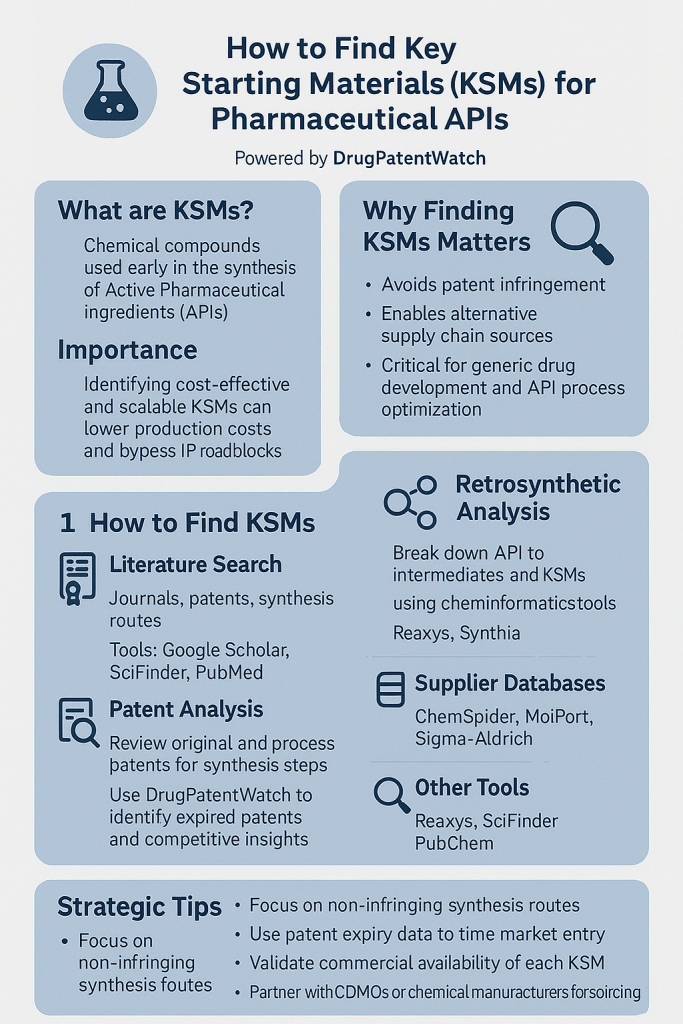
KSM designation is not a paperwork exercise. It is one of the earliest and highest-leverage decisions in pharmaceutical development, with direct consequences for patent strategy, regulatory timelines, COGS at commercial scale, and supply chain exposure. This guide covers the full technical, regulatory, and financial landscape, from ICH Q11 interpretation to Paragraph IV strategy, from Chinese API dependence to the IP valuation of synthetic route choices.
1. What Are Key Starting Materials and Why the Definition Has Legal Teeth
The Regulatory Definition and Its Structural Consequences
A Key Starting Material (KSM) is the point in a pharmaceutical synthesis at which Good Manufacturing Practice (GMP) controls formally begin. The ICH Q7 guideline defines an API starting material as ‘a raw material, intermediate, or an API that is used in the production of an API and that is incorporated as a significant structural fragment into the structure of the API.’ That phrase, ‘significant structural fragment,’ has generated more regulatory correspondence, complete response letters, and pre-IND meeting transcripts than almost any other term in pharmaceutical manufacturing guidance.
The definition has three functional consequences. First, everything upstream of the designated KSM is outside the GMP envelope and therefore outside the formal regulatory dossier for drug substance manufacturing. Second, the impurity profile of the KSM, whatever it contains, must be controlled with sufficient rigor to prevent unacceptable carryover into the final API. Third, every manufacturing change to the KSM or its supply chain after approval triggers a change control obligation that may require prior approval, CBE-30 notification, or at minimum an annual report, depending on the nature of the change and the target regulatory market.
Why the Designation Point Is a Business Decision, Not Just a Chemistry Decision
The economic logic is straightforward: every synthetic step above the KSM designation point operates outside GMP, which means lower documentation burden, less expensive contract manufacturing, and more flexibility in process optimization. A company that can justify designating a commercially available, structurally complex intermediate as its KSM is effectively arguing that it does not owe the FDA or EMA a detailed GMP account of the five synthetic steps that produced that intermediate.
The savings are real. GMP manufacturing adds 20 to 40 percent to the cost of a synthesis step versus non-GMP production, by industry estimates. For APIs requiring twelve or more synthetic steps, the difference between designating step three versus step seven as the KSM can represent tens of millions of dollars in annualized production cost at commercial scale.
Regulatory agencies understand this incentive. Both the FDA and EMA have, in the last decade, pushed back hard on KSM designations they consider too early, particularly for generic drug applications where the synthetic route is less complex and the commercial pressure to cut GMP compliance costs is acute.
Key Takeaways: Section 1
The KSM designation determines the GMP boundary and therefore the scope of the regulatory dossier. It carries direct cost implications, supply chain obligations, and change control triggers. The economic incentive to designate late in the synthesis is real, and regulatory agencies are aware of it. Justification must be scientific, not economic.
2. The Regulatory Architecture: ICH Q7, Q11, and the Q&A Documents That Actually Govern Practice
ICH Q7: The Foundational Standard
ICH Q7, published in 2000 and titled ‘Good Manufacturing Practice for Active Pharmaceutical Ingredients,’ established the first globally harmonized GMP framework for API synthesis. Its most consequential contribution was the acknowledgment that GMP requirements do not have to apply to the entire synthesis of a drug substance, only to those steps from the designated starting material forward. The guideline permits an API starting material to be ‘an article of commerce, a material purchased from one or more suppliers under contract or commercial agreement, or produced in-house.’ It does not, however, provide criteria for choosing where in a multi-step synthesis the starting material should sit.
ICH Q11: Where the Criteria Finally Appear
ICH Q11, finalized in 2012 and titled ‘Development and Manufacture of Drug Substances,’ substantially raised the bar on justification. The guideline introduced four criteria for evaluating a proposed starting material: the material should have defined chemical properties and structure; it should be incorporated as a significant structural fragment; manufacturing steps that impact the impurity profile of the drug substance should normally be included in the regulatory filing; and commercially available materials used in non-pharmaceutical markets receive less scrutiny than custom-synthesized compounds.
Q11 also addressed the ‘significant structural fragment’ concept directly, clarifying that reagents, solvents, and other raw materials that do not contribute structural content to the final API are not starting materials. A trivial alkylating agent that adds a single methyl group to a complex scaffold is a reagent. The complex scaffold itself, if commercially available and structurally proximate to the API, is a starting material candidate.
The Q&A Documents: Where Regulatory Intent Gets Specific
The ICH Q11 Q&A document, published in 2017, added further precision that the guideline itself lacked. It clarified that an applicant ‘does not have to justify the use of a commercially available substance as a starting material in the dossier,’ but it placed conditions on what ‘commercially available’ actually means. A substance must be offered and sold as a commodity in non-pharmaceutical markets. Custom synthesis performed exclusively for pharmaceutical use does not qualify, even if the supplier is willing to file it under a commercial catalog number.
The Q&A document also introduced the concept of ‘proposed acceptable ranges’ for starting material attributes, bringing KSM specification-setting closer to the design space framework of ICH Q8. This means that rather than writing fixed specifications, applicants can propose ranges for key quality attributes and justify those ranges using process characterization data.
EMA’s Reflection Paper: A Harder Line
The EMA published a reflection paper on starting material justification in 2021 that signaled a harder regulatory posture than the Q&A document. The EMA expects that steps with a significant impact on the impurity profile of the final drug substance be included in the regulatory filing even when the applicant argues those steps precede the KSM. The agency specifically flagged genotoxic impurities as a trigger for this expectation. If a step upstream of the proposed KSM can generate a potential mutagenic impurity that is not completely purged, the EMA’s position is that the step belongs inside the GMP boundary.
