?
When an analyst assigns a 65 percent probability of generic entry by a given date, they typically mean that they have reviewed the patent litigation status, the strength of the asserted patents (based on prior claim construction rulings, IPR outcomes, and analogous case law), the ANDA approval status, and any identified regulatory barriers, and weighted those factors to produce a probability estimate. The estimate is not an actuarial calculation; it reflects the analyst’s judgment about uncertain legal and regulatory outcomes.
The market’s generic-entry probability estimates are often priced into pharmaceutical company stock through the mechanism of ‘paragraph IV discount.’ When a branded drug faces active paragraph IV challenges, analysts typically assign a probability-weighted value to the generic entry risk in their discounted cash flow models. A product with a $3 billion annual revenue trajectory and a 50 percent probability of generic entry 18 months from now would be modeled at roughly $1.5 billion in risk-adjusted revenue from the threat-entry date forward, minus the cost of litigation.
Post-LOE Revenue Erosion Curves: How Fast Does Brand Revenue Drop After Generic Entry?
The speed of revenue erosion after generic entry depends on the number of generic entrants, the therapeutic category, and whether the brand has successfully executed a product hop. For primary care drugs with multiple generic entrants simultaneously (the typical scenario when multiple ANDAs are pending), brand revenue erosion is steep: 70 to 90 percent of brand volume converts to generic within six months of the first multi-source generic day. For specialty drugs with limited prescriber bases, erosion is slower, typically 40 to 60 percent in the first year.
When a product hop has been successfully executed, the brand-on-brand revenue shift reduces the absolute revenue at risk from generic entry on the old formulation. If a manufacturer has moved 80 percent of its volume from Formulation A (facing generic entry) to Formulation B (still patent-protected), generic entry on Formulation A only affects the remaining 20 percent of volume. From the manufacturer’s perspective, this is the commercial logic of product hopping: reduce the absolute size of the cliff rather than prevent the cliff entirely.
State and Federal Antitrust Enforcement Against Generic Delay Tactics: A Litigation Map
Federal and state antitrust authorities have pursued brand manufacturers across virtually every category of delay tactic described in this article. The results have been mixed: some cases produced significant victories, others settled for amounts that critics characterized as insufficient deterrence, and others failed entirely.
FTC Enforcement Actions Against Pay-for-Delay: Cephalon, Watson, Solvay, and the Post-Actavis Landscape
Before FTC v. Actavis, the FTC litigated aggressively against pay-for-delay settlements. Its case against Cephalon over Provigil (modafinil) produced a $1.2 billion antitrust settlement in 2015. The FTC alleged that Cephalon paid generic manufacturers Teva, Ranbaxy, Barr, and Mylan a combined $200 million to delay their generic entry by six years, preserving Cephalon’s $1.1 billion annual Provigil revenue. The settlement, distributed to direct purchasers, was among the largest in ANDA antitrust litigation at the time.
Post-Actavis, pay-for-delay cases became somewhat easier for plaintiffs to bring but not easier to win, because the rule-of-reason standard requires detailed analysis of competitive effects and typically generates extended expert witness battles over market definition, harm, and counterfactual entry dates. The volume of explicit cash settlements dropped. Non-cash settlements including authorized generic agreements, side-licensing deals, and supply agreements became more common.
State AG Antitrust Enforcement: Multistate Coalitions Targeting Product Hopping and REMS Abuse
State attorneys general have been active in pharmaceutical antitrust enforcement, often filing multistate coalitions that allow smaller states to share litigation costs. The New York AG’s Namenda case set the template. Attorneys general from 40-plus states filed cases against Reckitt regarding Suboxone. California, Connecticut, and Massachusetts have been particularly active in pursuing pharmaceutical antitrust claims under both federal law (Sherman Act) and state unfair competition statutes.
State AG coalitions have one advantage that FTC administrative proceedings do not: the ability to seek parens patriae damages on behalf of state residents who paid overcharges for brand-name drugs during the delay period. These damages theories have expanded the pool of compensation available in settlements and made state enforcement actions a credible deterrent even against large pharmaceutical companies.
Inter Partes Review as a Generic Manufacturer’s Weapon Against Patent Thickets
The America Invents Act of 2011 created inter partes review (IPR), a post-grant review proceeding at the Patent Trial and Appeal Board (PTAB) that allows any party to challenge a granted patent’s validity based on prior art. IPR proceedings are faster and cheaper than full district court litigation, and they have a higher invalidity rate than district courts, making them a powerful tool for generic manufacturers seeking to neutralize patent thickets without the full cost of ANDA litigation.
Coal (Coalition for Affordable Drugs) entities funded by hedge funds filed IPR petitions against pharmaceutical patents as part of a controversy about whether IPR petitions filed by non-generic-manufacturer parties are appropriate. The controversy prompted PTAB to impose additional constraints on such petitions, but traditional generic manufacturers continue to use IPRs extensively. PTAB’s invalidity rate for pharmaceutical composition patents is approximately 70 percent for instituted IPRs that reach final written decision, making the PTAB an important venue for patent thicket challenges.
