A single drug can generate $10 billion in annual revenue. The same drug, one day after its last relevant patent expires, can lose 80 percent of that revenue within twelve months. No other industry operates on timelines where a single legal document — or the absence of one — can move that magnitude of enterprise value so abruptly. That arithmetic is why patent strategy in pharmaceuticals is not a legal service line. It is the central mechanism of the entire business model.
This guide is written for the professionals who need to operate at that level of precision: IP counsel constructing and defending patent estates, R&D portfolio leaders deciding which programs to advance, and institutional investors modeling effective loss of exclusivity dates. It covers every phase of the pharmaceutical IP lifecycle — from the timing decision on a first patent filing through the mechanics of the BPCIA ‘patent dance’ for biologics and the inventorship questions raised by AI-assisted discovery. The goal is not to survey the landscape. It is to give you the technical tools to act in it.
Section 1: The Economics of Exclusivity — Why Drug Patents Are Corporate Strategy, Not Legal Overhead
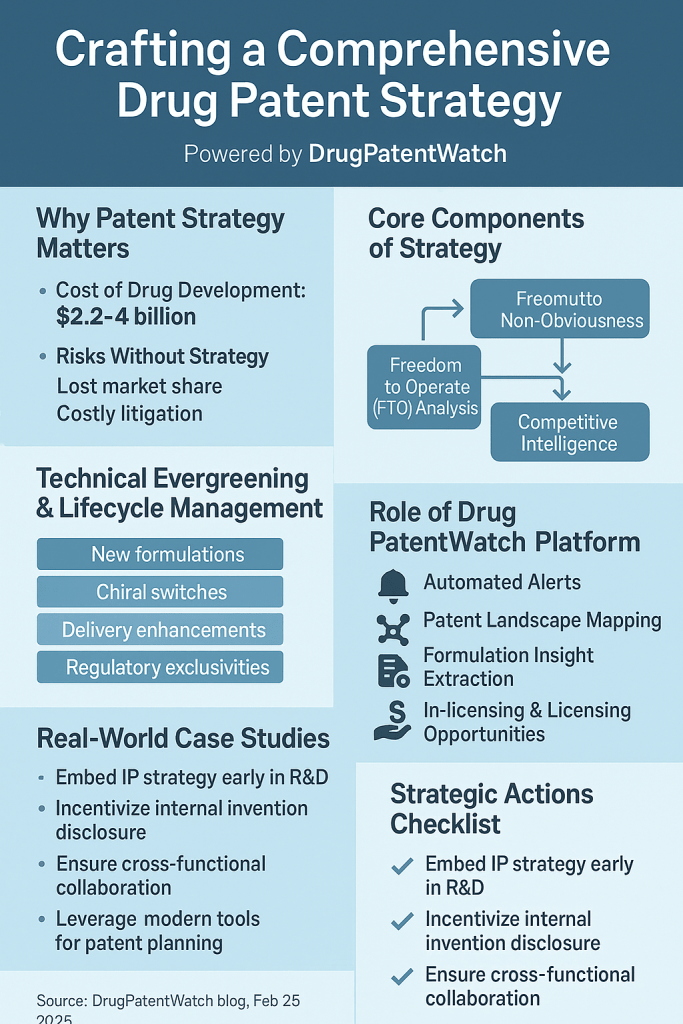
The Real Cost Equation: $2.2 Billion Per Asset, With Caveats
Deloitte’s 2024 analysis pegged the average cost to develop a new pharmaceutical asset at $2.2 billion — a figure that accounts for the capitalized cost of failures across a portfolio, not merely the direct cost of the drugs that succeed. Other analyses, including the Tufts Center for the Study of Drug Development’s methodology, produce estimates approaching $4 billion when failure-adjusted capital costs are fully included. Both figures are defensible depending on the model’s assumptions. The number that matters strategically is not which figure is correct, but what the figure implies for the length and quality of market exclusivity required to generate an acceptable return.
The math is unforgiving. A drug that takes 12 years from IND filing to approval and earns a five-year effective market exclusivity window must generate $440 million per year just to break even on a $2.2 billion capitalized cost, before SG&A, cost of goods, or tax. That window shrinks further in the most common scenarios: a key patent invalidated after Paragraph IV litigation, a Pediatric Exclusivity extension not obtained, or a biosimilar launch earlier than modeled because the secondary patent estate was thinner than it appeared.