Key Takeaways: Section 2
ICH Q7 set the framework; ICH Q11 set the criteria; the Q&A documents set the enforcement tone. The EMA’s 2021 reflection paper tightened expectations further, particularly around genotoxic impurity purge data. Teams writing Q11 justifications should treat the reflection paper as a de facto requirement, not aspirational guidance.
3. FDA vs. EMA: Where the Interpretational Gap Bites and How to Bridge It
The Structural Divergence
The FDA and EMA have operated from the same ICH text since Q7’s adoption, but they have arrived at meaningfully different default positions on what constitutes an acceptable KSM for a generic drug versus a new chemical entity. The divergence is clearest in two areas: the acceptable distance between the KSM and the final API (measured in synthetic steps), and the treatment of custom-synthesized compounds.
The FDA’s Division of Manufacturing Assessment, in post-approval inspections and complete response letters, has generally accepted KSM designations that are three to five synthetic steps from the final API for complex small molecules. The EMA has trended toward two to four steps in comparable molecules, with greater insistence on GMP controls for steps that generate or carry potential genotoxic impurities under ICH M7 assessment.
ANDA Filings vs. MAA Filings: Concrete Divergence Points
For generic drug ANDA filings in the U.S., the FDA accepts a Drug Master File (DMF Type II) or an Open Part/Closed Part dossier from the API supplier. The KSM designation appears in the manufacturing section of the DMF, and the FDA evaluator assesses whether the proposed starting material meets Q11 criteria based on the submitted justification. The FDA has been relatively transparent about its expectations through warning letters and process validation guidance, though it rarely issues formal guidance specific to KSM selection in ANDA review.
For Marketing Authorization Applications (MAAs) in Europe, the EMA reviews the complete drug substance dossier in Module 3 Section 3.2.S.2.3 and expects a more structured justification narrative with explicit references to Q11 criteria. The EMA has rejected KSM designations at Day 120 of the CHMP review cycle in cases where the justification narrative was present but the supporting impurity data was incomplete.
Practical Consequences for Global Development Programs
A company developing a generic API for simultaneous U.S. and European market entry should default to the EMA’s more conservative position when selecting and justifying its KSM. The practical reason is asymmetric: satisfying the EMA’s criteria almost always satisfies the FDA’s, but the reverse is not reliably true. The additional GMP compliance cost of applying the EMA standard to an extra synthetic step typically costs less than the delay from a Day 120 objection that requires a full response cycle.
When the two agencies have formally diverging requirements, which does happen for certain complex API classes, the applicant faces a genuine strategic choice. Some companies file different starting material designations in the two dossiers, which is permissible but creates ongoing manufacturing complexity. Others file the conservative designation globally and absorb the GMP cost. A third approach involves proposing the aggressive designation in the U.S. while conducting pre-submission meetings with the EMA to gauge its receptiveness before filing.
WHO and Other Reference Agencies
Teams preparing for markets that use WHO prequalification or national reference agency review (Brazil’s ANVISA, Canada’s Health Canada, Japan’s PMDA) face additional layers of interpretational variation. The WHO’s API GMP guidelines follow ICH Q7 but are applied by inspectors with varying levels of ICH Q11 familiarity. Health Canada has closely tracked the EMA’s position since 2019. PMDA has its own guidance document that in several areas imposes stricter requirements on genotoxic impurity control in starting materials than either the FDA or EMA.
Investment Strategy: Regulatory Arbitrage Is Not a Strategy
Analysts evaluating generic drug companies should treat KSM designation as an indicator of regulatory maturity. Companies that routinely push the most aggressive possible designation without adequate justification data are trading short-term COGS savings for regulatory delay risk. A single complete response letter requiring additional fate and purge data delays approval by 12 to 18 months. At a commercial-scale API worth $200 million per year in revenue, that delay costs more than the lifetime GMP compliance savings from the aggressive KSM designation.
Key Takeaways: Section 3
The FDA and EMA interpret ICH Q11 differently, with the EMA trending more conservative on step count and genotoxic impurity purge expectations. Filing to the EMA standard globally is the lower-risk strategy for most programs. Companies that use regulatory arbitrage, filing different designations in different markets, create ongoing manufacturing and change management complexity that investors should discount in their risk models.
4. KSM Designation as an IP Asset: Synthetic Route Patents, Trade Secrets, and Paragraph IV Exposure
The KSM Decision and the Patent Landscape
This is the section most KSM guides omit entirely: the starting material decision is not separable from the patent strategy, and the two must be developed in parallel. A KSM that sits upstream of proprietary synthetic chemistry is a trade secret. A KSM that sits downstream of patented intermediate synthesis exposes the generic manufacturer to infringement claims if the innovator holds process patents covering those steps.
Originator companies patent their synthetic routes aggressively. The Chemical Development and Process Chemistry patent families for major APIs frequently include composition-of-matter claims on key intermediates, process claims on the steps converting those intermediates to the final API, and product-by-process claims that cover the API itself when produced by a specified route. A generic company that designates a KSM at step five of a twelve-step synthesis, then contracts out steps one through four to a non-GMP supplier, must verify that steps one through four do not infringe live process patents belonging to the innovator.
How Innovators Use Process Patents to Extend Market Exclusivity
The process patent is one of the most potent evergreening tools available to brand-name pharmaceutical companies, and it operates at the synthesis level rather than the drug product level. When Gilead Sciences filed process patents covering the synthesis of tenofovir alafenamide (TAF) in the 2010s, the claims were specific enough to force generic manufacturers to develop alternative synthetic routes or wait for those patents to expire. The innovator’s ability to designate its KSM upstream of the patented chemistry effectively forced generic competitors to rethink their entire synthesis strategy.
For the IP team evaluating a new generic target, the starting point is a Freedom to Operate (FTO) analysis that maps every live process patent against each candidate synthetic route. The question ‘where should our KSM be?’ cannot be answered without first answering ‘which synthetic routes are clear of third-party IP?’ The Hatch-Waxman framework complicates this further: under 35 U.S.C. Section 271(e)(1), manufacturing for FDA submission purposes is exempt from infringement claims. But commercial manufacturing is not.
Paragraph IV Certification and Starting Material Disclosure
When a generic manufacturer files an ANDA with a Paragraph IV certification, it certifies that a listed patent is invalid, unenforceable, or will not be infringed by its product. The ANDA includes a detailed description of the proposed synthesis, including the designated starting material and the subsequent manufacturing steps. The innovator receives this information as part of the statutory notice requirement.
This creates a disclosure dynamic that many generic manufacturers underestimate. The innovator’s IP team will map the disclosed synthesis against its patent portfolio within 45 days of receiving the Paragraph IV notice. If the generic’s synthesis passes through chemistry covered by a listed process patent, litigation follows almost automatically. The KSM designation, and the synthetic route it anchors, is therefore a litigation risk assessment document as much as it is a chemistry document.
Trade Secret Protection for Synthetic Routes
The alternative to patent protection is trade secret. For proprietary synthetic chemistry that falls upstream of the KSM designation in the regulatory filing, a manufacturer may argue that those steps are outside the regulatory dossier and therefore not subject to disclosure requirements. This is partially correct. The FDA does not require detailed disclosure of non-GMP steps in the ANDA or NDA. But the regulatory agency can and does request this information during inspections or in the context of a complete response letter that questions the impurity control rationale.
The trade secret argument is also tested in litigation. When an innovator sues for trade secret misappropriation, which has happened in cases where former employees moved to generic companies and brought process knowledge with them, the courts look at whether the information was genuinely maintained as confidential and whether it had independent economic value. Synthetic route knowledge that stays upstream of the KSM and never appears in the regulatory filing has a stronger trade secret claim than chemistry disclosed in the DMF.
