1. The Anatomy of a U.S. Drug Patent: Knowing the Battlefield
The dominant narrative in pharmaceutical IP for most of the past three decades was one of inevitability. A brand company files its patents, runs its clinical trials, gets its drug approved, and then waits out the clock while competitors circle from a safe distance. When the key patent expired, everyone called it a ‘patent cliff’ and moved on. That model is functionally dead.
What replaced it is a continuous adversarial contest. Generic and biosimilar manufacturers no longer wait for expiration dates. They treat a brand’s patent portfolio as a structure to be analyzed, probed for weaknesses, and attacked at the most commercially efficient entry point. The metric has shifted from ‘patent expiration date’ to ‘Loss of Exclusivity’ (LOE), a date that can be moved years earlier through deliberate legal and regulatory strategy. Every day of accelerated market entry against a blockbuster generates measurable value, often millions of dollars.
This guide is built for challengers. Not the kind who file Paragraph IV certifications reactively, hoping the brand company will not sue. The kind who conduct forensic patent analysis before an ANDA is filed, who have a prior art theory in hand before submitting the notice letter, and who understand that the settlement negotiation begins with the quality of the invalidity case, not with the first court filing.
78%New drug patents cover existing drugs, not new molecules
66%Top-selling drug patent apps filed after market launch
76%Overall Para IV success rate (including settlements)
80%+PTAB IPR claim cancellation rate at institution (Jan 2025)
Understanding how to dismantle a drug patent requires first understanding how one is built. A U.S. patent is a property right, not a regulatory certificate. It is granted by the USPTO for inventions that meet a specific set of statutory criteria, not because a drug works or sells well. The commercial value of a patent depends entirely on the legal durability of the claims it contains, and that durability is the variable that challengers must accurately measure before committing capital to a litigation program.
Key Takeaway
The drug patent ecosystem in 2025 is characterized by deliberate obfuscation. Brand manufacturers list patents that are secondary to any original invention, often filed years after launch, specifically to create barriers to generic entry that go beyond the original molecule’s protection. The challenger’s job is to distinguish patents that represent genuine invention from patents that represent legal real estate built on weak foundations. That distinction drives every downstream strategic decision.
2. The Four Patentability Pillars and Their Exploit Surfaces
A U.S. patent claim is only as strong as the weakest statutory requirement it satisfies. The USPTO must, in theory, apply four requirements before granting a patent: novelty, non-obviousness, utility, and adequate written description and enablement. Each is a potential line of attack. Challengers who understand the specific legal standards, the governing case law, and the practical evidence required for each ground can construct invalidation arguments with precision rather than volume.
Novelty: 35 U.S.C. § 102
Novelty requires that the claimed invention was not publicly disclosed before the patent’s effective filing date. Any single piece of prior art that discloses every element of a patent claim destroys novelty outright, a theory called anticipation. The America Invents Act (AIA), effective March 2013, expanded the scope of prior art to a true global standard: a scientific paper published in any language in any country, a product sold anywhere in the world, or a public disclosure in any accessible form can qualify. Pre-AIA patents (filing date before March 16, 2013) use a more complex geographic standard, and many of the drug patents currently in active litigation fall under pre-AIA rules.
For pharmaceutical patents, the most commercially productive novelty challenges involve polymorphs. A compound that can crystallize in multiple solid-state forms is a common drug chemistry phenomenon. Brand companies often obtain separate patents on specific polymorphic forms discovered late in a drug’s development cycle. The challenge is that polymorph patent claims frequently face an inherent anticipation argument: if a prior art manufacturing process for the parent compound would inevitably produce the new polymorph as part of the process, even if the polymorph was not identified at the time, the prior art may anticipate the claim. This inherent anticipation theory does not require proving that the prior art author recognized the polymorph’s existence, only that the process necessarily produced it.
Non-Obviousness: 35 U.S.C. § 103
Non-obviousness is the highest-volume battleground in pharmaceutical patent litigation. The standard asks whether the differences between the claimed invention and the prior art would have been apparent to a person having ordinary skill in the art (POSITA) at the time of the invention. The POSITA is a legal construct calibrated to the field in question: for a small-molecule drug formulation patent, the POSITA might be a pharmaceutical scientist with a Ph.D. and five years of formulation experience. For a biologic dosing regimen, the POSITA might be a clinical pharmacologist with specific therapeutic area knowledge.
The Supreme Court’s 2007 decision in KSR International Co. v. Teleflex Inc. remains the controlling standard. KSR rejected the prior ‘teaching, suggestion, or motivation’ (TSM) test that required patent examiners and courts to find an explicit reason in the prior art to combine references. The KSR approach is more flexible and more demanding on patent applicants: it allows courts and the PTAB to use common sense and ordinary creativity in assessing whether a skilled artisan would have combined known elements to achieve a known result. For secondary pharmaceutical patents covering formulation improvements, dosage adjustments, or new routes of administration, KSR substantially expanded the scope of what counts as obvious.
Secondary considerations, sometimes called objective indicia of non-obviousness, include commercial success, long-felt but unresolved need, failure of others, and unexpected results. Brand companies invoke these regularly to defend secondary patents. The key countermove for challengers is to establish that commercial success was attributable to the original molecule’s therapeutic effect rather than the specific claimed modification, and that any ‘unexpected results’ were either expected in light of known science or not commensurate with the full scope of the claims.
Utility: 35 U.S.C. § 101
For approved pharmaceutical drugs, utility challenges are rarely the primary theory. The FDA’s approval for a specific indication establishes specific and credible utility as a matter of practical evidence. Utility arguments occasionally arise in early-stage biotech patents where a compound’s function is described in vague terms or where the claimed utility is speculative relative to the disclosed data, but for mature drug products already generating commercial revenue, § 101 utility is seldom the decisive battleground.
Subject matter eligibility under § 101 is a separate and more active issue for diagnostic method patents and certain biomarker-based claims, particularly after the Supreme Court’s decisions in Mayo Collaborative Services v. Prometheus Laboratories (2012) and Association for Molecular Pathology v. Myriad Genetics (2013). Companies with patent portfolios built heavily on diagnostic or biomarker correlations should map those claims against the Mayo/Alice framework carefully before asserting them in litigation.
Written Description and Enablement: 35 U.S.C. § 112
Section 112 contains two distinct requirements that are increasingly powerful against biologic and biosimilar patents. The written description requirement demands that the patent specification demonstrate that the inventor possessed the claimed invention at the time of filing. The enablement requirement demands that the specification teach a POSITA how to make and use the full scope of the claimed invention without undue experimentation.
The Supreme Court’s 2023 decision in Amgen Inc. v. Sanofi fundamentally recalibrated the enablement standard for broad functional claims in biologics. Amgen had claimed an entire genus of antibodies defined by their function, binding to a specific epitope on PCSK9 and blocking its interaction with LDL receptors, while disclosing only about two dozen specific examples. The Court found that the full scope of these functional claims encompassed potentially millions of antibodies that a POSITA could not make without undue experimentation. The patent was invalid for lack of enablement.
Amgen v. Sanofi did not only affect Repatha’s patent portfolio. It introduced a template for attacking any biologic patent that claims a large genus of antibodies by function rather than structure. Challengers developing biosimilars against antibody-based biologics should audit the originator’s patent claims against the Amgen v. Sanofi standard as a first screening step.
For PTAB proceedings, § 112 invalidity grounds are not available in an Inter Partes Review (IPR). They can only be raised in a Post-Grant Review (PGR), which requires filing within nine months of patent grant, or in district court litigation. This limitation has become a material strategic variable: a biologic patent with a clear Amgen-type enablement problem is a candidate for PGR, not IPR, and the nine-month window from patent grant is the operative deadline that determines whether the PGR route is available.
Key Takeaways — Section 2
Non-obviousness under the post-KSR standard is the dominant theory in most secondary pharmaceutical patent challenges, particularly for formulation, dosing, and method-of-use patents where prior art often supports an ‘obvious to try’ analysis. Enablement under the Amgen v. Sanofi standard is the emerging primary theory against broad functional claims in biologic patents and applies in PGR or district court, not IPR. Anticipation is the strongest single-theory ground when a clear prior art reference exists, because it requires no balancing of probabilities; one reference disclosing all claim elements ends the analysis.
Investment Strategy Note
Reading PTAB Petition Grounds as Signals
When a generic or biosimilar company files a PTAB IPR or PGR petition, the chosen invalidity grounds are public record and are informative about the challenger’s confidence level. An IPR petition restricted to obviousness arguments tells you the challenger’s best theory is prior art combination; it likely did not find a clean anticipation reference. A PGR petition invoking § 112 enablement against a broad biologic patent signals that the challenger reviewed the claim scope and concluded it fails the Amgen v. Sanofi standard. Tracking petitions filed against a brand’s key patents, the grounds asserted, and the PTAB’s institution decision gives equity analysts an early read on LOE probability years before any district court decision.
3. Patents vs. Regulatory Exclusivity: The Two-Layer Defense You Must Defeat Separately
The most expensive strategic error a generic company can make is treating patents and regulatory exclusivities as the same type of barrier. They are not. A successfully invalidated patent does not open the market if a regulatory exclusivity period is still running. The two systems are legally independent, operate on different timelines, are granted by different agencies, and require completely different response strategies.
Patents are property rights granted by the USPTO. They can be challenged, invalidated, designed around, or licensed. A patent’s effective protection period varies based on filing date, patent term adjustments for USPTO examination delays, and any Patent Term Extension (PTE) granted to compensate for FDA review time. The 20-year term is a starting point, not a guarantee.