The composition of that $2.2 billion is also worth scrutinizing. When independent analysts examine the full financial statements of the ten largest pharmaceutical companies, they consistently find that SG&A — which includes sales force deployment, physician marketing, and direct-to-consumer advertising — equals or exceeds R&D spend in most years. A 2020 AHIP analysis of the ten largest pharmaceutical companies found that seven spent more on selling and marketing than on R&D, with a collective gap of $36 billion. This does not invalidate the R&D cost argument for patent protection, but it refocuses the strategic question. The patent monopoly funds the entire commercialization infrastructure, not only R&D recovery. That reality drives the aggressive lifecycle management practices that define the modern pharmaceutical competitive landscape.
IP Valuation as a Core Asset: How Patent Portfolios Drive Pharma Valuations
For a pre-revenue biotech, the patent portfolio often accounts for 60 to 90 percent of total enterprise value. When Pfizer acquired Arena Pharmaceuticals for $6.7 billion in 2022, the headline asset was etrasimod, an S1P receptor modulator in Phase 3 for inflammatory bowel disease. But the acquisition price also reflected the breadth and defensibility of Arena’s composition of matter patents, the extent of method-of-use coverage across autoimmune indications, and the years of remaining exclusivity on those claims. An IP estate with two more years of primary patent protection carries a materially different valuation than one with eight years remaining, even if the underlying molecule is identical.
For established commercial franchises, patent portfolio analysis directly informs revenue modeling. The standard discounted cash flow model for a drug product requires an effective loss of exclusivity (LOE) date — the date when the first generic or biosimilar can realistically enter the market, accounting for all patent and regulatory exclusivity layers. Modeling that date with precision requires a full map of every Orange Book patent, its expiration date, its vulnerability to inter partes review (IPR) challenge, and the probability that any pending litigation results in settlement or judgment. This is the core of pharmaceutical equity research, not a peripheral legal analysis.
For early-stage biotechs raising capital or negotiating a licensing deal, the quality of the prosecution history matters as much as the number of granted patents. A portfolio where claim scope has been repeatedly narrowed to overcome prior art — visible in every amendment filed during examination — signals vulnerability. A portfolio with multiple pending continuation applications, each capturing additional claim scope as clinical data matures, signals a dynamic estate that will continue generating defensible rights through development. Acquirers and licensors read prosecution histories. Most management teams do not brief investors on them, which creates an information asymmetry worth exploiting.
Investment Strategy Note
When evaluating a pharma or biotech position, build a patent expiry waterfall for every material revenue asset. Map each patent type (composition of matter, formulation, method-of-use, process), assign a probability-weighted expiry date adjusted for litigation risk, and overlay applicable regulatory exclusivities. The delta between the statutory patent expiry and the effective LOE date is where valuation discrepancies concentrate. Companies with dense secondary patent estates and stacked regulatory exclusivities consistently trade at premiums to companies with bare-bones, single-layer protection.
Key Takeaways — Section 1
The $2.2 billion per-asset development cost is a failure-adjusted capitalized figure. Effective exclusivity duration directly determines whether development economics close at all.
Patent protection funds the commercial infrastructure — not only R&D recovery. This explains why lifecycle management is aggressive even for drugs where direct R&D costs were modest.
For pre-commercial biotechs, the patent estate is the primary asset. For commercial franchises, it is the primary driver of forward revenue modeling. Treating it as legal overhead is a material strategic error.
Prosecution histories contain more information about IP quality than the face of the granted patent. Repeated claim narrowing is a red flag that is rarely discussed in investor presentations.
Section 2: The Anatomy of a Drug Patent Portfolio
Composition of Matter Patents — The Foundational Asset
The composition of matter patent covers the active pharmaceutical ingredient (API) as a chemical or molecular entity. It claims the specific structural identity of the drug molecule itself. The U.S. Supreme Court has defined ‘composition of matter’ to include all compositions of two or more substances, whether the result of chemical union or mechanical mixture, across all physical states. In pharmaceutical practice, the base patent asserts exclusive rights to the exact chemical structure of the new molecular entity (NME) or new biological entity (NBE).