IP Valuation of KSM Choices: A Framework for IP Teams
An IP team valuing the synthetic route for a commercial API should assess four components. First, the composition-of-matter patent coverage on the API itself and how much remaining term it has, which sets the revenue protection window. Second, the process patent coverage on the synthesis, including any patents on key intermediates that might constrain the generic entry route. Third, the trade secret value of the non-GMP chemistry upstream of the KSM, which is protected as long as it remains confidential and economically valuable. Fourth, the regulatory data exclusivity periods that attach to the NDA or ANDA, which are independent of patent protection and attach to the drug product approval.
The total IP valuation of a synthetic API is the net present value of the revenue stream protected by all four components, discounted by the probability that each component withstands generic challenge. Starting material designation affects the second and third components most directly, and an IP team that treats the KSM decision as a chemistry matter rather than a legal strategy matter is leaving money on the table.
Key Takeaways: Section 4
KSM designation intersects with process patent strategy, Paragraph IV certification disclosure, and trade secret protection. Generic manufacturers must conduct a full FTO analysis before finalizing synthetic route and KSM designation. Innovators use process patents and intermediate composition-of-matter claims to constrain available generic routes. The trade secret value of upstream chemistry depends on maintaining it outside the regulatory dossier.
5. Technology Roadmap: Identifying KSMs Across Synthesis Complexity Classes
Class 1: Simple Linear Synthesis (2-5 Steps)
Simple linear APIs, including common analgesics, antacids, and low-complexity cardiovascular drugs, have short synthetic routes where regulatory agencies expect GMP controls to begin at a relatively early stage. For a three-step synthesis, designating the product of step one as the KSM effectively applies GMP to two of the three steps, which is the minimum that satisfies agency expectations in most cases.
The analytical challenge for simple synthetics is that the KSM is often a widely traded commodity chemical. Acetic anhydride, for example, is a starting material for aspirin synthesis, and its commercial availability in pharmaceutical and industrial markets is genuinely broad. Regulatory acceptance of commodity chemicals as starting materials is generally straightforward, but the quality implications are not trivial: commodity chemicals sold for non-pharmaceutical applications may carry trace impurities that are acceptable in industrial use but require specific limits for pharmaceutical synthesis. The specification-setting work for a commodity KSM must account for the full population of supplier batches, not just the best-performing lots.
Class 2: Moderate Complexity (6-10 Steps)
The majority of synthetic small-molecule APIs fall into this category. Metformin hydrochloride (five steps from guanidine carbonate, depending on the route), atorvastatin calcium (the Lipitor API, roughly eight steps from simple building blocks), and omeprazole (six to seven steps from pyridine precursors) all sit in this complexity class. The regulatory expectation for this class is that the KSM has meaningful structural complexity, contributes at least a primary ring system or chiral center to the final API, and is introduced no earlier than three to four steps from the final drug substance.
For atorvastatin, the IP history illustrates the point precisely. Pfizer’s original synthesis patents covering Lipitor included claims on the dihydroxy acid intermediate, a late-stage intermediate that contains both the beta-delta-dihydroxy acid side chain and the partially constructed HMG-CoA reductase inhibitor scaffold. Generic manufacturers developing atorvastatin calcium had to either license those process patents or develop alternative synthetic routes that bypassed the claimed intermediates. The KSM designation in generic atorvastatin ANDAs therefore varied considerably by manufacturer, reflecting different underlying synthetic strategies rather than different interpretations of Q11.
Class 3: High Complexity (10+ Steps, Multiple Chiral Centers)
High-complexity APIs include antiviral nucleotides, complex natural product derivatives, and many oncology small molecules. Ledipasvir (the NS5A inhibitor in Harvoni), sofosbuvir, and venetoclax all require synthesis routes of 12 steps or more from commercially accessible starting materials. For this class, regulatory agencies are more accepting of late-stage KSM designations, provided the proposed starting material has sufficient structural complexity.
The impurity control challenge for high-complexity APIs is that each synthetic step can introduce new impurity species, some of which may be genotoxic under ICH M7 assessment. The fate and purge study must be comprehensive and must address every step between the KSM and the final API, with particular attention to steps that introduce or risk generating potential mutagenic impurities (PMIs). A PMI that is introduced at step three of a twelve-step synthesis and purges to below the threshold of toxicological concern (TTC) by step eight is manageable. A PMI introduced at step ten, two steps from the final API, requires either a tight specification on the step-ten intermediate or a validated removal step before the final drug substance.
Technology Roadmap: Selecting a KSM Starting Point
The process for identifying a KSM candidate in a new synthesis should proceed in the following sequence. The chemistry team maps the complete synthetic route from commercially available raw materials to the final API, annotating each step with the reagents, solvents, and conditions involved. The regulatory team then overlays the ICH Q11 criteria: which intermediates have defined chemical structure? Which contribute a significant structural fragment? Which steps have the most significant impact on the impurity profile? The IP team maps the route against live third-party patents and identifies which intermediates must be avoided. The supply chain team identifies which candidate KSMs are commercially available from multiple qualified sources.
The intersection of these four assessments defines the viable KSM selection space. In most cases, two to four candidates emerge. The final selection balances structural complexity (to satisfy Q11), impurity controllability (to satisfy ICH M7 and agency expectations), IP clearance (to satisfy freedom to operate), and commercial availability (to minimize regulatory justification burden and supply risk).
Key Takeaways: Section 5
KSM identification is a multi-disciplinary activity, not a chemistry activity. The viable selection space is the intersection of Q11 regulatory criteria, ICH M7 impurity control capability, IP freedom to operate, and commercial availability. Simple linear APIs face earlier GMP start expectations; high-complexity APIs allow later designations. Chiral centers and fused ring systems in the KSM structure strengthen the justification.
Investment Strategy: Synthetic Route Complexity as a Competitive Moat
For institutional investors evaluating generic API manufacturers, the complexity of the synthetic route and the sophistication of the KSM justification are proxies for competitive moat. A manufacturer that has developed a proprietary, non-obvious synthetic route to a complex API, patented the key steps, and qualified unique KSM sources has built a 3 to 5 year competitive advantage over a later entrant that must develop and qualify an alternative route from scratch. This advantage shows up in gross margin (because the innovator route is typically optimized for COGS), in regulatory timeline (because the accumulated process knowledge is embedded in the DMF), and in supply chain stability (because KSM suppliers have been qualified through multiple commercial batches).
6. Impurity Fate and Purge Studies: The Analytical Work That Determines Regulatory Acceptance
Why Impurity Fate Is the Central Technical Question
The ICH Q11 position on impurity control is straightforward: if an impurity introduced at or before the KSM can persist through the synthesis to the final API at levels above acceptance criteria, the GMP boundary must move upstream. The entire scientific argument for a proposed KSM designation rests on demonstrating that the impurity profile of the proposed material is adequately controlled, either through specifications on the KSM itself or through purge during subsequent synthetic steps.
The FDA has reinforced this position in multiple complete response letters for ANDA submissions where the KSM justification narrative was present but the fate and purge data were insufficient. The most common deficiency is not a failure to identify potential impurities, but a failure to characterize their actual purge across the synthesis with quantitative data rather than theoretical arguments.
ICH M7 and the Genotoxic Impurity Overlay
ICH M7, ‘Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals,’ adds a separate and more stringent layer of control requirements for a specific subset of starting material impurities. Any compound with a structural alert for mutagenicity (identified by in silico Ames test prediction or confirmed mutagenic activity) that could be present in the KSM or introduced during subsequent synthesis must be assessed and controlled to below the acceptable intake limit. For most therapeutic indications, the threshold of toxicological concern (TTC) for a potential mutagenic impurity is 1.5 micrograms per day, which translates to an extremely low specification limit at commercial dose levels.
The N-nitrosamine problem that erupted in 2018 and 2019, when nitrosamine impurities were detected in valsartan, irbesartan, and other sartan APIs, illustrates what happens when the fate and purge framework is inadequate for genotoxic impurity control. The root cause in most sartan cases was a combination of process change (introduction of dimethylformamide as a solvent), inadequate risk assessment for nitrosamine formation, and failure to conduct fate and purge studies that would have detected the issue before commercial release. The subsequent recalls cost the affected manufacturers hundreds of millions of dollars and prompted FDA to issue a comprehensive nitrosamine guidance that now applies retroactively to virtually all marketed APIs.