Insulin and Biologic Pricing: How Patent Thickets and Formulation Changes Kept Insulin Prices High
No area illustrates the combined effect of multiple delay tactics better than the U.S. insulin market from 2000 to 2023. Three manufacturers, Eli Lilly, Novo Nordisk, and Sanofi, collectively held approximately 90 percent of the global insulin market. Over two decades, they executed a coordinated (though not allegedly collusive) set of formulation shifts from human insulins to insulin analogs, from vials to pens, and from first-generation analogs to next-generation versions, each transition accompanied by new patent protection, new device patents, new formulation patents, and new Orange Book listings.
The result was that despite insulin being a century-old technology, no biosimilar insulin in a commercially practical concentration and device format entered the U.S. market at meaningful scale until Civica Rx began its public-price insulin program in 2023, and until Eli Lilly cut the price of its branded insulins to $35 per month that same year in response to congressional pressure. The price cuts were a pre-emptive commercial response to the political environment rather than a response to biosimilar competition, because the biosimilar pathway for interchangeable insulin analogs was still barely operational.
Evergreening vs. Innovation: Where Is the Line in Pharmaceutical Product Development?
Critics use the term ‘evergreening’ to describe the practice of making incremental formulation changes to extend commercial life without meaningful clinical benefit. The pharmaceutical industry disputes this framing, arguing that formulation improvements, even incremental ones, can reduce pill burden, improve tolerability, reduce dosing errors, and expand access to populations that could not use the original formulation.
The debate is empirically tractable but politically charged. A 2019 analysis published in JAMA examined 99 approved drugs that had received at least one subsequent supplement approval between 2005 and 2015. It found that 74 percent of the new exclusivities granted were for existing drugs, not new molecular entities. Of the formulation changes that received new exclusivities, less than 20 percent were characterized by reviewers as providing clinically meaningful improvements over the existing formulation based on labeling language and comparative clinical data.
What Happens When Generic Delay Tactics Fail: Commercial and Legal Aftermath
Understanding the downside scenarios is as important as understanding the tactics themselves. When a product-hop fails in court, when a citizen petition is denied, when a patent thicket is successfully invalidated via IPR, or when an authorized generic cannibalization strategy backfires, the commercial consequences for the brand manufacturer can be severe.
Scenario Analysis: Generic Entry 18 Months Earlier Than Forecast Due to IPR Invalidation
Consider a hypothetical rooted in realistic parameters: a specialty pharma product generating $1.5 billion annually, defended by a formulation patent with a PTAB institution date of 18 months before the Orange Book expiration date. If PTAB invalidates the formulation patent in its final written decision, the relevant ANDA applicants can request FDA to remove the 30-month stay and seek final approval, potentially launching 18 to 24 months before the prior forecast. The revenue impact depends on whether the brand has successfully shifted patients to a follow-on formulation.
If the brand has executed a successful soft switch and moved 60 percent of its prescribers to the next formulation, the generic launch on the old formulation captures only 40 percent of the original revenue base, reducing the annual LOE impact from approximately $1.35 billion (90% erosion of $1.5B) to approximately $540 million. If the soft switch has not succeeded and patients remain on the old formulation, the full $1.35 billion cliff materializes 18 months earlier than projected, which can move the brand company’s stock price by 15 to 25 percent on the announcement date.
What Happens to Patients When a Product Hop Is Blocked by a Court?
The Namenda case provides a direct data point. When Forest was forced to keep Namenda IR on the market, Alzheimer’s patients who were switched to Namenda XR could remain there if they tolerated it well, while patients and physicians who preferred the IR formulation had the option to switch to generic memantine IR when it became available in mid-2015. Generic memantine IR launched at prices roughly 85 percent below Namenda IR’s list price. Payers, particularly Medicaid programs, benefited substantially.
The patient population most affected by successful product hops tends to be elderly patients with Medicare Part D coverage, where fixed co-pays for brand-name drugs at the preferred tier can be lower than the co-pays for non-preferred generic substitutes during the period when the product hop is being executed and the new formulation is being placed at preferred tiers. PBM formulary management can temporarily make the new brand cheaper out-of-pocket than the newly available generic of the old formulation, even while total system cost is higher.
The Role of DrugPatentWatch and Patent Monitoring in Generic Strategy
For anyone tracking pharmaceutical competitive intelligence, patent expiration monitoring is a core function. DrugPatentWatch maintains a comprehensive database of FDA Orange Book patent listings, Paragraph IV certifications, ANDA litigation filings, patent expiration dates, exclusivity periods, and ANDA approval statuses for both small molecules and biologics. Industry participants, from generic manufacturers evaluating their ANDA filing pipelines to institutional investors modeling revenue cliffs for brand companies, use resources like DrugPatentWatch to track these developments in real time.