Three requirements govern patentability under 35 U.S.C. sections 101 through 103: novelty, non-obviousness, and utility. Novelty requires that the molecule not appear in any prior art — any prior patent, published application, or scientific disclosure — before the priority date. Non-obviousness requires that the compound not be an obvious structural modification of a known compound to a person having ordinary skill in the art (PHOSITA). Utility, in the pharmaceutical context, requires a specific, substantial, and credible therapeutic application, not merely a theoretical one. A composition of matter patent on a drug candidate without demonstrated in vivo activity faces utility challenges during examination.
The composition of matter patent sets the initial 20-year term from the filing date. It is the broadest and most difficult claim for a generic challenger to design around, because any version of the molecule — any salt form, polymorph, or prodrug — that converts back to the same API will typically fall within the scope of an adequately drafted composition of matter claim. Securing broad claim scope at the composition of matter stage is worth the additional prosecution effort: every incremental unit of claim breadth translates directly into barriers for future competitors.
The Patent Thicket: A Technical Taxonomy of Secondary Patents
No single composition of matter patent is sufficient to maintain market exclusivity through the full commercial lifecycle of a successful drug. The patent thicket — a web of overlapping secondary patents covering different aspects of the drug product or its use — extends effective exclusivity beyond the primary patent term. The thicket’s deterrent value is cumulative. A generic company facing 80 patents must budget for the possibility of litigating all 80. That aggregate litigation cost can make settlement economically rational even when many individual patents are vulnerable — which is the outcome the thicket is designed to produce.
Formulation Patents
A formulation patent protects a specific pharmaceutical composition: the combination of the API with inactive ingredients (excipients), carriers, stabilizers, or delivery systems in defined ratios or structures. The strategic value of formulation patents operates in two directions simultaneously. A well-designed extended-release formulation can improve the drug’s therapeutic profile — smoother plasma concentration curves, reduced peak-trough variability, lower incidence of concentration-dependent adverse effects — generating a clinically differentiable product. Because a new formulation is patentable independent of the original API, it carries a fresh 20-year term from its own filing date. A formulation patent filed fifteen years after the composition of matter patent can extend effective exclusivity by a decade or more.
The legal durability of a formulation patent hinges on non-obviousness. Courts and the USPTO Patent Trial and Appeal Board (PTAB) have invalidated formulation patents where the selected excipient combination was routine and predictable to a skilled formulator. The strongest formulation patents claim a non-obvious selection — a specific polymer matrix composition achieving a release rate not predictable from prior art, or an excipient ratio that solves a specific stability problem in a non-intuitive way. Weak formulation patents claiming conventional techniques applied to a new API are low-value strategic assets and high-value targets for inter partes review (IPR) petitions.
Method-of-Use and New Indication Patents
A method-of-use patent claims the use of a compound to treat a specific condition by a defined mechanism or in a specific patient population. It does not require a new molecule — only a new, non-obvious therapeutic application. This makes it the primary vehicle for drug repurposing and for protecting the expansion of a drug’s approved label into new indications. When Pfizer’s sildenafil (Viagra) compound patent approached expiration, its method-of-use patent on the treatment of erectile dysfunction — filed after the initial cardiovascular indication — became the critical exclusivity asset for that indication. Generic companies filing ANDAs for sildenafil could only carve out the patented indication through a ‘skinny label’ (Section viii carve-out), limiting their product’s labeled uses. The legal standard requires that the use itself be novel and non-obvious. If prior art teaches that a compound of the same class has a specific therapeutic effect, a patent claiming that effect for a structurally similar compound faces serious obviousness challenges.
Process Patents
A process patent protects a novel and non-obvious manufacturing method for producing the API or drug product. For small molecules, process patents cover synthetic routes — specific reaction sequences, catalysts, reagents, or purification steps. For complex biologics, where the manufacturing process is inextricable from product quality, process patents cover cell line construction, fermentation parameters, purification train design, and formulation processing conditions. Process patents are not the strongest barrier because a generic manufacturer can often develop an alternative, non-infringing synthetic route, but the time and cost of developing and validating an alternative under GMP conditions — with the full documentation package required for an ANDA or 505(b)(2) filing — creates meaningful competitive delay. In biologics, the barrier is substantially higher because biosimilar manufacturers must demonstrate that their product is ‘highly similar’ to the reference biologic despite inherent process variability.