The lesson for KSM selection: any proposed starting material that contains structural motifs susceptible to nitrosamine formation, or that introduces reagents that could generate nitrosamines in subsequent steps, must be assessed under the M7 framework before the KSM designation is finalized.
Conducting a Fate and Purge Study: Technical Requirements
A rigorous fate and purge study for a proposed KSM requires several elements. The team must first identify all potential impurities in the KSM, including known process impurities, regioisomers, stereoisomers, and potential degradation products. For each impurity, the team then runs it through the subsequent synthesis, either in spiked laboratory experiments or through mechanistic modeling, to determine what happens at each step. Does it react away? Does it co-purify or reject during crystallization? Does it carry through unchanged?
The study should generate quantitative purge factor data: what is the ratio of impurity concentration at the KSM stage to impurity concentration at the final API stage? A purge factor of 1000x or greater is generally robust. A purge factor of 10x requires tight specification on the KSM and validated process controls. A purge factor of less than 5x, without validated remediation, is a serious weakness in the KSM justification.
For M7 impurities, the acceptance criteria are so tight (typically sub-ppm at the API stage) that the purge factor requirements are correspondingly demanding. Fate and purge data for an M7 impurity must be generated with validated analytical methods capable of quantifying at the sub-ppm level, using representative manufacturing process conditions rather than laboratory idealizations.
Analytical Method Requirements for KSM Impurity Testing
The analytical methods used to assess KSM impurities must be fit for purpose, which is a higher standard than commonly assumed. A method that was developed to release the final API is not automatically suitable for testing an early-stage starting material. The matrix is different, the impurity profile is different, and the acceptance limits may be more or less stringent depending on the fate and purge factor.
For starting materials tested at the supplier, the analytical method may need to be transferred to a contract laboratory and validated there. ICH Q2(R2), the revised analytical procedure validation guideline, requires that method validation demonstrate specificity, limit of detection, limit of quantitation, linearity, accuracy, precision, and robustness. For impurity methods on KSMs, specificity is the most critical parameter: the method must resolve the target impurity from all matrix components at the expected concentration range.
Key Takeaways: Section 6
Fate and purge studies are not optional supporting data. They are the core scientific justification for the KSM designation. M7 genotoxic impurity assessment must overlay the fate and purge work for any impurity with a structural mutagenic alert. The nitrosamine crisis of 2018-2019 demonstrated that inadequate impurity risk assessment at the starting material level produces commercial-scale recalls. Purge factor quantification must use validated analytical methods, not theoretical arguments.
7. Commercial Availability: The Regulatory Shortcut and How Agencies Have Tightened Its Use
What ‘Commercially Available’ Actually Means
The ICH Q11 Q&A document defined a commercially available substance as one ‘offered and sold as a commodity in the non-pharmaceutical market in addition to its use as a starting material.’ The key phrase is ‘in addition to’: the material must have genuine non-pharmaceutical market demand, not merely a catalog listing from a chemical supplier that happens to serve pharmaceutical customers.
This definition excludes several categories of material that generic manufacturers have historically tried to position as commercially available. A compound that is listed in the Sigma-Aldrich catalog at gram scale for research purposes, but has no documented industrial or non-pharmaceutical commercial application, is not commercially available in the regulatory sense. A compound synthesized under a confidential supply agreement with a single API manufacturer, and given a trade name to suggest commercial status, is not commercially available in the regulatory sense.
The EMA has been particularly active in scrutinizing commercial availability claims. In scientific advice responses and during Module 3 review, EMA assessors have asked applicants to provide evidence of non-pharmaceutical sales volumes, multiple supplier qualifications, and market data demonstrating genuine commercial status. Claims unsupported by this evidence have been rejected.
The Strategic Advantage of Genuine Commercial Availability
When a proposed KSM is genuinely commercially available, with documented non-pharmaceutical applications, multiple qualified manufacturers, and transparent market pricing, the regulatory justification burden drops substantially. The Q11 Q&A position that applicants ‘do not have to justify the use of a commercially available substance’ is a meaningful concession. It allows the applicant to focus the justification narrative on impurity control and structural fragment contribution, rather than defending the commercial status of the material itself.
This advantage is most accessible for commodity chemicals, widely traded intermediates in bulk chemical markets, and natural product precursors with established agricultural or industrial applications. Pioglitazone’s synthesis, for instance, uses pyridine-derived intermediates that are traded in large volumes for agrochemical applications. A generic pioglitazone manufacturer designating one of these intermediates as its KSM has a straightforward commercial availability argument.
Documentation Strategy for Commercial Availability
Teams seeking to establish commercial availability should collect and maintain a specific set of evidence. They need business-to-business pricing data or distributor catalogs showing the material sold at commercial scale. They need documented customer base information showing sales to multiple buyers in non-pharmaceutical industries, whether agrochemical, polymer, dye, or other markets. They need proof of multiple suppliers, ideally with qualification data from at least two. Regulatory agencies have become sophisticated enough to distinguish genuine commercial availability from engineered appearances of it.
Key Takeaways: Section 7
Commercial availability is a regulatory advantage, not merely a sourcing preference. The standard is genuine non-pharmaceutical market existence with multiple suppliers and documented industrial demand. Agencies scrutinize commercial availability claims and reject those unsupported by market evidence. The documentation package for commercial availability should be assembled as early as the material sourcing assessment, not retrofitted during the regulatory submission.
8. KSM Selection for Small Molecules, Semi-Synthetics, and Botanically Derived APIs
Synthetic Small Molecules: The Majority Case
For the roughly 85 percent of APIs that are fully synthetic small molecules, the KSM selection criteria apply in their most straightforward form. The material must contribute a significant structural fragment, must have defined chemical properties, and must be introduced at a point in the synthesis where GMP controls on all subsequent steps are feasible and proportionate to the regulatory expectation.
The practice varies considerably by therapeutic class. Oncology APIs, many of which are structurally complex and manufactured in small quantities, routinely have KSM designations 5 to 8 steps from the final API, supported by detailed fate and purge data. High-volume generics like metformin, which is manufactured at thousands of tons per year globally, have KSM designations at simpler intermediates, partly because the synthesis is genuinely short and partly because the global API supply network for metformin is so mature that agency expectations are well-established.
Semi-Synthetic APIs: Natural Precursors and Their Unique Challenges
Semi-synthetic APIs begin from a natural product substrate and complete the synthesis through chemical modification. Paclitaxel (Taxol) has historically been sourced from plant extraction (Taxus species) followed by semi-synthesis from 10-deacetylbaccatin III (10-DAB), the naturally occurring precursor. The 10-DAB serves as the starting material for the semi-synthetic route, and its quality attributes, specifically the profile of co-extracted taxane analogs, directly influence the impurity profile of the final API.
For semi-synthetic APIs, the ICH Q11 framework acknowledges that the natural product precursor often serves as a logical starting material, but it requires appropriate controls on the natural variability of that precursor. 10-DAB from Taxus needles varies in taxane analog composition based on species, geographic origin, and harvest timing. The specification for 10-DAB used in paclitaxel synthesis must control the relevant co-occurring taxanes, and the fate and purge study must address which of those taxane impurities can carry through the semi-synthetic steps to the final product.
The erythromycin-to-azithromycin conversion, which is a five-step semi-synthesis, presents similar challenges. Erythromycin itself is a fermentation product with a complex impurity profile including erythromycin B, C, and several other analogs. The semi-synthetic steps convert erythromycin A to azithromycin, but the process does not selectively convert the analog impurities in the same way. Manufacturers must either tightly control erythromycin A purity before the semi-synthesis begins, or demonstrate that analog impurities are purged during the conversion.