The practical value of systematic patent monitoring extends beyond individual product decisions. Tracking the pattern of continuation patent filings, Orange Book additions, and ANDA challenges across a manufacturer’s portfolio reveals the company’s competitive defense strategy in ways that are not apparent from any single patent or filing. A manufacturer that consistently adds Orange Book patents on aging products in the 18-to-24-month window before expected generic entry, for example, is executing a deliberate thickening strategy that can be identified and anticipated by generic manufacturers and their investors.
How to Use Patent Expiry Data to Build a Drug LOE Calendar for Investment Research
An LOE calendar for investment research purposes combines Orange Book patent expiration dates with exclusivity expiration dates and Paragraph IV challenge data to produce a timeline of expected generic entry events. The key steps are: identify all patents listed in the Orange Book for each target product, sort by expiration date to find the last patent standing (the one whose expiration most plausibly triggers generic entry), overlay any active Paragraph IV certifications with their litigation status to assess whether patents could be invalidated before expiration, and add any FDA exclusivity periods that extend beyond the patent dates.
The result is a range of LOE scenarios: an optimistic case (all patents are challenged successfully, generic entry occurs at the earliest Paragraph IV settlement date or ANDA approval date), a base case (patents hold until their last expiration date), and a pessimistic case (a formulation change or other tactic extends effective exclusivity by 2 to 5 years beyond the base case). Scenario probabilities are then assigned based on litigation status, patent quality, and commercial reform signals. This framework is used routinely by healthcare investment teams at major asset managers and hedge funds.
Regulatory and Legislative Responses to Generic Delay: What Has Actually Changed
Congressional and regulatory responses to generic delay tactics have been incremental and incomplete. Several proposals have become law; many others have stalled. Understanding what has changed, and what has not, helps in assessing which tactics remain fully operational and which carry increased legal risk.
The CREATES Act: What It Changed for REMS Sample Access and What Remains Unresolved
As described earlier, the CREATES Act of 2019 created a federal cause of action for generic manufacturers denied product samples under the guise of REMS compliance. Early court decisions under the CREATES Act have generally allowed cases to proceed past motions to dismiss, finding that brand manufacturers cannot simply assert REMS safety requirements as an absolute defense without demonstrating that the sample sharing they refused to undertake would actually have violated the REMS. Whether the Act has substantively reduced the incidence of REMS-based sample blocking is not yet clear from published litigation data, though FDA has noted a reduction in reported REMS sample access complaints since the Act’s enactment.
The FTC’s 2023 Campaign Against Improper Orange Book Listings: What It Means for Patent Thickets
The FTC’s 2023 initiative on Orange Book patent listings was the most aggressive direct challenge to the patent thicket mechanism in recent years. The FTC sent letters to multiple drug manufacturers, including those making respiratory inhalers and auto-injectors, challenging the legitimacy of device patents listed for drug-device combination products. The letters were accompanied by a policy statement arguing that these listings delay competition in violation of the FTC Act.
Several manufacturers responded to FTC letters by withdrawing patents from Orange Book listings. Sanofi withdrew certain device patents for Lantus SoloStar and Toujeo. Other companies delisted device patents from combination inhaler products. FDA’s own review process for the withdrawn patents’ appropriateness remained pending as of mid-2025. Whether the voluntary delistings will accelerate generic entry on the affected products is not yet clear, because ANDA applicants still need to design around or invalidate any remaining listed patents.
Proposed Legislation: Protecting Drug Innovation Act, Drug Price Transparency Laws, and LOE Acceleration Proposals
Congress has considered dozens of bills targeting pharmaceutical patent abuse since 2016, with most failing to advance past committee hearings. The most credible active proposals as of 2025 include a proposal to require FDA to review Orange Book patent listings for compliance with listing standards (rather than relying solely on sponsor certifications), a proposal to eliminate pediatric exclusivity for drugs where no pediatric indication exists, a proposal to require disclosure of all settlement agreements in ANDA litigation, and a proposal to expand the FTC’s authority to challenge pay-for-delay settlements involving non-cash compensation.
None of these proposals had been enacted as of the time this article was written. The pharmaceutical industry’s lobbying presence in Congress, combined with genuine policy debates about the effect of patent reform on drug development incentives, has produced a pattern of legislative activity without legislation. The Biden administration’s drug pricing provisions in the Inflation Reduction Act of 2022 focused primarily on Medicare price negotiation rather than patent reform, leaving the generic delay machinery largely intact.
Supply Chain and Manufacturing Considerations After Patent Expiry: Why Generic Entry Still Takes Time
Even when all legal barriers to generic entry have been cleared, practical supply chain and manufacturing constraints add months to the timeline. Understanding these constraints is essential for anyone forecasting actual generic availability, as opposed to legal eligibility for approval.