Regulatory exclusivities are statutory marketing rights granted by FDA upon drug approval. They cannot be challenged on validity grounds because they are not based on any claimed invention. They run from the FDA approval date for a fixed statutory period that varies by exclusivity type. The only ways to address a regulatory exclusivity are to wait it out, qualify for the statutory exception that allows ANDA filing during NCE exclusivity after year four, or argue that the brand did not properly qualify for the claimed exclusivity category.
Statutory criteria met at approval (NCE, orphan, pediatric, etc.)
Duration
20 yr from filing date; subject to PTE, PTA, and terminal disclaimers
Fixed: 3, 5, 7, 12 yr from approval date (by type)
Can be challenged?
Yes: via IPR, PGR, district court, reexamination
No: cannot be invalidated; must be waited out or navigated
Key types
Composition of matter, formulation, method of use, process, polymorph
NCE (5 yr), Orphan (7 yr), New Clinical Investigation (3 yr), Pediatric (+6 mo), Biologic (12 yr)
Strategic implication
Patent validity is the target of Paragraph IV and PTAB proceedings
Exclusivity type determines when ANDA/BLA filing is permitted; drives timeline planning
The Hatch-Waxman Act’s five-year NCE exclusivity includes a specific exception that allows a generic company to file its ANDA after four years, triggering the Paragraph IV litigation process during that fourth year. This design is not accidental. It ensures that the 30-month stay resulting from patent litigation can run concurrently with the final year of NCE exclusivity, theoretically allowing both barriers to expire at approximately the same time if the challenger times its filing precisely.
Pediatric exclusivity is the regulatory barrier most frequently underestimated by challengers. It adds six months to every existing patent and exclusivity listed for the drug, not just to specific patents. For a drug with multiple late-expiring formulation patents and an ongoing patent litigation program, a six-month pediatric extension can shift the entire commercial timeline and materially affect the value of a first-to-file ANDA position.
Orphan drug exclusivity (ODE) is particularly complex for combination products and drugs with multiple indications. ODE protects the drug only for the specific orphan indication for which it was designated. A generic seeking approval only for the non-orphan indication of a drug with an active ODE is not necessarily blocked. The FDA’s application of ODE to combination indications and subsequent approvals has been contested in litigation, and challengers should analyze the ODE scope carefully rather than treating it as a blanket barrier.
Key Takeaways — Section 3
Patent invalidation and regulatory exclusivity navigation are parallel workstreams, not sequential ones. A Paragraph IV challenge filed against the patent stack of a drug still three years into orphan drug exclusivity will resolve the patent dispute years before it can generate any commercial benefit. Challengers must map the complete exclusivity timeline before committing to a litigation strategy, because the commercially relevant LOE date is the later of all patent and exclusivity barriers, not just the soonest patent expiration.
4. Secondary Patents: Identifying the Weakest Links in the Patent Thicket
A composition-of-matter patent covering the API is almost always the most legally durable protection in a drug’s portfolio. It was filed earliest, covers the broadest subject matter, and was subjected to the most rigorous scrutiny during examination. Everything filed after it, the formulation patents, the polymorph patents, the dosing regimen patents, the device patents, occupies legally weaker territory. The statistical evidence is consistent: approximately 78% of new drug patents cover existing drugs rather than new molecules, and for the top-selling drugs, 66% of patent applications are filed after the drug is already on the market. These are the targets.
Formulation Patents
Formulation patents claim a specific physical presentation of a known active ingredient: an extended-release matrix using a particular polymer, a microencapsulated suspension with defined particle size ranges, a liposomal formulation with a specific lipid composition. The obviousness attack on a formulation patent requires showing that the general approach was well-known in pharmaceutical formulation science, that the specific excipients or techniques used were standard tools in the field, and that the result (e.g., extended release of the drug) was a predictable and routine outcome of applying those known techniques. The POSITA definition is critical here: a formulation scientist in the relevant subfield has a deep knowledge of the prior art, and what might seem novel to a layperson is often a textbook technique to the POSITA.
The most effective prior art for formulation patent attacks is non-patent literature: pharmaceutical formulation textbooks (Remington’s Pharmaceutical Sciences is a standard reference), product monographs for older formulations using the same techniques, and published research articles documenting the specific release mechanisms and excipient combinations used. The patent’s prosecution history is equally important: if the applicant distinguished prior art by arguing a specific technical feature (e.g., a particular polymer concentration range), that argument creates prosecution history estoppel limiting the claim scope and potentially supports a non-infringement design-around.
Polymorph Patents
A compound’s polymorphic landscape is a function of its crystalline chemistry, not a commercial decision. Manufacturers often discover new polymorphs during scale-up or optimization of their synthesis process, and they patent these forms as late-stage lifecycle management. The inherent anticipation theory is the primary challenge vehicle: if the prior art manufacturing process for the parent compound would necessarily produce the new polymorph as part of normal synthesis, even if not recognized at the time, the prior art anticipates the polymorph claim. Proving inherent anticipation requires expert witness testimony establishing that the prior art process inevitably generated the claimed form.
The non-obviousness attack on polymorph patents relies on the ‘routine screening’ argument: pharmaceutical solid-state chemistry practice routinely includes polymorph screening as a standard step in drug development, making it obvious for a skilled formulation scientist to screen for and characterize polymorphic forms of a known API. Post-KSR, this obvious-to-try argument is particularly strong when there are a finite number of known crystalline screening conditions and a reasonable expectation that the process would yield novel polymorphs.
Dosage Regimen Patents
Dosing schedule patents cover how often a drug is administered, by what route, in what amount. They are a standard late-stage lifecycle management tool because they are cheap to obtain (no new synthesis required), extend beyond the compound patent, and can support formulary arguments for brand retention after generic entry on the original schedule. The Copaxone 40mg three-times-weekly patent is the canonical example of how far this tactic can be pushed, and the canonical example of how thoroughly it can fail when challenged.
The obvious-to-try attack on dosing patents argues that when there are a finite number of dose levels or schedules that a skilled clinician would test, and when the prior art establishes motivation to optimize (e.g., improving patient adherence, reducing injection-site reactions), selecting one of those finite options is not an inventive step. The post-KSR standard is notably flexible on this point, and the Federal Circuit has affirmed multiple dosing patent invalidations on obvious-to-try grounds since 2007.
Method-of-Use Patents
A method-of-use patent covers the therapeutic application of a drug for a specific disease or condition. The generic challenger’s primary tool against method-of-use patents is the ‘skinny label,’ officially called carve-out labeling: FDA allows an ANDA approval that omits the patented indication from the generic label if the generic seeks approval only for the non-patented uses. This strategy avoids infringement of the method-of-use patent without requiring its invalidation. The limitation is that if physicians prescribe the generic for the patented indication off-label in significant volume, the brand company may argue induced infringement. The viability of the skinny label strategy requires careful analysis of prescribing patterns and the specific claim language of the method-of-use patent.
Device and Delivery System Patents
For combination products, the delivery device can become the effective monopoly long after the drug itself is off-patent. Epinephrine, the API in the EpiPen, has been freely available for decades. The auto-injector device is where all commercially meaningful IP resided. Inhaled corticosteroids, insulin pen injectors, and nasal spray devices follow the same pattern. Device patents tend to be mechanical or engineering patents rather than pharmaceutical chemistry patents, and they often require a different expert witness profile and prior art search strategy than compound or formulation patents. Industrial design patents and utility patents on device modifications are both in scope.
Key Takeaways — Section 4
Secondary patents are the operational target of most generic patent challenge programs. The specific vulnerability depends on the patent type: inherent anticipation for polymorphs, routine formulation science for formulation patents, obvious-to-try for dosing regimens, skinny labeling for method-of-use patents, and engineering prior art for device patents. Identifying the patent type correctly determines the expert witness profile needed, the prior art search strategy, and the appropriate invalidity theory. Conflating these types leads to misdirected litigation effort and higher costs.
5. Conducting a Lethal Prior Art Search: Methodology and Practice
Prior art searching for invalidation is different from prior art searching for patentability. A patentability search is defensive: find the landscape, assess risk, carve out claims. An invalidity search is offensive: find the one document or set of documents that destroys the asserted claims. The goal is not comprehensive landscape mapping but targeted destruction of specific patent elements. That difference in objective changes the search strategy, the resource allocation, and the criteria for success.
Claim-Level Analysis Before the Search Begins
An invalidity search that proceeds without first mapping the claim language is wasted effort. Every asserted claim must be parsed element by element before a single search query is run. Each element is a requirement that prior art must disclose; finding a reference that discloses nine of ten elements in an independent claim has no anticipation value and limited obviousness value. The claim chart, documenting which element each prior art reference addresses, is the working document that drives the search and ultimately drives the expert’s invalidity declaration.
For claims using functional language, the meaning of the claim must be construed before searching. A claim requiring a formulation that ‘provides a pharmacokinetic profile substantially similar to the reference product’ needs a construction of ‘substantially similar’ before you can assess whether prior art meets it. The prosecution history is the first place to look for that construction, because the applicant may have defined terms during examination in ways that are now binding on the patent owner.
The Three-Channel Search Strategy
A comprehensive invalidity prior art search uses three parallel channels: text and keyword searching, classification-based searching, and citation network analysis. Running all three independently, then merging and deduplicating results, maximizes coverage and minimizes the risk of missing the decisive reference.