For APIs extracted directly from plant material, cannabis (for cannabidiol), Rauwolfia serpentina (for reserpine), or Catharanthus roseus (for vincristine), the regulatory framework distinguishes between the plant biomass, the crude extract, and the API starting material. GMP requirements apply from the API starting material forward; the collection and initial processing of plant material follows agricultural and good agricultural and collection practices (GACP) standards rather than pharmaceutical GMP.
The ICH Q7A position is that for API extracted from plant sources, the API starting material is generally ‘the material obtained from the first extractions.’ This is logical because plant biomass is inherently variable in ways that cannot be controlled to pharmaceutical GMP standards. A field of Catharanthus roseus plants will produce vincristine at varying alkaloid concentrations depending on rainfall, soil composition, and harvest timing. GMP cannot control weather. What GMP can control is the extraction and purification process applied to the crude plant material.
The quality control challenge for botanical starting materials is the complexity of the co-extracted material. A crude alkaloid extract from Catharanthus roseus contains dozens of alkaloid species, most of which are process impurities relative to the target vincristine. The specifications for the botanical starting material must control the most significant co-extracted alkaloids, particularly those that are structurally similar to vincristine and may not be fully resolved during purification.
Key Takeaways: Section 8
Semi-synthetic APIs require fate and purge data covering natural product precursor analog impurities, not just process-generated impurities. Botanical APIs have a defined regulatory boundary between agricultural practice and GMP: the first extraction product. GACP compliance at the plant collection stage does not substitute for GMP at the extraction stage. Impurity complexity is highest for botanicals and must be addressed with appropriately sophisticated analytical methods.
9. Biologics and Biosimilars: Where ‘Starting Material’ Means Something Entirely Different
The Definitional Shift for Biologics
For biologics, the ICH Q11 framework’s focus on significant structural fragments and defined chemical properties does not apply in the conventional sense. A monoclonal antibody does not have a synthetic starting material the way atorvastatin does. Its ‘starting material’ is the cell line, specifically the Master Cell Bank (MCB) from which the production cell bank is derived. The MCB is the foundational IP asset for a biologic, not a chemical intermediate.
ICH Q5B (Analysis of the Expression Construct in Cells Used for Production of r-DNA Derived Protein Products) and ICH Q5D (Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biological Products) govern the characterization and qualification of cell banks for biologic production. The level of characterization required for an MCB is substantially greater than for a pharmaceutical KSM: identity, purity, stability, absence of adventitious agents, and genetic stability over the expected production scale are all required.
Biosimilar Development and Starting Material Equivalence
For biosimilar development, the starting material question takes on a different dimension. The biosimilar manufacturer cannot access the originator’s MCB. It must independently derive a cell line that expresses the same protein sequence, then demonstrate through comparative analytical studies that the resulting product is highly similar to the reference biologic in structure, function, and clinical behavior. This is the biosimilar interchangeability standard under FDA’s Purple Book framework.
The biosimilar cell line is itself a significant IP asset. Several biosimilar manufacturers, including Celltrion, Amgen, and Sandoz, have filed patents on their proprietary cell lines and upstream bioprocess conditions, creating a secondary IP estate around the biosimilar’s ‘starting material’ even when the reference product’s composition-of-matter patents have expired. These cell line patents can extend the effective exclusivity period of a biosimilar by three to five years after approval if competitors cannot design around the claimed cell culture conditions.
IP Valuation of Biologic Cell Banks
The MCB is a core IP asset with valuation characteristics distinct from a chemical synthesis route. A fully characterized, GMP-qualified MCB with demonstrated genetic stability over 200 population doublings represents several years of cell line development work and regulatory dossier preparation. The cost to independently derive an equivalent MCB is typically $10 to $30 million for a monoclonal antibody, depending on the target and the expression system. This cost creates a natural barrier to entry that, combined with the biosimilar regulatory pathway requirements under 42 U.S.C. Section 262(k), effectively limits the number of biosimilar competitors that can mount a credible challenge to a major biologic within the first five years after reference product patent expiry.
For institutional investors, the cell bank valuation question matters most when evaluating biosimilar pipeline companies. A company with six products in its biosimilar pipeline, each backed by a proprietary GMP MCB and a comparative analytical data package, has a significantly different risk profile than a company that has licensed cell lines from a third-party developer and does not control its own upstream manufacturing IP.
Key Takeaways: Section 9
For biologics, the Master Cell Bank is the functional equivalent of the KSM: it is the foundational production asset and a primary IP holding. Biosimilar manufacturers build proprietary cell line IP to create barriers to later biosimilar entrants. MCB characterization requirements under ICH Q5B and Q5D are substantially more demanding than KSM specifications. Investors should assess cell bank ownership, not just product pipeline, when valuing biosimilar-focused companies.
10. Supply Chain Architecture: Dual Sourcing, Geographic Diversification, and the China Dependence Problem
The Structural Dependence on Chinese API Manufacturing
China produces approximately 40 percent of the world’s active pharmaceutical ingredients by volume and a higher proportion by value for certain generic drug categories. For penicillin intermediates, vitamin C, heparin sodium, and a long list of other APIs and key starting materials, Chinese manufacturers hold a near-monopoly on global supply. The concentration is the result of three decades of deliberate industrial policy, including energy subsidies, environmental compliance deferrals, and state-directed capacity investment that allowed Chinese API manufacturers to undercut Western producers on price by margins that eliminated most domestic manufacturing in the U.S. and Europe by the early 2000s.
The U.S. Biomedical Advanced Research and Development Authority (BARDA) estimated in 2019 that 80 percent of the active ingredients used to make drugs sold in the United States come from abroad, with China and India as the primary sources. For China specifically, the combination of geographic concentration and single-supplier risk creates a category of supply chain vulnerability with no short-term mitigation other than inventory buffering.
The HHS white paper on pharmaceutical supply chain vulnerabilities published in January 2024 recommended geographic diversification as the primary structural mitigation strategy, explicitly endorsing ‘diversification of supply, both in overall redundancy of manufacturing capability and geographic diversity.’ The paper also discussed Medicare reimbursement mechanisms to support buffer stock programs for essential medicines, which is the first time the U.S. government has proposed using reimbursement policy to address KSM supply risk.
India’s Role: Scale Manufacturing With Its Own Concentration Risks
India produces approximately 20 percent of global API volume and is the dominant supplier for many generic drug categories, particularly oral solid dosage forms. But India’s API manufacturing base is itself partially dependent on Chinese KSMs. It is estimated that 60 to 70 percent of India’s API manufacturing capacity relies on Chinese starting materials, intermediates, or bulk chemicals at some stage of the synthesis. When China restricted chemical exports during COVID-19 in 2020, Indian API manufacturers faced immediate input shortages even as they were being asked to produce record volumes of generic drugs for global markets.
This upstream dependence means that geographic diversification at the API level is insufficient if the KSM supply chain has not also been diversified. A U.S. generic drug company that sources its finished API from an Indian CDMO, believing it has diversified away from China, may in practice still have 70 percent of its supply chain value at risk from Chinese market disruptions.
Dual Sourcing: Implementation Requirements
Dual sourcing for a critical KSM requires more than identifying a second supplier. Both suppliers must be qualified to identical specifications, meaning the second supplier’s material must pass all acceptance criteria established for the primary supplier. This qualification typically involves paper assessment of the supplier’s quality system, a site audit against applicable GMP standards (even for non-GMP KSMs, basic quality standards apply), testing of multiple batches against specifications, and an assessment of the second supplier’s capacity to scale to required volumes on short notice.
The regulatory implications of a secondary supplier also need management. If the primary and secondary KSM sources are listed in the regulatory filing (as required in the EU and increasingly expected by the FDA), adding a new supplier after approval is a change that requires notification. In some cases, depending on the change classification under FDA’s post-approval changes guidance or EMA’s variation regulations, prior approval may be required before the secondary supplier’s material can be used in commercial API manufacturing.