API Manufacturing Lead Times and ANDA Approval-to-Launch Gaps
A generic manufacturer whose ANDA is approved by FDA still needs to have its active pharmaceutical ingredient (API) supply chain operational at commercial scale before it can launch. For complex APIs, particularly those requiring specialized synthetic chemistry or controlled substance precursor licenses, establishing commercial-scale API production can take 12 to 24 months after FDA approval. This creates a gap between ANDA approval and actual launch that the brand company’s commercial team tracks carefully, because it determines how long the brand has before meaningful supply of the generic is available to pharmacies.
For biologic products, the manufacturing lead time for biosimilars is substantially longer. Commercial biologic manufacturing requires validated cell banks, bioreactor scale-up, downstream purification process validation, and product characterization studies that collectively take 18 to 36 months to establish at commercial scale. The total capital investment required for a biosimilar commercial manufacturing program is typically $150 million to $300 million, which limits the number of companies that can realistically launch biosimilars of any given reference product and influences the degree of price competition that follows biosimilar entry.
FDA ANDA Approval Backlogs and the Impact on Generic Entry Timing
The FDA’s Office of Generic Drugs manages a review queue that can extend ANDA approval timelines beyond what the legal patent and exclusivity picture would otherwise allow. Complete response letters requiring additional data or manufacturing corrections, plant inspections at foreign manufacturing facilities, and information requests on bioequivalence methodologies all add time to the ANDA review process. For complex generics, particularly those involving modified-release formulations, inhalation products, or topical products with complex bioequivalence standards, FDA review can take 4 to 6 years from initial filing.
FDA’s Generic Drug User Fee Act programs, renewed most recently in 2022, have reduced median ANDA approval times substantially from the 4-to-5-year cycle times that prevailed before 2012. But the agency still faces challenges with complex generics, and brand manufacturers who succeed in obtaining complex product designations for their reference drugs can rely on the FDA review complexity as an indirect barrier to generic entry that no antitrust law directly addresses.
International Perspective: How Generic Delay Tactics Differ Outside the United States
The generic delay tactics described in this article are primarily U.S.-specific in their mechanics, though analogous strategies exist in other jurisdictions. Several features of U.S. law make it uniquely hospitable to patent thickets and product hops: the continuation patent system, the Orange Book linkage mechanism that automatically stays generic approvals upon patent assertion, the 180-day exclusivity incentive that creates value in paying off the first generic filer, and the REMS system that can be exploited for sample blocking.
European Generic Entry Timelines vs. U.S.: Why the Same Drug Loses Exclusivity Faster in Europe
European patent systems allow fewer continuation-style applications than the U.S. system, limiting the accumulation of large patent portfolios around a single drug. The European Medicines Agency’s regulatory data protection system is 8 to 11 years (compared to 5 years of data exclusivity plus potential extensions in the U.S.), but European patent courts have been more willing to invalidate secondary pharmaceutical patents and less tolerant of Orange Book-style linkage mechanisms that automatically delay regulatory approvals.
The practical result is visible in biosimilar penetration statistics. European biosimilar markets for infliximab (Remicade), etanercept (Enbrel), adalimumab (Humira), and rituximab (Rituxan) all achieved 40 to 70 percent market penetration within two years of first biosimilar entry. U.S. biosimilar market penetration for comparable products ran at 10 to 20 percent two years post-entry through most of the 2018-2022 period, reflecting the combined effect of patent thickets, formulary exclusion contracts, and the absence of an interchangeability mechanism for most of that period.
Case Studies: Five Branded Drugs That Used Delay Tactics and What Happened
Case Study 1: Nexium (Esomeprazole) and the ‘Purple Pill’ Enantiomer Strategy
AstraZeneca’s Nexium is among the most analyzed examples of enantiomer-based product hopping. Omeprazole (Prilosec) is a racemic mixture of two mirror-image molecules. Esomeprazole (Nexium) is the S-enantiomer alone. AstraZeneca filed new composition-of-matter patents on the S-enantiomer as Prilosec’s primary patents were approaching expiration, obtained FDA approval for Nexium as a new chemical entity, and migrated its prescriber base from Prilosec to Nexium before Prilosec generics arrived.
The clinical differentiation argument for Nexium was always thin: meta-analyses consistently found minimal clinical differences between Nexium and generic omeprazole for most patients at equivalent doses. But the commercial migration was successful enough that Nexium became a $7 billion annual product by 2005, generating enormous revenue even after generic omeprazole had captured most of the Prilosec market. Nexium itself lost exclusivity in 2014 when its own composition patents expired, and generic esomeprazole entered the market rapidly.