Polymorph Patents
Many APIs exist in multiple crystalline forms (polymorphs) or as amorphous solids. Different polymorphs can have substantially different solubility, bioavailability, and stability profiles. Polymorph patents are among the most heavily litigated in pharmaceutical IP. Their vulnerability stems from the difficulty of establishing that a specific polymorph is non-obvious when other polymorphs are already disclosed in prior art. The PTAB and district courts have frequently invalidated polymorph patents on grounds of obviousness-type double patenting (ODP) or because prior art taught routine screening methods that would have led a skilled crystallographer to the claimed form. The AstraZeneca v. Ranbaxy litigation over rosuvastatin (Crestor) calcium salt polymorphs is a well-documented example of the complex interplay between composition of matter claims, salt form patents, and polymorph patents in high-value patent thickets.
Chiral Switch Patents
A chiral switch involves the development and commercialization of a single enantiomer from a previously marketed racemic drug. Because the single enantiomer is a structurally distinct compound from the racemate, it qualifies for a new composition of matter patent, provided it was not previously disclosed or rendered obvious by the prior art on the racemate. Chiral switch patents can be accompanied by new data exclusivity if the single enantiomer receives its own NDA approval with new clinical investigations. AstraZeneca’s transition from Prilosec (omeprazole, a racemate) to Nexium (esomeprazole, the S-enantiomer) is the canonical case. AstraZeneca’s clinical data showed that esomeprazole achieved higher plasma levels than equivalent doses of omeprazole due to differences in CYP2C19 metabolism between the two enantiomers. The IP strategy successfully transferred a large fraction of the Prilosec patient base to the new, protected product before generic omeprazole was available. The most defensible chiral switch patents rest on unexpected advantages of the single enantiomer not predicted by prior art: a different metabolic profile extending half-life, different tissue distribution reducing a class-effect adverse event, or a superior therapeutic window not predictable from racemate data.
Delivery Device Patents
For drugs requiring a physical delivery system — auto-injectors, prefilled syringes, inhalers, nasal delivery devices — the device itself can be patented independently of the drug formulation it contains. Device patents are examined against a medical device and engineering prior art landscape rather than a chemical or pharmaceutical one, which creates a distinct set of patentability arguments. A biosimilar manufacturer that secures FDA approval for a biosimilar molecule cannot immediately use the reference product’s proprietary auto-injector if that device is separately patented. Sanofi’s litigation strategy around the Lantus SoloSTAR insulin pen device — involving multiple device patents filed after the insulin glargine composition of matter patent — delayed biosimilar market entry and is a frequently cited example of device IP commercial utility.
IP Valuation: Contribution of Each Patent Type to Portfolio Value
High (obviousness, prior art on excipient combinations)
LOE date extension; depends on clinical differentiation of the formulation
Method of Use (New Indication)
Varies; tied to indication-specific filing date
Moderate-High (obviousness over prior class data)
New market access; enables skinny-label carve-out strategy
Process
Up to 20 years from filing date
Low-Moderate (can often be designed around)
Increases generic development cost and timeline; high value in biologics
Polymorph
Up to 15+ years beyond primary patent
Very High (routine screening arguments, ODP challenges)
Secondary defense layer; frequently invalidated under IPR
Chiral Switch
Up to 20 years from filing; new data exclusivity possible
Moderate (must establish unexpected superiority)
Patient migration to new protected product; fresh data exclusivity stack
Delivery Device
Device-specific; can extend 10 to 15+ years
Low-Moderate (distinct technical domain)
Biosimilar entry barrier; protects administration experience and brand loyalty
Key Takeaways — Section 2
A pharmaceutical patent portfolio is a collection of legally distinct assets with different scopes, terms, vulnerabilities, and valuations. Treating it as a monolithic ‘drug patent’ systematically misprices the company’s IP position.