Keyword searching starts with the exact claim language and expands systematically to synonyms and alternative technical terms. For a formulation patent claiming a ‘hydrophilic matrix sustained-release system,’ the search must include ‘hydroxypropyl methylcellulose matrix,’ ‘HPMC matrix,’ ‘controlled-release hydrogel,’ and every other standard descriptor for the technology in pharmaceutical sciences literature. Non-English language searches in German, Japanese, and Chinese are necessary for AIA patents because prior art is global; for pre-AIA patents, the geographic scope of prior art depends on the disclosure type (patents are global; prior public use was limited to the U.S. under pre-AIA law). Japanese patent literature and German pharmaceutical chemistry literature are particularly rich sources of overlooked prior art for compound and formulation patents.
Classification-based searching uses the Cooperative Patent Classification (CPC) system’s hierarchical structure to browse the technological neighborhood around the target patent. A drug formulation patent classified under CPC code A61K 9/20 (tablets) can be cross-searched with A61K 31/[relevant compound class] to find prior formulation art for the same or related compounds. Classification searching captures references that use different terminology for the same technology, which keyword searches systematically miss. The USPTO’s Patent Full-Text and Image Database (USPTO PatFT) and Espacenet support classification search, and Derwent Innovation provides more sophisticated cross-classification queries.
Citation analysis examines both the backward citations in the patent itself (references the examiner considered) and the forward citations to the patent from later publications. The examiner’s considered references are an explicit acknowledgment of the closest prior art as evaluated during prosecution. If the examiner reviewed a reference and still allowed the claims, the challenger needs to understand why, which informs the argument needed to overcome the prosecution’s prior art evaluation. Forward citations can reveal scientific publications or competing patents that characterize the same technology in ways the original examiner did not see, opening new invalidity avenues.
Non-Patent Literature: The Most Productive Search Space
For pharmaceutical patents on formulations, dosing regimens, and methods of use, non-patent literature is frequently more productive than the patent database. Scientific journals in pharmaceutical sciences (Journal of Pharmaceutical Sciences, European Journal of Pharmaceutics and Biopharmaceutics, AAPS Journal), clinical pharmacology (Clinical Pharmacology and Therapeutics), and the relevant therapeutic area (New England Journal of Medicine, The Lancet, specialty journals) are primary resources. Textbooks, particularly Remington’s Pharmaceutical Sciences for formulation and Goodman and Gilman’s for clinical pharmacology, document standard techniques in forms that qualify as prior art for any publication date before the patent’s priority date.
Academic dissertations and theses are systematically overlooked and represent some of the most valuable prior art. ProQuest Dissertations and Theses Global indexes over 5 million dissertations, many of which contain original pharmaceutical research that preceded corresponding patents by years. A dissertation describing a formulation technique that the patent applicant later ‘invented’ is devastating prior art, and it is frequently missed by applicants who searched only patent databases during prosecution.
Conference proceedings and abstracts are public prior art once presented, even before formal journal publication. The effective date of prior art from conference presentations is the presentation date, provided the abstract or proceeding was publicly accessible. Pharmaceutical sciences conferences (American Association of Pharmaceutical Scientists, American Chemical Society, Drug Information Association) and major therapeutic area meetings (ASCO, ASH, ADA) generate substantial prior art that predates many secondary pharmaceutical patents.
Key Takeaways — Section 5
The 80/20 rule applies to prior art searching: the first 20% of search time finds 80% of the references, but the last 20% of time is where decisive prior art often lives. Persistence in non-patent literature, particularly dissertations, foreign-language technical publications, and conference proceedings, consistently outperforms additional keyword patent searching after the initial broad patent search is complete. Every invalidity case that proceeds to PTAB or district court should have a documented search certification confirming that all three channels, text, classification, and citation analysis, were executed before concluding the search.
6. Mining the Prosecution History for Fatal Flaws: Estoppel, Disclaimers, and Contradictions
Prior art is the external weapon used to attack a patent. The prosecution history is the internal weapon that challengers often overlook or undervalue. The complete record of communication between a patent applicant and the USPTO examiner during prosecution, the file wrapper, is public and permanent. Every claim amendment made to overcome an examiner rejection, every argument made to distinguish prior art, and every statement made about the invention’s scope is part of the legal record that governs the patent’s interpretation in every subsequent proceeding.
Prosecution History Estoppel
Prosecution history estoppel prevents a patent owner from reclaiming in court subject matter that was surrendered during prosecution to obtain the patent. If an applicant narrowed a claim to overcome a prior art rejection, they cannot later invoke the doctrine of equivalents to extend the claim back to its original scope. The Festo Corporation v. Shoketsu Kinzoku Kabushiki Co. decisions established a rebuttable presumption that any claim narrowing creates estoppel over the surrendered territory, which the patent owner must overcome by proving the narrowing was made for a reason unrelated to patentability, was unforeseeable, or tangentially related to the equivalent at issue.
The practical application in pharmaceutical patent challenges is direct. Consider a claim that originally required a pharmaceutical composition with ‘an effective amount of drug X.’ The examiner rejects it over prior art disclosing the same drug in any therapeutic dose. The applicant amends to ‘a therapeutically effective amount of 10 to 50 mg of drug X.’ In subsequent litigation, the patent owner cannot use the doctrine of equivalents to argue that a generic product containing 55 mg infringes. They surrendered the scope above 50 mg to get the patent allowed, and estoppel bars its recapture. This is a non-infringement argument, not an invalidity argument, but it can be equally dispositive for achieving market entry without the burden of proving invalidity by clear and convincing evidence in district court.
Argument-Based Disclaimers
Formal claim amendments are not the only source of prosecution history estoppel. Arguments made during prosecution, without any claim amendment, can create a disclaimer of claim scope. If an applicant argues that their formulation is different from a prior art formulation because the prior art ‘lacks the critical pH adjustment step that provides the claimed stability improvement,’ that argument is a disclaimer: the claim now implicitly requires that pH adjustment step, even if the claim language does not explicitly state it. In infringement litigation, the generic can argue that a product without a pH adjustment step does not infringe, relying on the applicant’s own prosecution argument to narrow the claim’s scope.
Finding argument-based disclaimers requires reading every office action response in full, not just the claim amendment section. Applicants frequently make arguments in the body of a response that their attorneys did not intend to be claim-limiting, but courts apply the legal standard based on how a reasonable examiner would have understood the statement, not on the applicant’s subjective intent. For major pharmaceutical patents with complex prosecution histories spanning multiple continuations and continuation-in-part applications, this analysis can take weeks and requires patent counsel with both prosecution and litigation experience.
Cross-Portfolio Contradictions
Brand pharmaceutical companies file dozens or hundreds of patents related to a single drug across its development lifecycle. Each patent has its own prosecution history. The strategic opportunity is that statements made in one prosecution to obtain a claim can directly contradict statements made in another prosecution to obtain a different claim, or arguments made in litigation to defend a different patent. The Humira IPR cases illustrate this precisely: AbbVie argued in one IPR that Humira’s commercial success was attributable to its dosing regimen’s novelty, while in a separate IPR for a different Humira patent, AbbVie attributed commercial success to the formulation. The PTAB used those contradictory positions to undermine AbbVie’s credibility and the weight of its commercial success argument in both proceedings.
Mining cross-portfolio contradictions requires aggregating prosecution histories across the entire patent family, which for major biologics can include dozens of related U.S. applications, corresponding PCT applications, and national phase applications in key jurisdictions (EPO, Japan, China). Statements made to the EPO during prosecution of a European counterpart patent are admissible in U.S. proceedings to inform claim interpretation. Foreign prosecution statements can be devastating prior art for claim construction if they reveal admissions about the invention’s scope that the U.S. applicant preferred to avoid in the domestic prosecution.
Key Takeaways — Section 6
Prosecution history analysis is forensic work. It requires reading the entire file wrapper, not just the claims and drawings, and it requires cross-referencing the prosecution histories of related patents in the same family. The most commercially productive outputs are argument-based disclaimers that support non-infringement positions, prosecution history estoppel that blocks doctrine of equivalents claims by the brand, and cross-portfolio contradictions that undermine the brand’s litigation credibility. These findings frequently determine whether a generic product can be designed around the asserted patents entirely, which is a lower-risk outcome than full-scale invalidity litigation.
7. Orange Book and Commercial Intelligence Infrastructure
The FDA’s Orange Book is the legal document that connects approved drug products to their associated patents and regulatory exclusivities. It is also, by design, an incomplete and imperfect map. Understanding what it tells you, what it conceals, and how to use commercial intelligence platforms to fill the gaps is a foundational operational requirement for any generic or biosimilar challenge program.
What the Orange Book Reveals and What It Hides
The Orange Book discloses every patent that the brand manufacturer has listed with FDA as covering the approved product. FDA does not verify these listings for accuracy or relevance; it accepts them as submitted. The listing is the brand’s self-assessment of which patents cover the product, and it is binding on the brand in one specific way: the brand can only assert in Hatch-Waxman litigation the patents it has listed. A patent not listed in the Orange Book cannot trigger the 30-month stay if a generic files a Paragraph IV certification against it.
Patent Use Codes in the Orange Book describe the specific method of use or indication that each listed patent covers. These codes are critical for evaluating skinny label strategies: if a use code covers only a specific indication (e.g., U-123: treatment of moderate-to-severe plaque psoriasis in adults), a generic seeking approval only for other non-coded indications may carve that use from its label and avoid the listed patent entirely. The scope and accuracy of use codes are contested. Brand companies have been accused of listing overly broad or inaccurate use codes to expand the apparent scope of their patent protection, and FDA has increased scrutiny of these listings following FTC challenges.