Case Study 2: Concerta (Methylphenidate OROS) and the Bioequivalence Controversy
ALZA Corporation (later acquired by Johnson & Johnson’s Janssen division) developed Concerta using OROS (Osmotic Release Oral System) technology, a sophisticated drug delivery system that released methylphenidate in a specific ascending-dose pattern over 12 hours. Concerta’s OROS patents were listed in the Orange Book, and generic manufacturers filed Paragraph IV challenges. After litigation settled and generic ANDAs were approved, FDA subsequently determined that several approved generic versions of Concerta were not bioequivalent to the branded product, triggering a rare FDA action in 2014 recommending that prescribers not switch patients to those generic versions.
The FDA bioequivalence issue, later resolved through revised bioequivalence standards, created years of confusion in the market. Some generic versions were pulled from pharmacy shelves. Others remained available but with FDA labeling reservations. J&J’s authorized generic of Concerta maintained significant market share during the confusion period. The episode illustrated how complex drug delivery technology can create post-approval bioequivalence issues that benefit the brand, even absent any deliberate anticompetitive conduct by the manufacturer.
Case Study 3: Revlimid (Lenalidomide) and Celgene’s Volume-Restricted License Strategy
Bristol Myers Squibb (through its acquisition of Celgene) agreed to settlement-based authorized generic entry for Revlimid (lenalidomide) beginning in 2022, but with a highly unusual volume restriction: the authorized generic would be limited to a small fraction of total U.S. lenalidomide volume for several years, gradually increasing until full generic competition was permitted around 2026. The FTC challenged the volume-restriction structure as an anticompetitive agreement to restrain generic supply, arguing that it was functionally equivalent to a delayed entry date and therefore subject to Actavis scrutiny.
The FTC filed suit in 2023 against BMS over the Revlimid settlement structure. The case, pending in the D.C. Circuit as of mid-2025, could determine whether volume-restricted authorized generic settlements constitute illegal pay-for-delay arrangements. Revlimid generated approximately $12 billion in 2021 global revenues, and the volume restrictions in the settlement agreements, if upheld by courts, could defer full price competition on lenalidomide until the mid-2020s.
Case Study 4: Glumetza (Metformin Extended Release) and the 500 Mg vs. 1000 Mg Exclusivity Dispute
Assertio Therapeutics and Valeant (later Bausch Health) prosecuted Glumetza, an extended-release metformin formulation, through a series of settlements with Paragraph IV ANDA filers that contained provisions restricting the volume of generic metformin ER available in the market. A class action antitrust suit filed in 2019 alleged that the settlements were pay-for-delay arrangements that maintained Glumetza’s price at many times the cost of generic metformin IR.
The case settled for $300 million in 2020 before trial, with the defendants neither admitting nor denying liability. The settlement amount was notable for a product that was not a blockbuster by top-tier pharmaceutical standards, suggesting that courts and defendants alike recognized the structural vulnerability of the settlement structure under Actavis.
Case Study 5: Restasis (Cyclosporine Ophthalmic) and the IPR Jurisdiction Dispute
Allergan’s Restasis (cyclosporine 0.05% ophthalmic emulsion) faced multiple IPR petitions from generic manufacturers including Teva and Mylan challenging the validity of its formulation patents. In a controversial 2017 maneuver, Allergan assigned its Restasis patents to the Saint Regis Mohawk Tribe, a federally recognized sovereign Native American tribe, and then licensed them back. The strategy was designed to exploit the doctrine of tribal sovereign immunity to shield the patents from PTAB IPR proceedings, which are administrative rather than judicial proceedings from which sovereign entities can claim immunity.
The Federal Circuit rejected the strategy in 2018, holding that tribal sovereign immunity does not apply to inter partes review proceedings because IPR is not an action against the tribe but a proceeding to determine the validity of a patent grant that the federal government has authority to review. The Restasis patents were ultimately held unpatentable in IPR proceedings, and generic cyclosporine ophthalmic entered the market in 2022 at prices substantially below Allergan’s list price.
What This Means for Generic Manufacturers, Investors, and Health Systems
What This Means for Generic Pharmaceutical Manufacturers
Generic manufacturers need to build patent challenge capability that extends well beyond simple Orange Book expiration calendars. A product hop that moves 70 percent of a market to a new formulation can make a successful ANDA approval on the original formulation commercially worthless within two years of launch. Generic manufacturers who monitor formulation change filings, track physician prescription pattern shifts, and assess the commercial viability of ANDA targets in the context of the full competitive landscape will allocate their ANDA pipelines more efficiently than those who focus solely on patent expiration dates.
The IPR pathway at PTAB has become a critical tool for thinning patent thickets before launching full district court ANDA litigation. A generic manufacturer that systematically files IPR petitions on the weakest patents in a thicket, wins PTAB invalidations, and then uses those outcomes as leverage in settlement discussions has a qualitatively different strategic position than one that files a Paragraph IV certification and waits passively for the litigation to resolve.