Strategic Inventory: Buffer Stocks as Risk Mitigation
The logistics of buffer stock management for pharmaceutical KSMs depend on stability. A KSM with a 24-month retest period under controlled storage conditions can be safely stockpiled for 12 to 18 months without quality risk, allowing adequate response time for most supply disruptions. A KSM with a 6-month retest period requires either accelerated replenishment logistics or a more active dual-supplier rotation strategy to avoid stability-driven write-offs.
The cost of maintaining a six-month buffer stock of a high-value KSM must be modeled against the cost of a supply disruption. For a generic drug with $500 million in annual U.S. revenue and a sole-source KSM, a three-month supply disruption translates to $125 million in lost revenue plus back-order penalties and potential market share loss to competitors. The carrying cost of six months of buffer stock for the same product’s KSM is typically $5 to $20 million depending on the material’s cost and storage requirements. The risk-adjusted economics strongly favor buffer stock investment for high-revenue products with concentrated KSM supply.
Investment Strategy: Supply Chain Resilience as a Valuation Premium
Institutional investors evaluating generic API manufacturers should treat supply chain architecture as a valuation input. Companies with full dual-sourcing across their top five KSMs, qualified secondary suppliers in geographically distinct regions, and 4-to-6 month buffer stock programs for strategic materials carry lower supply disruption risk than companies relying on sole-source Chinese KSM suppliers. The valuation premium for this supply chain resilience is difficult to quantify precisely but becomes very visible during supply disruption events, when companies with resilient supply chains gain market share from competitors experiencing shortages.
The BIOSECURE Act, passed by the U.S. House of Representatives in 2024 and pending Senate consideration, would restrict U.S. government contracting with companies using Chinese biotechnology service providers. While the Act targets biologics-focused CMOs rather than small-molecule API manufacturers directly, it reflects a legislative trend toward structurally mandated supply chain diversification that will eventually affect KSM sourcing requirements for any manufacturer seeking government procurement contracts.
Key Takeaways: Section 10
China’s dominance in KSM supply represents a structural vulnerability for the global pharmaceutical supply chain. India’s API manufacturing base is partially dependent on Chinese inputs, making simple API-level diversification insufficient. Dual sourcing requires full qualification of secondary suppliers to identical specifications and may involve regulatory change notification. Buffer stock economics favor investment for high-revenue products. Legislative trends in the U.S. are moving toward mandated supply chain diversification, which will affect KSM sourcing strategy.
11. Supplier Qualification: From Paper Assessment to Full GMP Audit
The Qualification Framework
Supplier qualification for KSMs follows a tiered risk-based approach. The depth of qualification required scales with the material’s criticality, its position in the synthesis relative to the final API, and whether it is a commercially available commodity or a custom-synthesized compound. For a commodity chemical KSM purchased from a single supplier with no viable alternatives, the qualification effort is intensive. For a commodity chemical available from many suppliers with established pharmaceutical use, the qualification requirements are lighter.
A complete supplier qualification program has four phases. The initial paper assessment covers quality management system documentation, regulatory inspection history, environmental and safety compliance, and financial stability. The on-site audit verifies that documented quality systems are operationally effective, that manufacturing equipment is appropriate for the material’s synthesis and handling requirements, and that the facility’s environmental controls are adequate for the material’s stability requirements. Batch testing of representative lots against full release specifications confirms that the supplier can consistently produce material meeting requirements. Ongoing performance monitoring, including Certificate of Analysis trending and periodic re-qualification audits, maintains the qualified status of the supplier relationship.
Auditing Non-GMP Suppliers
A common misconception is that KSM suppliers operating outside the GMP envelope do not require auditing. This is wrong in both the regulatory and practical sense. ICH Q7 requires that API manufacturers assess whether suppliers provide ‘material of appropriate quality.’ For a KSM that is the GMP entry point, the supplier’s manufacturing process and quality controls directly determine the consistency and quality of the material entering the GMP process. An unaudited supplier with poorly controlled manufacturing can produce batches with significant lot-to-lot variability in impurity profile, which then propagates through the GMP synthesis and potentially creates API batches that fail release testing or, worse, pass release testing but carry undetected impurities.
The audit scope for a non-GMP KSM supplier focuses on process control consistency, analytical capability, and documentation quality. The auditor should verify that the supplier uses fixed raw material sources (not opportunistically substituting alternative chemicals), that analytical methods are validated to detect relevant impurities, and that batch documentation is complete and traceable. The supplier need not operate to pharmaceutical GMP in its own manufacturing, but it must demonstrate that its process is controlled well enough to produce material consistently meeting the pharmaceutical specification.
Change Control and Notification Obligations
Once a KSM supplier is qualified and listed in the regulatory filing, any material change to the supplier’s manufacturing process creates a change control obligation. The notification requirements vary by market and by change classification. In the FDA system, post-approval manufacturing changes are classified as major, moderate (CBE-30), or minor (annual report) based on their potential to affect product quality. A change to the KSM synthesis route that could affect the impurity profile is typically a CBE-30 or major change. A change to the packaging or storage conditions for the KSM is typically a minor change.
In the EU, post-approval changes to drug substance manufacturing, including KSM-related changes, are governed by variation regulations. Type II variations (major changes) require prior EMA assessment before implementation. Type IB variations (moderate changes) can be implemented 30 days after notification. Type IA variations (minor changes) can be implemented with simultaneous notification. Determining the correct variation type for a KSM supplier change requires analysis of EMA’s classification guidelines and, frequently, regulatory agency advice.
Key Takeaways: Section 11
Supplier qualification for KSMs requires audit depth proportional to the material’s criticality and synthetic proximity to the final API. Non-GMP suppliers still require process consistency audits because their material quality directly determines GMP manufacturing input quality. Post-approval supplier changes trigger regulatory notification obligations that vary by market; teams should classify changes under FDA’s SUPAC-like guidance and EMA’s variation regulations before implementing.
12. Quality Agreements, Change Control, and the Regulatory Notification Cascade
Quality Agreement Architecture for KSM Suppliers
A quality agreement between a pharmaceutical manufacturer and its KSM supplier defines the responsibilities, obligations, and expectations of both parties. For a KSM that sits at the GMP entry point, the quality agreement must be specific about what the supplier controls, what the pharmaceutical manufacturer tests on receipt, and how changes are communicated and managed.
The quality agreement should address specification enforcement, specifying whether the supplier is responsible for testing and releasing material against specifications or merely for manufacturing to a process, with testing performed by the pharmaceutical manufacturer. It should address notification requirements for process changes, with defined lead times that give the pharmaceutical manufacturer sufficient time to evaluate the change’s impact and, if necessary, notify regulatory agencies before implementing. It should address the supplier’s obligation to notify the pharmaceutical manufacturer of quality deviations and the expected response timeline.
Quality agreements are legally binding contracts in most jurisdictions, and their enforceability has been tested in supply disputes. A pharmaceutical manufacturer that suffers an API recall due to a KSM quality deviation, and that had a quality agreement requiring the supplier to notify of deviations within 48 hours, has a contractual basis for damages if the supplier delayed notification. Conversely, a supplier that changed its manufacturing process without notifying the pharmaceutical manufacturer, and that did not have a quality agreement requiring such notification, may face limited legal exposure.
The Regulatory Notification Cascade
A KSM-related quality deviation or supplier change can trigger a cascade of regulatory notifications. If the deviation affects the quality of released API batches, the pharmaceutical manufacturer must assess whether a product recall is required. If API was incorporated into drug product that was distributed to patients, the drug product manufacturer must notify its NDA or ANDA holder. The NDA or ANDA holder must notify the FDA if the situation meets the threshold for a field alert report or a recall notification.
This cascade can move very fast. The FDA expects initial notification of a Class I recall situation within three days of determination. For a situation that starts with a KSM supplier discovering a manufacturing deviation, the time from discovery to FDA notification may be less than one week. This timeline requires that quality agreements include clear escalation paths and that quality systems are designed for rapid impact assessment.