What the Orange Book does not tell you is equally important. Manufacturing and process patents are not listable in the Orange Book under the Hatch-Waxman regulations. A generic company that limits its patent landscape analysis to Orange Book listings will miss these patents entirely. They cannot trigger a 30-month stay, but they can be asserted in a separate infringement action after the generic launches if the generic’s manufacturing process infringes the process patent. Process patents can be substantial barriers to market even for a generic that has resolved every Orange Book patent, making their identification a necessary part of any complete IP landscape analysis.
Commercial Intelligence Platforms
DrugPatentWatch and comparable commercial patent intelligence platforms address the Orange Book’s blind spots by aggregating data from the Orange Book, USPTO patent database, PCT filings, PTAB proceedings, district court dockets, and international patent office records. The integrated view allows challengers to identify all patents related to a target drug, including process patents absent from the Orange Book, litigation history against related patents, PTAB petitions filed by prior challengers, and patent expiration timelines that account for patent term adjustments, patent term extensions, and terminal disclaimers.
Historical litigation data available through these platforms enables probability-weighted assessment of a specific patent challenge. If the same assignee has previously litigated and lost on obviousness grounds against formulation patents with similar claim structures, that track record informs the current program’s risk assessment and settlement leverage. If a prior challenger’s IPR petition against the same patent was denied institution, the petition contents are public record and reveal both the theory attempted and the PTAB’s specific reason for denial, which directly guides the current challenger’s petition strategy.
Real-time monitoring capabilities allow challengers to track new patent filings by a brand company against a target molecule, new Orange Book listings, ANDA filings by competitors, and PTAB petition filings by other challengers. Being the second filer in a Paragraph IV race means losing the 180-day exclusivity. The monitoring function is not supplementary; it is operational infrastructure for any company with an active ANDA pipeline.
Key Takeaways — Section 7
The Orange Book is the starting point, not the complete picture. Process patents, unlisted formulation patents in foreign jurisdictions, and PTAB petition histories require commercial intelligence platforms to capture. A generic company that makes filing decisions based only on Orange Book data is working with a systematically incomplete view of the IP landscape. For any ANDA program with 180-day exclusivity value exceeding $50 million, the investment in comprehensive intelligence infrastructure is a rounding error against the potential return.
8. The Invalidation Toolkit: Comparing Your Venues
U.S. law provides three primary venues for challenging the validity of a pharmaceutical patent: district court litigation under the Hatch-Waxman framework, Inter Partes Review before the PTAB, and Post-Grant Review before the PTAB. Each venue has different procedural rules, cost profiles, timelines, burden of proof standards, decision-maker expertise, and strategic consequences. Choosing the correct venue, or the correct combination of venues, is the most consequential early-stage decision in any challenge program.
Feature
District Court (Hatch-Waxman)
Inter Partes Review (IPR)
Post-Grant Review (PGR)
Timing
45 days from Para IV notice triggers stay; anytime otherwise
After 9 months from patent grant; anytime
Within 9 months of patent grant only
Invalidity grounds
Any: §§ 101, 102, 103, 112, double patenting
§§ 102 and 103 only, based on patents and printed publications
Any statutory ground: §§ 101, 102, 103, 112
Decision-maker
District court judge; jury for some issues
Panel of 3 Administrative Patent Judges (APJs)
Panel of 3 Administrative Patent Judges (APJs)
Burden of proof
Clear and convincing evidence
Preponderance of the evidence
Preponderance of the evidence
Presumption of validity
Yes; patent is presumed valid
No presumption of validity
No presumption of validity
Timeline
2 to 4+ years plus appeals
Final decision within 1 year of institution (18 months from petition)
Final decision within 1 year of institution (18 months from petition)
Estimated cost
$1M to $4M+
$300K to $600K
$350K to $700K+
Estoppel scope
Res judicata on litigated issues
Grounds raised or reasonably could have been raised in IPR are estopped in other proceedings
Grounds raised or reasonably could have been raised in PGR are estopped in other proceedings
Primary use case
Required for 180-day exclusivity eligibility; full invalidity and infringement resolution
Cost-effective challenge on prior art grounds; parallel to district court
Broad attack on newly issued patents; § 112 enablement challenges against biologic patents
The estoppel consequence of IPR filing is the most underappreciated strategic risk in the venue selection analysis. If a petitioner raises or ‘reasonably could have raised’ a ground of invalidity in an IPR, and the PTAB institutes review and issues a final decision, the petitioner is estopped from raising those grounds in any later district court or ITC proceeding. This means a petitioner who runs an IPR on the best prior art references and loses cannot then use those references in district court infringement litigation. The IPR’s cheaper cost must be weighed against this estoppel risk for each invalidity theory.
Key Takeaways — Section 8
IPR and district court litigation are complementary, not mutually exclusive. The standard practice for generic defendants in Hatch-Waxman litigation is to file a parallel IPR against the asserted patents, request a district court stay pending the PTAB’s decision, and use the PTAB’s faster and more defendant-friendly forum to invalidate claims before the district court must rule. PGR is a separate tool for newly issued biologic patents with § 112 enablement vulnerabilities under Amgen v. Sanofi, and the nine-month window requires monitoring patent issuance dates to deploy it in time.
9. Hatch-Waxman Paragraph IV: The Full Operational Guide
The Paragraph IV certification is the formal mechanism by which a generic company asserts that a brand’s Orange Book-listed patent is invalid, unenforceable, or will not be infringed by the generic product. By statute, this certification constitutes an artificial act of patent infringement, giving the brand company the right to sue before any generic product reaches the market. The entire process is a carefully sequenced series of legal triggers, each with specific timelines and strategic consequences.
The ANDA Filing Decision and Paragraph IV Certification
The decision to include a Paragraph IV certification in an ANDA filing is not made at the time of filing; it is made months earlier, during the patent landscape analysis and prior art development phase. By the time an ANDA is submitted, the generic company should have a fully developed invalidity theory for each patent it is certifying against, a draft notice letter framework, and a litigation team with identified outside counsel experienced in Hatch-Waxman patent litigation. Filing a Paragraph IV without this preparation is the equivalent of declaring a war without knowing the terrain.
For each Orange Book-listed patent, the ANDA filer must choose among four certifications. Paragraph I certifies that no patent information has been filed with FDA. Paragraph II certifies that the patent has expired. Paragraph III certifies that the patent will expire before the date of generic market entry. Paragraph IV is the challenge certification. A generic company can file a Paragraph III certification for patents it cannot challenge and a Paragraph IV for patents it believes are vulnerable, creating a mixed certification ANDA that sequences market entry based on the results of the challenge litigation and the remaining term of the Paragraph III patents.
The 20-Day Notice Letter: Strategic Drafting
After FDA acknowledges receipt of the ANDA for filing (a separate determination from filing itself), the generic company has exactly 20 days to send a Paragraph IV notice letter to each patent owner and the NDA holder. The letter must include a detailed, claim-by-claim statement of the factual and legal basis for the invalidity or non-infringement position. This is not boilerplate; it is the opening statement of the litigation.
The letter must be detailed enough to satisfy the statutory requirement, which is interpreted as giving the brand company sufficient notice to evaluate whether to file suit. But the drafting carries strategic trade-offs: a letter that reveals the precise prior art references the generic intends to use in litigation gives the brand company months to prepare its counter-arguments before the 30-month stay clock begins. A letter that describes the invalidity theories more generally satisfies the statutory requirement but provides less intelligence to the opponent.
Courts have occasionally ruled that a notice letter was legally deficient, which can have severe consequences including the loss of first-filer status or the invalidation of the Paragraph IV certification itself. The legal standard for letter sufficiency requires claim-level specificity: for each challenged patent, the letter must specify which claims are challenged and the factual and legal basis for the challenge to each claim. Generic companies with active Paragraph IV programs maintain developed letter templates and quality control processes to ensure compliance with this requirement under tight timelines.
The 45-Day Window and 30-Month Stay
After receiving the Paragraph IV notice letter, the brand company has 45 days to file a patent infringement lawsuit in federal district court. If it files within 45 days, an automatic 30-month stay is triggered, during which FDA cannot grant final approval to the ANDA. The stay runs from the date the brand received the Paragraph IV notice, not from the suit filing date. It can be shortened by court order if the district court rules the patent invalid or not infringed before the 30 months expire, or lengthened by court order if the generic failed to reasonably cooperate in discovery or scheduling.
If the brand does not file within the 45-day window, FDA can proceed to approve the ANDA without any stay, subject only to the remaining patent and regulatory exclusivity periods. A brand’s decision not to sue within 45 days is strategically significant: it signals that the brand has assessed the invalidity or non-infringement arguments in the notice letter and concluded that litigation is not worth the cost or the risk of a judicial invalidity ruling that would be binding in future litigation against other filers. Challengers should monitor the 45-day window carefully and document whether a suit was filed.
The 180-Day Exclusivity: Mechanics and Forfeiture Risks
The first generic company to file a substantially complete ANDA containing a Paragraph IV certification earns the right to 180 days of marketing exclusivity upon approval and commercial marketing. ‘First to file’ is determined at the date of ANDA filing, down to the time of receipt; two companies filing on the same date share the exclusivity. FDA does not award exclusivity to a single filer among simultaneous filers.
Forfeiture of the 180-day exclusivity is one of the most heavily litigated procedural issues in generic drug law. The statutory forfeiture events include failure to market the generic drug within 75 days of receiving final FDA approval (or 30 months after filing, whichever is earlier), withdrawal or abandonment of the ANDA, failure to receive tentative approval within 30 months of ANDA filing, and amendment of the Paragraph IV certification to a different paragraph. Brand companies and subsequent filers monitor first-filers’ ANDA status actively, looking for forfeiture events that would open the market to their own ANDAs without waiting for the 180 days to run.