What This Means for Healthcare Investors and Analysts
For investors in branded pharmaceutical companies, the key analytical question is no longer ‘when does the patent expire?’ but ‘which of the delay tactics are in play, how durable are they legally, and what is the probability distribution of effective LOE dates?’ A company that has successfully executed a product hop on its flagship product, maintained formulation patent protection on the new version, and obtained pediatric exclusivity has a qualitatively different risk profile than one relying solely on a single compound patent.
For investors in generic pharmaceutical companies, the question is symmetric: which branded products have delay tactics that are legally vulnerable, and what is the cost-benefit of challenging them? Resources like DrugPatentWatch allow systematic screening across the Orange Book for products where the gap between nominal patent expiration and the last-standing patent in a thicket is large, where continuation patent applications are still pending, and where no Paragraph IV challenge has yet been filed. Those gaps represent pipeline opportunities.
What This Means for Hospital Systems and Pharmacy Benefit Managers
Hospital formulary committees and PBM formulary designers need specific policies for evaluating brand-to-brand switches when the new formulation is introduced as a product hop rather than a genuine clinical advance. Developing clinical criteria for formulary tier placement of new formulations relative to the pending generic of the original formulation, and maintaining the original formulation on formulary through the generic entry period, can capture generic savings that would otherwise be lost to a successful soft switch.
PBM contracts with rebate provisions that favor new formulations should be examined carefully when those formulations are being introduced by manufacturers who are simultaneously seeking to reduce generic substitution of the prior formulation. Rebate optimization models that ignore the generic savings opportunity on the original formulation will systematically understate the total cost impact of product hops.
Key Takeaways
Product hopping, in both its hard-switch and soft-switch variants, is the most commercially significant generic delay tactic, and courts have begun drawing clearer lines between legitimate product reformulation and anticompetitive market manipulation. Hard switches that pull the original product before generic entry are most vulnerable to antitrust challenge after the Second Circuit’s Namenda ruling.
Patent thickets work by accumulating continuation patents that individually may be weak but collectively require generic challengers to conduct parallel litigation across dozens of patents, each triggering its own 30-month FDA approval stay. AbbVie’s Humira thicket delayed U.S. biosimilar competition by approximately five years relative to European markets.
Reverse payment settlements are not per se illegal under FTC v. Actavis but are subject to rule-of-reason antitrust scrutiny. Non-cash settlements including authorized generic rights, supply agreements, and co-promotion deals have replaced explicit cash payments as the primary vehicle for pay-for-delay arrangements.
REMS programs have been systematically exploited to deny generic manufacturers product samples needed for bioequivalence testing. The CREATES Act created a federal cause of action for sample-blocking, but enforcement data on the Act’s effectiveness is still accumulating.
Pediatric exclusivity adds six months to all unexpired Orange Book exclusivities and is routinely obtained for high-revenue products without meaningful clinical benefit to pediatric patients. The financial return on investment for the required studies typically exceeds 10:1.
Orange Book listing abuse, particularly of device patents for drug-device combination products, is now explicitly on the FTC’s enforcement radar, with multiple manufacturers having voluntarily delisted patents following FTC challenge letters in 2023 and 2024.
Biosimilar-specific barriers include the 12-year BPCIA exclusivity period, the patent dance disclosure mechanism, interchangeability designation requirements, and formulary exclusion contracts funded by manufacturer rebates to PBMs. Each layer independently reduces biosimilar price competition below the level European markets have achieved.
Effective LOE analysis requires integrating patent expiration calendars, Paragraph IV challenge tracking, IPR status, regulatory exclusivity stacks, and commercial signals of product switching, not just reading a single patent expiration date from the Orange Book.
FAQ: Brand Generic Delay Tactics, Patent Evergreening, and Market Exclusivity
What is product hopping in the pharmaceutical industry?
Product hopping is a strategy where a brand-name drug manufacturer introduces a reformulated version of an existing product, such as an extended-release capsule version of a twice-daily tablet, then shifts prescriber and patient volume to the new version before the original formulation faces generic competition. Because state pharmacy substitution laws require therapeutic equivalence for automatic generic substitution, a generic of the original formulation cannot automatically substitute for the reformulated brand, effectively stranding the market away from generic savings.
How does a patent thicket delay generic drug entry?
A patent thicket is a cluster of overlapping patents covering different aspects of a drug, from its chemical structure to its coating formulation to its dosing device. Each patent can be listed in the Orange Book, and asserting even one listed patent against an ANDA filer triggers a 30-month stay on FDA generic drug approval. A generic manufacturer facing a thicket of 20 patents must either challenge all of them simultaneously (expensive and time-consuming) or wait for all of them to expire (sometimes decades).
What is a pay-for-delay settlement and is it legal?