Key Takeaways: Section 12
Quality agreements must be specific about change notification lead times, deviation reporting requirements, and testing responsibilities. The regulatory notification cascade following a KSM quality deviation can be compressed to days. Quality systems at both the KSM supplier and the pharmaceutical manufacturer must support rapid impact assessment and regulatory notification.
13. Continuous Manufacturing and Its Effect on Starting Material Designation
How Continuous Manufacturing Disrupts the Discrete Step Model
Traditional batch manufacturing has discrete steps with physical product isolation between them. The KSM designation makes intuitive sense in this context: it is the step where GMP begins, and it is associated with a material that is isolated, tested, and released before the next step proceeds. Continuous manufacturing removes many of these discrete isolations. A continuous API manufacturing process may run the KSM through conversion, intermediate synthesis, and final drug substance production in a single uninterrupted flow, without the intermediate testing checkpoints that traditional batch manufacturing provides.
This creates a regulatory question that ICH Q11 does not fully resolve: where is the GMP boundary in a continuous process where the KSM feeds directly into the reaction cascade without isolation? The FDA’s guidance on continuous manufacturing, finalized in 2019, addressed this partially by acknowledging that real-time release testing (RTRT) and process analytical technology (PAT) can substitute for traditional end-of-step testing. But the question of where the starting material designation sits in a continuous flow process remains an area of evolving guidance.
PAT Tools and Their Role in KSM Quality Monitoring
Process analytical technology has direct relevance to KSM quality management. Rather than testing the KSM at receipt and releasing it before use, PAT-enabled processes can monitor KSM quality attributes in-line as the material enters the continuous flow. Near-infrared (NIR) spectroscopy, Raman spectroscopy, and inline mass spectrometry can monitor identity and purity attributes in real time, allowing the process control system to divert out-of-specification material before it enters the main synthesis stream.
This capability allows tighter integration between KSM quality monitoring and process control, and may support reduced end-of-batch testing requirements under RTRT frameworks. Regulatory agencies have been receptive to PAT-based release strategies for continuous manufacturing, and several companies have received FDA approvals for PAT-enabled RTRT in drug product manufacturing. The extension of these approaches to API starting material monitoring is a natural next step.
Key Takeaways: Section 13
Continuous manufacturing challenges the discrete step model that underlies traditional KSM designation frameworks. PAT tools allow real-time KSM quality monitoring that can substitute for traditional receipt testing in integrated continuous processes. Regulatory guidance on KSM designation in continuous manufacturing is evolving; teams implementing continuous API manufacturing should engage with FDA and EMA during process development to align on starting material designation expectations before filing.
14. Building the Regulatory Package: Section 3.2.S.2.3 and the ICH Q11 Justification Narrative
What Section 3.2.S.2.3 Must Contain
The regulatory package for a KSM sits primarily in Module 3 Section 3.2.S.2.3 of the Common Technical Document (CTD). This section covers the ‘Control of Materials’ used in the drug substance manufacturing process, and for the designated starting material it must include the material’s identity and chemical structure, its specification with justified limits, analytical methods and their validation summaries, supplier information, a brief description of the synthesis if custom-made, and the scientific justification for its designation as a starting material.
The justification narrative in 3.2.S.2.3 must be cross-referenced with the drug substance manufacturing description in 3.2.S.2.2 and the impurity profile discussion in 3.2.S.3.2. Regulatory reviewers read these sections together, and an inconsistency between the KSM justification in S.2.3 and the impurity discussion in S.3.2 is a reliable source of Day 120 objections in MAA review or information requests in ANDA review.
The Justification Narrative Structure
A well-constructed KSM justification narrative addresses four elements in sequence. First, it establishes that the proposed material meets the Q11 definition: it has defined chemical properties and structure, and it contributes a significant structural fragment to the final API. This section should include a structural overlay showing which atoms from the KSM are present in the final API structure, making the ‘significant structural fragment’ argument visual and unambiguous.
Second, it addresses commercial availability or, if the material is custom-synthesized, provides the scientific rationale for the late-stage designation. Third, it summarizes the impurity control strategy: which impurities are controlled by specification on the KSM, which are controlled by process, and which fate and purge data support the conclusion that impurities are adequately managed throughout the synthesis. Fourth, it addresses the regulatory history: have other companies designated this material as a starting material for the same or analogous API? What was the outcome? Precedent, while not binding on the reviewing agency, is persuasive.
Pre-Submission Engagement
The regulatory package for a novel or complex KSM designation should not be submitted cold. Pre-submission engagement, whether through a Type B meeting with FDA (typically a pre-IND or pre-NDA meeting), a scientific advice request with EMA, or an informal meeting with a national competent authority in a reference market, allows the applicant to gauge agency receptiveness before committing to the designation in a formal filing.
The value of pre-submission engagement is asymmetric: the cost of a meeting is low, the time investment is moderate, and the benefit of receiving early negative feedback is substantial if it prevents an expensive post-submission back-and-forth. The FDA’s commitment in the Prescription Drug User Fee Act (PDUFA) performance goals includes a 90-day response time for Type B meeting requests. This timeline is adequate for development programs that plan 12 to 18 months ahead for regulatory submissions.
Key Takeaways: Section 14
Section 3.2.S.2.3 is the primary regulatory home for the KSM designation and must cross-reference consistently with S.2.2 and S.3.2. The justification narrative should address structural fragment contribution, commercial availability or custom synthesis rationale, impurity control strategy, and regulatory precedent. Pre-submission engagement with FDA or EMA is a low-cost risk reduction step that is underutilized in generic drug development.
15. KSM Strategy Through the Drug Development Lifecycle
Discovery and Early Research: Setting the Strategic Parameters
The KSM question during discovery is primarily a synthetic route question. Medicinal chemists are not thinking about ICH Q11 when they design the first milligram-scale synthesis of a new lead compound, nor should they be. But the process chemistry team that takes over during lead optimization and candidate selection should begin mapping the synthesis with KSM designation in mind, because synthetic route decisions made during optimization can foreclose or constrain KSM options during development.
Specifically, routes that rely on highly specialized, custom-synthesized intermediates at early stages create KSM challenges later. A route where step three produces a novel intermediate with no commercial analog is one where the KSM will almost certainly need to be step three or later, which means GMP controls on 80 percent of the synthesis. A route designed around commercially available building blocks at steps one and two, with proprietary chemistry only in steps seven through ten, allows a much more economical KSM designation at a commercially available intermediate.
IND-Enabling Studies Through Phase 2: Developing the Justification
During IND-enabling studies and early clinical development, the synthesis is refined, the impurity profile is characterized, and the KSM designation is tested against regulatory expectations in early agency interactions. Phase 1 and Phase 2 clinical material is typically manufactured to GMP standards throughout the synthesis, because the volume requirements are low and the cost of full GMP compliance at this scale is manageable. The flexibility to operate with a less conservative KSM designation at commercial scale is developed during this period, as fate and purge data are generated and process understanding deepens.
Pre-IND meetings with the FDA are the primary forum for testing the KSM designation during this phase. The FDA’s guidance on pre-IND meetings for new molecular entities (NMEs) recommends that applicants include drug substance manufacturing information in the briefing document, and KSM designation is an appropriate topic for discussion at this stage.
Phase 3 and Pre-NDA/ANDA: Locking the Strategy
By the time Phase 3 clinical studies are underway, the KSM designation should be finalized and the regulatory package substantially complete. Changes to the KSM designation after Phase 3 manufacturing begins are possible but risky. If the Phase 3 clinical batches were manufactured under a specific KSM designation and GMP controls, the regulatory submission will describe those manufacturing conditions. A post-Phase-3 change to the KSM designation would require a comparability assessment or, in some cases, bridging studies to confirm that the drug substance manufactured under the new designation is comparable to the clinical trial material.
For ANDA applications, the KSM designation must be consistent between the API DMF and the ANDA drug substance section. Inconsistencies between the DMF-listed starting material and the ANDA manufacturing description are a common source of information requests during review.