Key Takeaways — Section 9
The Paragraph IV process is a legal machine with precise procedural requirements at each step, and a procedural failure at any point can forfeit the 180-day exclusivity that justified the entire program. The 20-day notice letter timeline, the claim-level specificity requirement, the 45-day monitoring window, and the 75-day marketing obligation are all operational compliance requirements that require dedicated internal process management, not ad hoc handling. Generic companies with multiple simultaneous ANDA programs need formalized workflows and calendar management systems for each of these triggers.
Investment Strategy Note
ANDA Pipeline Analysis for Equity Research
For analysts covering generic pharmaceutical companies, the ANDA pipeline is the primary driver of near-term revenue visibility. The key variables are: number of first-to-file Paragraph IV positions held, the revenue of the reference listed drugs at risk, the litigation status and timeline for each challenge, and the probability of 180-day exclusivity being awarded versus shared with co-filers. An ANDA pipeline concentrated in five or fewer first-to-file positions against high-revenue brands is a higher-variance asset than one with twenty positions across a broader revenue range. Both can be valuable; the risk profile differs materially.
10. Inter Partes Review: The PTAB Advantage in Detail
The America Invents Act of 2011 created the Patent Trial and Appeal Board (PTAB) as an administrative tribunal within the USPTO with jurisdiction to review the validity of issued patents. The IPR proceeding, which replaced the prior inter partes reexamination process, is the most heavily used PTAB proceeding in pharmaceutical patent challenges and has fundamentally altered the litigation economics of the generic industry.
Why PTAB Panels Outperform District Court Juries for Technical Arguments
An IPR petition is decided by a panel of three Administrative Patent Judges (APJs). These are experienced patent attorneys, most with graduate degrees in science or engineering. They understand technical arguments, follow complex prior art combinations, and apply the legal standards for obviousness with more consistency than a general-jurisdiction district court jury that has no pharmaceutical science background and may be encountering the concept of a POSITA for the first time during voir dire.
The practical consequence is that technical invalidity arguments work better at the PTAB than in front of a district court jury. Complex prior art combinations, multiple-reference obviousness arguments, and technical claim construction disputes all benefit from a technically sophisticated fact-finder. For pharmaceutical formulation patents, polymorph patents, and biologic dosing regimen patents, the PTAB’s technical competence is a structural advantage for challengers.
Institution Threshold and Petition Strategy
The PTAB institutes an IPR only if the petition establishes a ‘reasonable likelihood’ that the petitioner would prevail on at least one challenged claim. This threshold is lower than the ‘clear and convincing’ standard required to actually prevail on the merits, but it is a meaningful filter: roughly half of all filed IPR petitions are instituted for review. For pharmaceutical patents specifically, the institution rate has varied by patent type, with method-of-use and dosing regimen patents showing higher institution rates than compound patents.
Petition quality is the primary determinant of institution success. A petition must present all arguments and evidence within the page limits (currently 14,000 words for the petition itself) without any supplemental argument permitted before institution. Every argument must be supported by an expert declaration explaining the technical basis, the POSITA analysis, and the application of the invalidity theory to each challenged claim element. Weak petitions that present arguments at a high level without claim-specific technical support are denied institution at rates substantially higher than well-developed petitions.
The page limit constraint requires prioritization. A petition that spreads its argument across eight patents and twenty invalidity theories gives each theory insufficient development. The most effective IPR petitions target no more than three to five claims with two or three well-developed invalidity grounds per claim, supported by a comprehensive expert declaration. Depth of argument on fewer theories consistently outperforms breadth of theories in institution decisions.
PTAB Statistics: Current Performance Data
As of January 2025, the PTAB cancelled over 80% of all claims for which it instituted review in that month’s final written decisions. For context, historical data from 2012 through 2017 showed biopharma IPR claim invalidity rates of approximately 36%, substantially below the chemical sector rate of 62% and the overall average across all technology areas. The biopharma rate has increased since then, partly due to improved petition quality as practitioners developed experience with the forum and partly due to the PTAB’s evolving approach to pharmaceutical obviousness arguments post-KSR.
The gap between biopharma and other sectors historically reflected two factors: the complexity of pharmaceutical obviousness arguments and the active use of secondary considerations (commercial success, long-felt need) by brand manufacturers to defeat obviousness challenges. As the PTAB has developed more nuanced analysis of secondary considerations, particularly requiring a tight nexus between the commercial success and the specific claimed invention rather than the product broadly, the biopharma gap has narrowed.
An IPR petition is not a lawsuit. It is a technical brief to a panel of expert judges who will read every word and evaluate every claim element. The quality of the prior art and the quality of the expert witness analysis, not the volume of the arguments, determines the outcome. PTAB practice observation, pharmaceutical IPR context
The Parallel District Court and IPR Two-Front Strategy
Filing an IPR petition against patents asserted in concurrent district court Hatch-Waxman litigation, then moving the district court for a stay pending the PTAB’s decision, is the standard operating procedure for well-resourced generic defendants. District courts grant stays pending IPR in the majority of cases when the IPR covers the same claims at issue in the district court litigation, because a PTAB invalidity decision on the claims eliminates the need for the district court to adjudicate the same issues.
The timing of the IPR petition relative to the district court litigation requires coordination. An IPR petition must be filed within one year of service of the infringement complaint. Filing too early can waste petition page limits on claim interpretations that the district court will later adopt differently. Filing too late cuts into the petition preparation time. The standard approach is to file the IPR petition after receiving the brand’s infringement contentions in district court discovery, which reveal which claims the brand is asserting, allowing the petition to focus on exactly those claims.
Key Takeaways — Section 10
IPR is the highest-value lever in the pharmaceutical patent challenge toolkit for prior art-based invalidity. The combination of no presumption of validity, preponderance of evidence standard, technically sophisticated decision-makers, and a one-year timeline makes it structurally more favorable for challengers than district court on pure invalidity arguments. The estoppel consequence requires careful selection of which grounds to raise, and the petition quality requirement makes early investment in expert witness development and claim charting essential. IPR petition quality is not correlated with spending; it is correlated with specificity, technical precision, and depth of claim-level analysis.
11. Post-Grant Review: The Expanded Arsenal for New Patents
Post-Grant Review (PGR) is the PTAB proceeding that allows challenge on any statutory ground of invalidity, including § 112 written description and enablement, § 101 subject matter eligibility, and indefiniteness, in addition to the prior art grounds available in IPR. The trade-off for this expanded scope is a strict filing deadline: a PGR petition must be filed within nine months of a patent’s grant date. After nine months, the patent can only be challenged in IPR or district court.
The nine-month window makes PGR a time-critical, event-driven proceeding. Companies with active patent monitoring programs that track issuances by brand pharmaceutical companies in their therapeutic areas of focus are positioned to file PGR petitions. Companies without real-time patent issuance monitoring will miss the window entirely, because nine months from grant to filing is not a long runway when accounting for the time required to develop a comprehensive invalidity petition with supporting expert declarations.
PGR for Biologic Patents Post-Amgen v. Sanofi
The Supreme Court’s 2023 Amgen v. Sanofi decision created the most commercially significant PGR opportunity in recent years for biosimilar developers. Any biologic patent claiming a genus of antibodies or other biologics defined primarily by their function, rather than by structure or sequence, is now a PGR candidate under the enablement theory the Court applied. The analysis requires comparing the claim scope (how many antibodies or functional variants does the claim potentially cover?) against the number of specific examples disclosed in the specification (how many did the applicant actually make and test?).
When the ratio of claimed scope to disclosed examples is large, and when producing new members of the claimed genus would require ‘substantial time and effort’ by a skilled artisan rather than routine application of known techniques, the Amgen v. Sanofi standard supports a PGR enablement challenge. Biosimilar developers monitoring their reference biologic manufacturers’ patent issuances should have a standard workflow that automatically triggers an Amgen-type enablement analysis within 30 days of any new biologic patent grant, leaving sufficient time to develop and file a PGR petition within the nine-month window if the analysis warrants it.
PGR Institution Threshold
The institution threshold for PGR is higher than for IPR: the petitioner must show it is ‘more likely than not’ (greater than 50% probability) that at least one challenged claim is unpatentable, compared to the ‘reasonable likelihood’ (some probability above a minimal threshold) required for IPR institution. This higher threshold reflects PGR’s broader attack scope and reflects Congressional intent to prevent PGR from being used as a general-purpose attack on all issued patents regardless of merit.
Key Takeaways — Section 11
PGR is the correct venue when the invalidity theory is § 112 enablement or subject matter eligibility, grounds not available in IPR. The nine-month deadline makes it a time-critical proceeding requiring continuous patent monitoring infrastructure. For biosimilar developers, the Amgen v. Sanofi enablement standard has created a systematic screening test that should be applied to every new biologic patent grant by reference product manufacturers in their areas of focus. A PGR petition based on a strong Amgen-type analysis, filed within the nine-month window, is materially more valuable than an IPR petition on prior art when the target patent’s primary weakness is claim scope exceeding disclosed examples.
12. The Economics of a Paragraph IV Challenge: ROI Analysis
Generic companies invest in patent litigation for one reason: the financial return justifies the cost. The math is not complex at a high level. The 180-day exclusivity period creates a temporary duopoly between the brand and the first generic. During those six months, the generic can price at a modest discount to the brand and still capture most of the prescription volume through automatic substitution. The margin profile of that 180-day window is dramatically better than any subsequent period, because after the exclusivity expires, additional generic entrants drive prices down rapidly and margins compress toward manufacturing cost.