A pay-for-delay or reverse payment settlement occurs when a brand-name drug manufacturer pays a generic challenger to drop its patent challenge and delay market entry. After the Supreme Court’s 2013 ruling in FTC v. Actavis, these settlements are not immune from antitrust law and must be evaluated under a rule-of-reason standard. Large, unexplained payments to delay entry are more likely to be found anticompetitive. Non-cash compensation in the form of authorized generic rights or side agreements is now more commonly used.
What is the 180-day generic exclusivity period and how is it manipulated?
Under Hatch-Waxman, the first generic manufacturer to file a Paragraph IV ANDA and successfully challenge a brand patent earns 180 days during which no other generic can receive final FDA approval. Brand manufacturers deploy authorized generics, which are sold under the brand’s own NDA rather than an ANDA, to compete directly with the 180-day exclusivity holder during this period, halving the generic’s duopoly profit and reducing the commercial incentive for future patent challenges.
How does FDA’s Orange Book work and what is wrong with how patents are listed in it?
The Orange Book lists patents that the NDA holder certifies claim its approved drug product, its active ingredient, or its method of use. FDA historically accepted these certifications without substantive review. This allowed manufacturers to list patents of dubious relevance, including device patents, process patents, and patents covering metabolites rather than the drug itself, forcing generic manufacturers to file unnecessary Paragraph IV certifications and triggering unjustified 30-month stays. The FTC challenged numerous improper listings in 2023 and 2024.
What is the CREATES Act and how does it help generic drug manufacturers?
The Creating and Restoring Equal Access to Equivalent Samples Act (2019) created a federal cause of action allowing generic and biosimilar manufacturers to sue brand companies that refuse to provide the product samples needed for bioequivalence or comparative testing, when the refusal is unjustified by REMS safety requirements. Before the CREATES Act, brand manufacturers routinely denied samples by claiming REMS compliance constraints, delaying generic development by years.
How long is FDA exclusivity for biologics under the BPCIA and can it be extended?
The Biologics Price Competition and Innovation Act grants 12 years of reference product exclusivity to approved biologics, with a 4-year period during which no biosimilar application can even be filed. This exclusivity runs from the approval date of the reference biologic, not from initial clinical development or patent filing. Pediatric exclusivity can add six months to BPCIA exclusivity periods. Patent thickets and interchangeability requirements add effective commercial exclusivity beyond the statutory periods.
What is the difference between a biosimilar and an interchangeable biosimilar?
A biosimilar is a biologic product FDA has determined to be highly similar to an approved reference biologic with no clinically meaningful differences in safety and effectiveness. An interchangeable biosimilar has met additional requirements demonstrating that switching between the biosimilar and the reference product does not produce greater clinical risk than continued use of either product alone. Only interchangeable biosimilars can be automatically substituted at the pharmacy under most state pharmacy practice laws without a new physician prescription.
What happened in FTC v. Actavis and why does it still matter?
In FTC v. Actavis, Inc. (2013), the Supreme Court held that reverse payment settlements in ANDA patent cases are not automatically shielded from antitrust scrutiny by the existence of the underlying patent. Courts must evaluate them under the rule of reason, considering the size of the payment, the delay it produced, and the competitive harm. The decision ended the ‘scope of the patent’ immunity doctrine that had made pay-for-delay agreements legally safe in several circuits, though it fell short of declaring them per se illegal.
What data sources do analysts use to track pharmaceutical patent expirations and generic entry timelines?
Analysts use a combination of FDA Orange Book records (available at fda.gov), ANDA filing and approval data from FDA’s Drugs@FDA database, PTAB inter partes review petition records at USPTO, court docket databases for ANDA litigation, and commercial patent intelligence services. DrugPatentWatch integrates many of these sources and provides organized patent portfolios, exclusivity calendars, Paragraph IV certification records, and ANDA litigation tracking across the full landscape of FDA-approved drugs, making it a standard resource for pharmaceutical competitive intelligence work.
Sources
Association for Accessible Medicines. (2023). 2023 U.S. Generic & Biosimilar Medicines Savings Report. Association for Accessible Medicines.
Federal Trade Commission. (2011). Authorized Generic Drugs: Short-Term Effects and Long-Term Impact. U.S. Federal Trade Commission.
Federal Trade Commission. (2022). Fiscal Year 2022 Summary of ANDA Settlements: A Report on Pharmaceutical Patent Settlements. U.S. Federal Trade Commission.
Federal Trade Commission. (2023). FTC Policy Statement on Orange Book Listings. U.S. Federal Trade Commission.
Federal Trade Commission. (2024). Pharmacy Benefit Managers: The Powerful Middlemen Inflating Drug Costs and Squeezing Main Street Pharmacies. U.S. Federal Trade Commission.
FTC v. Actavis, Inc., 570 U.S. 136 (2013).
New York v. Actavis PLC, 787 F.3d 638 (2d Cir. 2015).