Commercial Phase: Change Management
After approval, the KSM designation is fixed in the regulatory dossier. Changes to the designated starting material, its supplier, its specifications, or its manufacturing process are post-approval changes subject to the notification requirements discussed in Section 11. The commercial phase requires systematic change management processes that can identify, classify, and notify regulatory agencies of KSM-related changes within the required timeframes.
Key Takeaways: Section 15
Early synthetic route design should consider KSM designation options. GMP is typically applied throughout the synthesis in early development, with KSM designation optimization reserved for commercial manufacturing. Pre-IND meetings are the appropriate forum to test KSM strategy with FDA. Post-Phase-3 KSM designation changes require comparability assessment. Commercial-phase change management must handle KSM-related changes within regulatory notification timeframes.
16. Investment Strategy: How KSM Choices Affect Valuation, Competitive Moats, and Risk Profiles
KSM Quality as a Leading Indicator of Regulatory Risk
For institutional investors and analysts covering pharmaceutical and generic drug companies, the KSM strategy is a leading indicator of regulatory execution quality. Companies with documented, compliant, well-justified KSM designations backed by comprehensive impurity fate and purge data are less likely to receive complete response letters, information requests, or post-approval compliance actions related to drug substance manufacturing. This translates directly to probability-adjusted pipeline valuation: a program with a strong KSM regulatory package carries a higher probability of first-cycle approval than a program with an aggressive, under-supported designation.
Analysts should ask, in earnings calls and investor presentations, whether the company’s key API programs have completed Phase 3 manufacturing under the commercial KSM designation, whether DMFs for critical KSMs are complete and cross-referenced in ANDA or NDA filings, and whether the supply chain has qualified secondary KSM sources for high-revenue products. These are specific, answerable questions that meaningfully differentiate companies in the generic and specialty pharmaceutical space.
KSM IP as a Balance Sheet Asset
For originator pharmaceutical companies, proprietary synthetic routes and their associated KSM IP represent balance sheet assets that are rarely valued explicitly but have quantifiable economic impact. A synthetic route patent covering a key intermediate with eight years of remaining term on a drug with $1 billion in annual U.S. sales creates a barrier to generic entry that is worth, on a probability-adjusted basis, several hundred million dollars to the originator. This value appears nowhere in standard GAAP financial statements, which treat patent costs as intangible assets amortized over their statutory life without regard to strategic or competitive value.
IP-focused investors and royalty-stream acquirers (Royalty Pharma, BCPE Nucleon-Pharma, DRI Healthcare) are increasingly sophisticated about the relationship between process IP, including synthesis route and intermediate patents, and the duration of effective market exclusivity. A drug that has lost its composition-of-matter patent but retains five additional years of process patent protection, because no generic competitor has developed a commercially viable non-infringing synthesis, is worth substantially more to these investors than a drug at the same development stage with no remaining process IP.
Supply Chain KSM Risk as a Discounting Factor
Conversely, unmitigated KSM supply chain risk is a discounting factor in pharmaceutical company valuation. A company with 60 percent of its API manufacturing dependent on a sole-source Chinese KSM supplier, without dual sourcing or buffer stock, carries tail risk that is not fully reflected in its stock price until a supply disruption actually occurs. The COVID-19 pandemic demonstrated this dynamic clearly: companies with resilient supply chains maintained manufacturing continuity and gained market share, while companies with concentrated supply chains absorbed revenue losses and one-time remediation costs.
Quantifying this risk for valuation purposes requires knowing which KSMs are sole-sourced, which are concentrated in high-risk geographies, what the buffer stock coverage is for each critical material, and what the product’s revenue contribution is at risk from a 3-month supply disruption. Most pharmaceutical companies do not disclose this level of supply chain detail in public filings. Analysts who develop proprietary KSM supply chain intelligence through supplier mapping, shipping data analysis, and primary research with contract manufacturers have a genuine informational edge.
M&A Implications: KSM Due Diligence
In pharmaceutical M&A transactions, KSM due diligence is a discrete workstream that is often underweighted relative to clinical, financial, and commercial due diligence. The acquirer should assess whether key APIs in the target’s portfolio have complete, compliant KSM regulatory packages; whether sole-source KSM dependencies create earnout risk; whether any KSM-related complete response letters or FDA warning letters are pending or recent; and whether the target’s DMFs for critical APIs are owned, controlled, and transferable. A target that manufactures its lead commercial product from a sole-source Chinese KSM with no secondary qualification and a three-week inventory buffer has a supply chain liability that should reduce the acquisition price, or at minimum trigger a post-closing integration requirement for dual-source qualification.
Key Takeaways: Section 16
KSM regulatory quality predicts first-cycle approval probability. Proprietary synthesis route IP is a balance sheet asset that standard GAAP does not capture adequately. Sole-source KSM dependencies are discounting factors in pharma company valuation. M&A due diligence should include a discrete KSM supply chain and regulatory workstream.
17. Frequently Asked Questions
What is the difference between a Key Starting Material and an API Starting Material?
They are the same thing described with different terminology. ‘API Starting Material’ is the official ICH regulatory term. ‘Key Starting Material’ is the industry operational term, emphasizing that not all starting materials carry equal strategic weight. Both terms refer to the designated point where GMP controls begin in API synthesis.
How early in drug development should my company finalize the KSM designation?
The designation should be stable by the time Phase 3 manufacturing begins, because the regulatory submission will describe commercial manufacturing conditions that must be consistent with Phase 3 production. The designation can and should be refined during Phase 1 and Phase 2, as process understanding develops and fate and purge data are generated. Pre-IND meetings are the appropriate forum to test the designation with FDA before Phase 3.
Can the same KSM designation be used in FDA and EMA submissions?
Yes, and it is the preferred approach for global development programs. When the two agencies have meaningfully different expectations, the conservative approach is to satisfy the more stringent agency (typically EMA) and use that designation globally. Filing different designations in different markets is permissible but creates manufacturing and change management complexity.
What are the most common reasons a proposed KSM designation fails regulatory review?
The most frequent causes of regulatory rejection or objection are: insufficient structural complexity relative to the final API; inadequate fate and purge data for impurities, particularly genotoxic impurities under ICH M7; a commercial availability claim that agencies investigate and find unsupported; limited process understanding for synthetic steps proposed to be outside the GMP boundary; and inconsistency between the KSM justification narrative and the impurity profile discussion elsewhere in the dossier.
How do continuous manufacturing processes affect the starting material designation?
Continuous manufacturing without intermediate isolation complicates the discrete step model that traditional KSM designation assumes. PAT tools and real-time release testing can substitute for traditional receipt testing. Regulatory guidance on starting material designation in continuous API manufacturing is still developing; early regulatory engagement is essential for programs pursuing this approach.
What IP protection is available for proprietary KSMs and synthetic routes?
Proprietary synthetic routes are protectable through process patents (covering the synthesis steps), composition-of-matter patents on novel intermediates, and trade secret protection for non-GMP steps that do not appear in the regulatory filing. Process patents are effective against generic competitors because generic ANDAs must describe their synthesis, which Paragraph IV litigation will then evaluate for infringement of listed process patents.
How should investors assess KSM supply chain risk in pharma companies?
Assess whether key APIs have dual-sourced KSMs qualified in geographically distinct regions; whether buffer stocks cover 4 to 6 months of demand for high-revenue products; and whether any sole-source dependencies are in high-risk jurisdictions. Companies that disclose supply chain resilience metrics in investor presentations and annual reports are more likely to have mature programs than those that do not disclose at all.
This analysis is based on publicly available regulatory guidance, industry data, and patent filings. It does not constitute legal, regulatory, or investment advice. Readers should consult qualified pharmaceutical regulatory counsel and financial advisors before making decisions based on this material.
Originally derived from source material at DrugPatentWatch.com. All expansions, analysis, investment commentary, and technical elaboration are original.