15–25%Typical generic discount during 180-day exclusivity
70–90%Price erosion 12–24 months after full generic entry
$78BEstimated net social gain from Para IV challenges, hypertension market study
48%Generic win rate in contested district court decisions
For a brand drug generating $3 billion in annual U.S. revenue, a 180-day exclusivity period represents approximately $1.5 billion in gross generic market access. At the 15% to 25% price discount typical during the exclusivity window, and accounting for rapid market share capture through formulary substitution, a first-filer generic can realistically generate $400 million to $700 million in revenue during that period at margins substantially above its post-exclusivity run rate. A full Hatch-Waxman litigation program against that drug, including district court, potential appellate proceedings, and parallel IPR petitions, might cost $5 million to $10 million. The ROI calculation is unambiguous.
The cost-benefit calculation changes materially when the 180-day exclusivity is expected to be shared with co-filers or preempted by an authorized generic. An authorized generic launched simultaneously by the brand effectively creates a three-party market during the 180-day period, compressing the first-filer’s revenue capture significantly. Some branded companies have pre-committed to authorized generic launches through licensing agreements with generic subsidiaries. Others have agreed in patent settlement negotiations not to launch an authorized generic as a concession to secure the generic’s agreement to a specific market entry date. Evaluating the authorized generic probability for any target drug is a required component of the challenge ROI analysis.
The post-exclusivity market structure matters for the duration component of the ROI calculation. In markets where only two or three generics ultimately enter (due to complex bioequivalence requirements, narrow therapeutic index, or specialized manufacturing), price erosion is slower and margins remain elevated for longer. In markets where ten or more generics enter within 24 months of the exclusivity window closing, the first-filer’s post-exclusivity business is essentially a commodity business with margins near manufacturing cost. The competitive density projection for the post-exclusivity market belongs in the financial model from the program’s outset.
Investment Strategy Note
Para IV Filing Density as a Revenue Signal
The number of Paragraph IV filers against a specific drug is publicly trackable through FDA’s ANDA database. A large number of Para IV filers indicates broad market consensus that a patent challenge is viable and that the drug represents a commercially attractive generic target. It also signals that the 180-day exclusivity will be shared if multiple companies filed on the same date, which reduces per-filer value. An analyst tracking first-to-file date density can estimate the market’s view of exclusivity sharing probability. A brand drug with a single Para IV filer holding a clear first-to-file position represents maximum value for that filer. A drug with ten simultaneous filers on the same date represents shared value with compressed per-filer returns.
13. Settlement Strategy and Antitrust Guardrails
The majority of Paragraph IV patent disputes settle before reaching a final court verdict. This is not a failure of the litigation system; it is the predictable outcome of a framework that assigns asymmetric financial stakes to the parties. The brand faces catastrophic loss if a key patent is invalidated: an immediate, court-sanctioned conclusion to exclusivity and the arrival of multiple generic competitors within months. The generic faces a delayed but certain opportunity loss if the litigation drags for years past a patent’s effective expiration date. Both parties have strong incentives to negotiate a known outcome rather than litigate to a binary one.
What a Favorable Settlement Looks Like
A settlement in Paragraph IV litigation is, fundamentally, a negotiated launch date. The generic agrees to delay market entry beyond what would be immediately possible if it prevailed on the merits; the brand agrees to allow market entry before the patent’s natural expiration. The specific launch date negotiated reflects the relative perceived strength of the invalidity case, the remaining patent term, and the commercial value of the drug.
A generic company with strong invalidity evidence, a well-developed prior art case, and credible PTAB IPR petitions pending has substantial settlement leverage. The brand faces potential immediate LOE from an adverse PTAB decision that would be binding on all subsequent ANDAs. A generic company with weak evidence, a notice letter that revealed the full theory before suit was filed, and no parallel PTAB proceedings has little settlement leverage and will likely not obtain a meaningful early launch date.
Settlement agreements in pharmaceutical patent cases typically include license grants (allowing the generic to sell during a defined period before the patent expires), covenants not to sue, and in some cases, supply agreements or other commercial arrangements. The FTC reviews major pharmaceutical patent settlements and requires notification of agreements meeting specific thresholds.
Pay-for-Delay: The Antitrust Red Line
The Supreme Court’s 2013 FTC v. Actavis decision established that ‘reverse payment’ settlements, in which the brand company pays the generic challenger to delay market entry beyond the litigation’s natural resolution, are subject to antitrust scrutiny under the rule of reason. A large, unexplained reverse payment from a brand to a generic is circumstantial evidence that the parties recognized the weakness of the brand’s patent position and chose to divide the monopoly profits rather than litigate to a decision that would have eliminated them.
Post-Actavis, brand companies rarely pay cash directly to generic challengers to settle. Instead, alternative value exchanges are used: authorized generic agreements giving the challenger a portion of the market, supply agreements where the brand manufactures product for the generic, licenses to other drugs in the brand’s portfolio, and co-promotion arrangements. The FTC analyzes all of these structures for antitrust implications, and settlements that transfer value from the brand to the generic in amounts disproportionate to any legitimate business purpose remain vulnerable to FTC challenge and potential treble damages under the antitrust laws.
European regulators have been more aggressive than their U.S. counterparts in pursuing pay-for-delay cases. The European Commission fined Teva 462.6 million euros in 2024 related to Copaxone patent enforcement practices that included misuse of divisional patents and disparagement of generic competitors, a regulatory enforcement action that extended well beyond simple pay-for-delay allegations into a broader theory of anticompetitive conduct by a patent holder.
Key Takeaways — Section 13
Settlement negotiations in Paragraph IV cases are commercial negotiations about launch dates, conducted with invalidity evidence as the primary currency. The strength of the invalidity case, the quality of the prior art, and the status of parallel PTAB proceedings determine the generic’s leverage. Pay-for-delay settlements are antitrust-vulnerable under Actavis and face active FTC scrutiny; settlement structures that transfer significant value from brand to generic without legitimate business justification should be reviewed by antitrust counsel before execution. The goal is a settlement that delivers a commercially valuable launch date without creating legal risk that exceeds the value of the settlement itself.
14. Case Studies: Four Battles That Defined Modern Pharmaceutical Patent Strategy
Case Study 1
AbbVie Humira (Adalimumab): The 247-Patent Thicket and the IPR Precision Tool
Humira’s original composition-of-matter patent expired in the U.S. in 2016. AbbVie did not wait passively for the competitive landscape to open. It constructed what became the largest patent thicket in pharmaceutical history, accumulating over 247 patents related to adalimumab’s formulation, dosing regimens, manufacturing processes, and methods of use in specific indications. Approximately 89% of those patents were filed after Humira received FDA approval in 2002, a figure that encapsulates the evergreening strategy’s scale. The result was that no U.S. biosimilar launched until 2023, seven years after the compound patent expired, despite European biosimilar competition beginning in 2018.
Coherus Biosciences chose Inter Partes Review as its primary challenge vehicle, targeting specific dosing regimen patents one at a time rather than attempting to litigate the entire thicket simultaneously. In a key 2017 IPR victory, Coherus successfully invalidated all claims of AbbVie’s U.S. Patent No. 8,889,135, which covered a 40mg every-other-week subcutaneous dosing regimen. The PTAB found the claims obvious based on two prior art references, Kempeni (1999) and van de Putte (2004), which collectively disclosed the drug’s efficacy, its mechanism, and prior dosing exploration. Combining those references in the way a skilled rheumatologist would was not an inventive step under the post-KSR standard.
The commercial success secondary consideration argument AbbVie mounted was undermined by its own prior PTAB arguments. In a separate IPR for a different Humira formulation patent, AbbVie had attributed the drug’s commercial success to its formulation, not its dosing schedule. The PTAB found these positions irreconcilable and gave the commercial success argument minimal weight in the dosing regimen IPR. This outcome illustrates the danger of patent thicket strategies with inconsistent prosecution narratives: the volume of patents creates volume of potential contradictions.
Despite specific IPR victories by Coherus and others, the thicket’s breadth ultimately forced every major biosimilar developer into settlement agreements that included licensing the contested patents in exchange for specific U.S. launch dates, all beginning in January 2023. Amgen, Sandoz, Boehringer Ingelheim, Mylan, Samsung Bioepis, and Coherus all settled on negotiated dates rather than litigating through the remaining contested patents.
IP Valuation Analysis: Humira Patent Portfolio
The Thicket’s Financial Architecture
At its U.S. revenue peak, Humira generated approximately $21 billion annually. The compound patent expired in 2016. Without the secondary patent thicket and the settlement agreements it forced, U.S. biosimilar competition would likely have begun in 2017 or 2018, consistent with European market experience. The thicket successfully delayed U.S. competition by approximately five years from the compound patent’s expiration date, preserving an estimated $75 billion to $90 billion in additional protected U.S. revenue across that period.
The per-patent value of this thicket was not uniform. Most of the 247 patents contributed marginal incremental protection in isolation; their value was collective, creating the cost and complexity of a multi-patent litigation that most generic challengers could not sustain. The dosing regimen and formulation patents that Coherus and others challenged in IPR were individually the weakest, but their role in the thicket was to increase the cost and complexity of any challenge program rather than to independently defend market access.
Valuing the Humira IP portfolio for a potential acquirer or licensing transaction required distinguishing between patents whose invalidation would immediately open the market (high-criticality, high-value individually) and patents whose invalidation was a prerequisite for any other market entry (blocking patents). The compound patent was the most critical single asset. After its expiration, the thicket’s value was its aggregate barrier function, not any individual component’s legal strength. This distinction matters in portfolio M&A: the price should reflect the thicket’s aggregate deterrence value, discounted for its vulnerability to precisely the kind of coordinated IPR campaign that Coherus, Amgen, and Sandoz executed.