Sandoz Inc. v. Amgen Inc., 582 U.S. 1 (2017).
Alpern, J. D., Stauffer, W. M., & Kesselheim, A. S. (2014). High-cost generic drugs: Implications for patients and policymakers. New England Journal of Medicine, 371(20), 1859-1862.
Beall, R. F., Nickerson, J. W., Kaplan, W. A., & Attaran, A. (2016). Is patent ‘evergreening’ restricting access to medicine/device combination products? PLOS ONE, 11(2), e0148939.
Bawa, R. (2011). Protecting innovation and enabling competition: The Hatch-Waxman Act as a model for biosimilar regulation. Drug Discovery Today, 16(15-16), 607-609.
Feldman, R., & Wang, E. L. (2017). A new pharmaceutical patent settlement database sheds light on potential anticompetitive practices. Stanford Technology Law Review, 20, 339-373.
Government Accountability Office. (2020). Drug Pricing: Research on Savings From Generic Drug Use. GAO-21-21. U.S. Government Accountability Office.
Kesselheim, A. S., Avorn, J., & Sarpatwari, A. (2016). The high cost of prescription drugs in the United States: Origins and prospects for reform. JAMA, 316(8), 858-871.
Kessler, R. (2019, June 17). ‘CREATES Act’ Passes Congress, Blocks Drug Firms From Withholding Samples. Reuters Health.
Laleh, R. (2016). Patient access to biosimilars: The interchangeability question. The Journal of Law and the Biosciences, 3(2), 272-285.
Lipsky, J. J. (2013). Authorized generics. Nature Reviews Drug Discovery, 12(10), 728-729.
Mulcahy, A. W., Hlavka, J. P., & Case, S. R. (2018). Biosimilar cost savings in the United States: Initial experience and future potential. RAND Health Quarterly, 7(4), 3.
Sagonowsky, E. (2021, June 2). How AbbVie uses patent thickets and lawsuits to fight off Humira biosimilar competition. FiercePharma.
U.S. Senate Finance Committee. (2023). Insulin: Examining the Factors Driving the Rising Cost of a Century-Old Drug. U.S. Senate Finance Committee.
Van Arnum, P. (2022, August 15). Tracking LOE events: A look at major patent cliff products. DCAT Value Chain Insights.
Wouters, O. J., McKee, M., & Luyten, J. (2020). Estimated research and development investment needed to bring a new medicine to market, 2009-2018. JAMA, 323(9), 844-853.
DrugPatentWatch. (2024). Patent and exclusivity data for FDA-approved drugs. Retrieved from https://www.drugpatentwatch.com
Carrier, M. A. (2011). Why pay-for-delay agreements are anticompetitive and what courts can do about it. Yale Journal of Health Policy, Law, and Ethics, 11, 367-398.
Congressional Budget Office. (2021). Research and Development in the Pharmaceutical Industry. U.S. Congressional Budget Office.
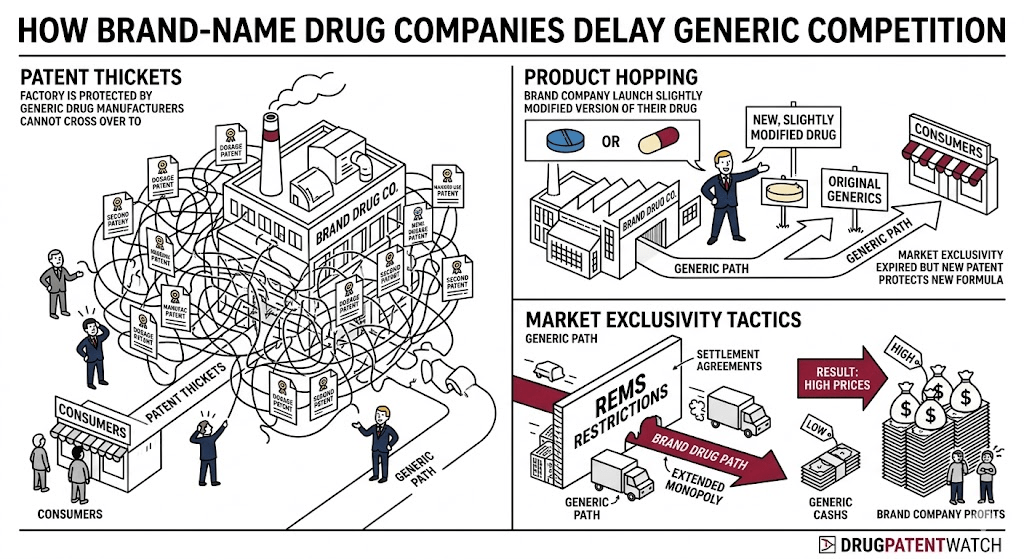


")