Case Study 2
Teva Copaxone (Glatiramer Acetate): Obviousness Defeats a Dosing Regimen at $4.3 Billion Peak Revenue
Copaxone’s original 20mg daily formulation had its core patents expire in the 2013 to 2015 timeframe, and multiple generics entered that market. Teva’s lifecycle management strategy was to develop and patent a 40mg three-times-weekly formulation, arguing that this new dosing schedule required inventive skill and produced unexpected results in patient adherence and tolerability. The secondary patents on the 40mg regimen were expected to defend a substantial portion of Copaxone’s revenue, which peaked at $4.3 billion annually, well into the next decade.
Sandoz and Mylan challenged the 40mg dosing patents on obviousness grounds. The core argument was direct and clinically grounded: any physician treating multiple sclerosis patients with an injectable drug would recognize the medical rationale for exploring less frequent dosing schedules. Reducing injection frequency from daily to three times weekly, with a compensating dose increase, was a routine clinical optimization strategy aimed at improving adherence and reducing injection-site reactions, both well-documented problems with daily glatiramer acetate. This was not an inventive step. It was clinical medicine.
The Federal Circuit agreed. The patents were invalidated on obviousness grounds. Teva’s commercial success argument failed because the improved adherence observed with the 40mg three-times-weekly regimen was attributable to the reduced injection frequency, which itself was the thing alleged to be obvious, creating a circular argument that the court rejected. This case established that clinical optimization of dosing schedules, even if commercially successful, does not generate patentable subject matter when the optimization direction is obvious from the medical and pharmacological literature.
Teva’s strategic response, executing a ‘product hop’ that converted nearly 70% of Copaxone prescriptions to the 40mg formulation before generic competition could be established, partially mitigated the financial damage. But the intellectual and legal premise of the 40mg strategy, that the dosing regimen was patentably non-obvious, was comprehensively rejected.
IP Valuation Analysis: Copaxone 40mg Patent Portfolio
Secondary Patent Value When the Primary Thesis Fails
The Copaxone 40mg dosing patents were valued by Teva’s management and equity analysts at the time of their filing as representing three to five years of additional protected revenue at approximately $4 billion annually, implying an aggregate IP value of $12 billion to $20 billion for those specific patents if they survived legal challenge. That valuation assumed the patents would prevail in litigation, which they did not.
The correct ex-ante valuation of the dosing regimen patents required assigning a probability weight to the invalidity risk. An informed analysis in 2010 to 2012, when the 40mg patents were being listed and the litigation was anticipated, would have required assessing the strength of the ‘obvious to try’ argument given the prior clinical literature on glatiramer acetate dosing exploration and the post-KSR standard for dosing regimen patents. That analysis, conducted rigorously, should have produced an invalidity probability substantially above 50%, which would have discounted the patent portfolio value by more than half against any undiscounted calculation. Financial models that treated the dosing patents as certain protection systematically overstated Copaxone’s LOE-adjusted revenue profile.
The European Commission’s separate antitrust enforcement action against Teva in 2024, resulting in a 462.6 million euro fine related to Copaxone patent practices, added a retrospective liability component to the portfolio’s net value. Aggressive patent thicket strategies that cross into anticompetitive conduct generate contingent liabilities that offset the IP’s market protection value in any comprehensive portfolio valuation.
Case Study 3
Mylan EpiPen (Epinephrine): When the Device Is the Drug and Antitrust Becomes the Battlefield
Epinephrine has been off-patent for decades. The active pharmaceutical ingredient in an EpiPen costs approximately $1 per dose to manufacture. The auto-injector device that makes epinephrine accessible for emergency self-administration is what carried all commercially meaningful IP protection. Mylan, which acquired the EpiPen from Merck KGaA in 2007, and its manufacturing partner Pfizer (through the King Pharmaceuticals acquisition), built patent protection around successive iterations of the auto-injector device, extending market exclusivity to approximately 2025.
The pricing strategy that accompanied this IP position became politically and legally untenable. A two-pack of EpiPens rose from approximately $100 in 2007 to over $600 by 2016, a price increase of more than 500% over nine years during which the cost of the API and basic manufacturing remained essentially flat. Congressional hearings, intense media coverage, and FTC scrutiny followed. The litigation against Mylan ultimately centered not on patent invalidity but on antitrust claims: allegations that Mylan used exclusionary contracts with pharmacy benefit managers, a ‘hard switch’ to a two-pack only to prevent dose-splitting, and ‘sham’ patent litigation to maintain the device monopoly illegally.
The antitrust class action settled with Mylan paying $264 million and Pfizer paying $345 million. No patent was judicially invalidated in the primary litigation. The lesson is specific: aggressive price increases on combination products protected by device patents, combined with exclusionary contracting practices, can transform a patent enforcement dispute into an antitrust case that generates damages on an entirely different scale than any patent invalidity proceeding.
IP Valuation Analysis: EpiPen Device Patent Portfolio
Combination Product IP and Antitrust Contingent Liability
The EpiPen’s device patent portfolio was, from a pure IP validity perspective, legally defensible. Auto-injector design improvements, needle cover mechanisms, and electronic dose-confirmation systems can represent genuine engineering innovation that satisfies patentability criteria. The patent portfolio was not the legal problem.
The legal problem was the pricing behavior enabled by the patent portfolio and the conduct used to defend the market position it created. Any valuation of the EpiPen IP assets that did not account for the antitrust contingent liability generated by the pricing and contracting strategy around those assets was structurally incomplete. The $609 million in combined settlements represents real cash paid against conduct enabled by the IP position, which means the IP portfolio’s net value to Mylan/Pfizer was reduced by that amount at minimum, not counting legal fees, reputational costs, and regulatory relations costs.
For institutional investors evaluating combination product companies with significant device patent protection around commodity APIs, the EpiPen case establishes a monitoring requirement: track the pricing trajectory relative to the IP exclusivity runway and the contracting practices used to defend market position. When pricing substantially exceeds cost inflation, exclusionary contracting has been implemented, and the device patent thicket has delayed multiple potential competitors, the antitrust contingent liability embedded in the IP position requires explicit quantification in the asset valuation.
Case Study 4
Amgen vs. Sandoz (Neupogen & Enbrel): Procedural Wins, Substantive Losses, and the BPCIA’s Complexity
The Amgen-Sandoz biosimilar litigation encompassed two distinct battles with opposite outcomes, offering a study in how BPCIA procedural strategy and substantive patent strength interact in ways that can determine decade-long competitive outcomes.
For Neupogen (filgrastim, G-CSF), the key dispute was procedural. The BPCIA requires a biosimilar applicant to provide 180 days’ notice of commercial marketing before launching. Amgen argued this notice could only be given after FDA biosimilar approval, effectively adding a mandatory six-month delay post-approval to every biosimilar launch. Sandoz argued the notice could be given before approval, allowing launch immediately upon FDA action. The Supreme Court’s unanimous 2017 ruling in Sandoz Inc. v. Amgen Inc. sided with Sandoz. Biosimilar developers can now give the 180-day commercial marketing notice before FDA approval, eliminating the mandatory six-month post-approval delay that Amgen sought to impose. This procedural win was worth hundreds of millions of dollars across the biosimilar industry.
For Enbrel (etanercept), the outcome was the opposite. Sandoz received FDA approval for Erelzi, its etanercept biosimilar, in 2016. It has still not launched commercially in the U.S. as of 2025. Amgen successfully defended two key secondary patents covering the etanercept fusion protein and its manufacturing process in district court and in the Federal Circuit. The courts rejected Sandoz’s invalidity arguments for obviousness-type double patenting and lack of written description, and the Supreme Court declined to hear Sandoz’s appeal, leaving those patents in force through their expiration in 2028 to 2029. Sandoz’s invalidity theories were technically complex doctrines that carry high evidentiary burdens, and Amgen’s patent positions were substantively stronger than many secondary pharmaceutical patents.
The Enbrel case is important specifically because it demonstrates that secondary patents can survive challenge when they represent genuinely non-obvious inventive steps. The manufacturing process patent covering Enbrel’s production from a specific CHO cell system with defined process parameters was not a cosmetic modification of a known process. The Federal Circuit found it represented a genuine technical contribution that satisfied the patentability requirements. Not every secondary patent is weak. The commercial premise of the biosimilar challenge, that all late-filed secondary patents are vulnerable, is an empirical generalization with real exceptions.
IP Valuation Analysis: Enbrel Manufacturing Process Patent
When Process Patents Are the Real Barrier
Enbrel’s compound and biologic exclusivity protections expired years before the manufacturing process patent that has actually kept Sandoz’s biosimilar off the U.S. market. The process patent covers the specific CHO cell culture and purification process used to produce etanercept with the quality characteristics required for therapeutic use. This is not a trivially incremental process patent; it covers the means of production for a biologically complex fusion protein in ways that a generic cell line and fermentation process cannot simply replicate.
For biosimilar portfolio valuation and competitive intelligence purposes, the Enbrel case requires a fundamental expansion of the patent analysis scope. IP due diligence for a biosimilar program that reviews only the reference biologic’s Orange Book-listed patents misses the category of manufacturing process patents entirely, because these are not Orange Book-listable. The relevant question is not only ‘what does the reference biologic company claim in its composition and method patents?’ but also ‘what does it claim in its manufacturing patents, and can a biosimilar manufacturer produce the protein using an alternative process that does not infringe?’
For investors evaluating biosimilar company valuations, the Enbrel case establishes that a single manufacturing process patent can create a market barrier worth over $4 billion in annual brand revenue (Amgen’s Enbrel U.S. revenues in the 2017 to 2023 period) and can extend that barrier nearly a decade past the compound patent expiration. A biosimilar pipeline asset valued on the assumption of U.S. launch in year X, without a specific FTO analysis for process patents, carries unquantified but material risk of that assumption being wrong by years.
Key Takeaways — Section 14
These four cases do not describe a uniform playbook. Humira required IPR precision targeting against a broad thicket, with the strategic goal of creating settlement leverage rather than litigating to comprehensive invalidity findings. Copaxone required straightforward obviousness litigation against a dosing regimen patent that the clinical literature effectively pre-invalidated. EpiPen required antitrust litigation when the patent position itself was legally defensible but the conduct surrounding it was not. Enbrel required honest recognition that strong manufacturing process patents can survive well-funded biosimilar challenge programs. The right strategy is context-specific, and the wrong strategy against the wrong target costs years and hundreds of millions of dollars.
15. FTC Scrutiny, Legislative Reform, and AI in Patent Analysis
FTC Enforcement: Orange Book Challenges and Patent Thicket Scrutiny
The Federal Trade Commission has become substantially more aggressive in pharmaceutical patent enforcement since 2021. The Commission initiated a systematic challenge to Orange Book patent listings it considers improperly listed, filing disputes with FDA against patents for dozens of branded drugs across multiple therapeutic areas. The FTC’s theory is that brand companies have listed patents that do not qualify for Orange Book listing under FDA’s regulations, particularly patents that cover manufacturing processes (not listable) or peripheral product features that do not satisfy the ‘claims the drug product or a method of using the drug product’ requirement.
When the FTC challenges an Orange Book listing and FDA agrees that the patent does not qualify, the brand company must withdraw the listing. A withdrawn listing eliminates the patent’s ability to trigger the 30-month stay in future Paragraph IV proceedings, materially reducing its defensive value. Generic companies should monitor FTC Orange Book challenge proceedings actively, because a successful FTC challenge can create a litigation pathway that the 30-month stay would otherwise have blocked.
Legislative proposals addressing patent thickets and evergreening have been introduced in the U.S. Congress repeatedly since 2020. Bills proposing to limit the number of Orange Book-listable patents per product, to impose heightened patentability requirements for secondary pharmaceutical patents, and to create stronger FTC enforcement authority against pay-for-delay settlements have been proposed, with none yet enacted. The legislative environment warrants monitoring but does not yet represent a structurally different legal landscape.
AI-Assisted Patent Analysis: Capabilities and Limitations
Artificial intelligence tools for patent analysis have matured substantially over the past three years. Large language model-based systems can assist with claim charting (mapping claim elements against prior art disclosures), prosecution history analysis (identifying relevant office actions and applicant arguments across large file wrappers), and prior art categorization (organizing large prior art sets by claim element relevance). These capabilities accelerate the early stages of patent analysis and can reduce the cost of initial patent landscape assessments for ANDA screening programs.
The limitations are equally important. AI tools as of 2025 cannot reliably perform the legal judgment components of patent analysis: determining whether a prior art reference anticipates a claim requires a legal conclusion about claim construction that exceeds current AI reliability. Determining whether a combination of references would have been obvious to a POSITA requires the contextual judgment of a patent attorney with domain expertise in pharmaceutical sciences. AI accelerates the data collection and organization phases; it does not replace the expert analysis phase that determines whether a litigation-ready invalidity case exists.
The practical integration of AI into a patent challenge program works as follows: AI tools run the initial prior art searches, categorize and rank results by claim element relevance, and extract key passages from file history documents. Human patent counsel then reviews the AI’s outputs to perform claim construction, assess legal relevance of prior art combinations, and develop the invalidity narrative. This human-AI workflow can reduce the cost of early-stage invalidity assessment by 30% to 50%, which is material for companies with large ANDA screening programs that require initial patent analysis across dozens of potential targets before committing to full challenge programs on selected candidates.
Key Takeaways — Section 15
The FTC’s Orange Book challenge program and its continued scrutiny of pay-for-delay settlements represent the most significant regulatory shift affecting pharmaceutical patent litigation since the Actavis decision in 2013. Generic companies should integrate FTC Orange Book dispute monitoring into their intelligence infrastructure, because successful FTC challenges can open Paragraph IV filing opportunities on previously protected patents. AI-assisted patent analysis is a legitimate cost-reduction tool for screening large ANDA pipelines, but it does not replace patent counsel expertise on the substantive invalidity and non-infringement questions that determine litigation outcomes.
Investment Strategy Note
Legislative Risk Modeling for Pharma Patent Portfolios
Legislative proposals to limit Orange Book listings or impose heightened secondary patent standards are structural risks to branded pharmaceutical company valuations that equity models rarely quantify explicitly. The probability of any specific bill passing in the current Congress is low, but the directional regulatory trend, FTC enforcement, congressional attention, and ongoing litigation around patent thickets, is consistently toward more restrictive pharmaceutical IP enforcement. Pharmaceutical company patent portfolio valuations built on the assumption that the current enforcement environment is stable through the next patent cycle (five to ten years) are implicitly discounting a non-trivial legislative risk. That risk warrants explicit scenario modeling rather than assumption away.
Master Summary: The Challenger’s Complete Decision Framework
Patent invalidation in the pharmaceutical industry is not a single legal event. It is an integrated program that begins with commercial opportunity identification, runs through IP landscape analysis and prior art development, executes through carefully sequenced legal proceedings, and concludes with either a market entry decision or a negotiated launch date. Each phase has specific analytical requirements, and a failure in any phase undermines the value of the others.
The commercial goal determines everything that follows. A company filing a Paragraph IV ANDA because a drug’s Orange Book patents look challengeable, without first confirming that the commercial opportunity justifies the litigation investment and that first-to-file status is achievable, has inverted the decision sequence. The right starting point is the market: the brand’s U.S. revenue, the reimbursement landscape for the generic, the likely competitive entry density, and the authorized generic probability. If those variables generate a positive expected value on the 180-day exclusivity scenario, proceed to patent analysis. If they do not, the strongest invalidity case in the world does not create a viable program.
Secondary patents are the primary target population. The composition-of-matter patent on the original molecule is rarely the first target of a well-resourced challenger because it is the hardest to invalidate. Formulation patents, polymorph patents, dosing regimen patents, device patents, and method-of-use patents filed after the drug’s original approval are the vulnerable components of any patent thicket. Identifying which specific patents represent the weakest legal foundations, not which patents look most important to the brand, is the operative screening question.
The prior art search must be exhaustive, cross all three channels (keyword, classification, citation), and invest disproportionate effort in non-patent literature including dissertations, conference proceedings, and foreign language technical publications. The prosecution history analysis must cover the full file wrapper for all patents in the challenge target set, with cross-portfolio comparison to identify contradictions that undermine the brand’s credibility across proceedings.
Venue selection is a legally consequential decision with long-term consequences. IPR’s estoppel provisions mean that grounds raised in an IPR cannot be re-raised in district court after a final decision. PGR’s nine-month window closes permanently after the grant anniversary. District court’s 30-month stay is only triggered by timely Paragraph IV litigation. These are not administrative choices; they are strategic commitments with binding downstream consequences that must be mapped against the full litigation program before any single proceeding is initiated.
Settlement is not failure. For most commercially significant patent disputes, a negotiated launch date that captures multiple years of pre-expiration revenue is worth more than a decade of litigation culminating in a judicial invalidity finding that arrives after the drug’s commercial peak. The goal is winning the market, not the lawsuit. A competent invalidity case is the instrument; market entry is the objective.
Data Sources and Legal References
Patent filing statistics (78% secondary patents, 66% post-approval filings): I-MAK analysis, ‘Overpatented, Overpriced’ (2018 and updates). PTAB claim cancellation data: Finnegan PTAB Statistics Report, January 2025. Paragraph IV success rate (76%): PMC, Grabowski et al.; NBER Working Paper 17188, Lee et al. KSR International Co. v. Teleflex Inc., 550 U.S. 398 (2007). Amgen Inc. v. Sanofi, 598 U.S. 594 (2023). Sandoz Inc. v. Amgen Inc., 582 U.S. 1 (2017). FTC v. Actavis, Inc., 570 U.S. 136 (2013). Festo Corp. v. Shoketsu Kinzoku Kabushiki Co., 535 U.S. 722 (2002). PTAB IPR cost estimates: UpCounsel; Congressional Research Service R48016. European Commission Teva Copaxone fine: EC Case AT.40588, 2024. EpiPen settlement amounts: Proskauer litigation alerts; MDL 17-2785 (D. Kan.). Humira patent count: Harvard DASH, ‘A Case Study of Humira’s Patent Extension Strategies’; AbbVie 10-K filings. Coherus IPR victory: PTAB IPR2016-00127, Final Written Decision (2017). Enbrel biosimilar status: Amgen press releases; Federal Circuit No. 18-1551 (2019); Supreme Court certiorari denial (2021).
This document is prepared for informational purposes for pharmaceutical and biotech professionals. It does not constitute legal advice. Patent challenge decisions require qualified patent litigation counsel with pharmaceutical industry experience.
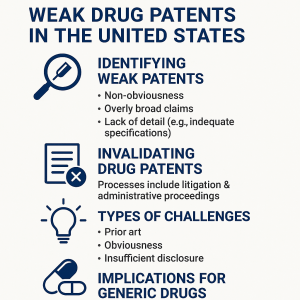















")