Composition of matter patents are the strongest asset but expire first. The strategic objective is to build a secondary patent estate with later expiry dates that extends effective exclusivity beyond the primary patent term.
The patent thicket’s deterrent value is cumulative. A generic company facing 80 patents must budget for the possibility of litigating all 80. That aggregate litigation cost can make settlement economically rational regardless of the individual strength of each patent.
Polymorph and weak formulation patents are high-cost maintenance items with limited durability against well-funded PTAB petitioners. Method-of-use and chiral switch patents resting on genuine clinical differentiation are significantly more durable assets.
Section 3: Integrating Patent Strategy Across the Drug Development Lifecycle
Discovery and Preclinical: The Patent Landscape Analysis as a Capital Allocation Tool
Before committing meaningful capital to a drug candidate, conduct a patent landscape analysis — a systematic search and map of existing patents and published applications in the target therapeutic area, covering the relevant chemical or biological space. A rigorous landscape analysis addresses four questions. First, does the candidate itself, or a structurally obvious modification, already appear in a competitor’s patent? Second, are there dominant blocking patents that would prevent commercialization even if the company’s own composition of matter patent is granted? Third, where are the ‘white space’ areas in the landscape — therapeutic uses, patient sub-populations, or delivery approaches for the compound class that are not yet claimed? Fourth, what is the density of competitor patent activity, and what does its recent trajectory indicate about their pipeline direction?
A 2022 study found that pharmaceutical companies using systematic patent landscape analyses before committing to a development candidate avoided infringement disputes at a significantly higher rate, with average savings of $15 million per avoided infringement case. At a development cost of $2.2 billion per asset, the primary value of the landscape analysis is actually the early identification of a blocking patent or overcrowded space that allows the company to redirect capital to a more defensible position before Phase II costs accumulate. The $15 million avoided litigation figure is real but secondary to the capital redeployment value.
The Filing Timing Dilemma: Every Month of Delay Is Future Revenue
The 20-year patent term begins at the filing date, not the approval date. A composition of matter patent filed in Year 1 of preclinical development and not reaching market approval until Year 13 has only seven years of effective post-approval exclusivity. The filing date decision is one of the highest-stakes calculations in pharmaceutical strategy, and most companies handle it suboptimally by defaulting to the earliest defensible date rather than the financially optimal one.
The ‘file early’ imperative is driven by the United States’ first-to-file system adopted under the America Invents Act of 2013, and by the risk that a competitor will file a similar compound first. Both concerns are real. But they must be weighed against the value of the delayed filing. If filing six months later would push the effective patent expiry date back six months, and the drug eventually generates $3 billion per year at peak, each month of delay in filing that converts to a month of additional exclusivity at peak commercial scale is worth $250 million in gross revenue. The expected value calculation must account for the probability that a competitor files first during the delay period and the probability that the delay creates public disclosure issues.
A key insight from pharmaceutical patent law is that Phase I clinical trials — which test safety and tolerability in healthy volunteers without generating meaningful efficacy data — typically do not constitute ‘public use’ or ‘on sale’ activity that starts the one-year grace period under 35 U.S.C. section 102. This suggests that in many cases, a company can safely delay filing beyond Phase I initiation, capturing more clinical development data to file a better-supported application, without triggering a prior art bar. Phase II initiation, which involves human efficacy testing in patients, is a more aggressive boundary. Companies that have modeled this tradeoff explicitly — running scenarios for what each additional year of effective exclusivity is worth at modeled peak sales — consistently find that the optimal filing date is later than instinct suggests.
Building a ‘patent-aware’ scientific culture is the prerequisite for executing this strategy correctly. Every laboratory notebook entry, every conference presentation, and every preprint submission is a potential public disclosure event that can start or bar the patent clock. The most successful companies run ongoing IP education for their scientific staff and require invention disclosure forms before any external communication of research results. The research scientist who presents unpublished data at a small specialist conference without notifying the patent team can start a one-year clock that management will not discover until it is too late to file.
Clinical Development: Systematic IP Mining
Clinical trials generate data at a rate that typically outpaces the organization’s capacity to evaluate it for patentable inventions. The Phase II and Phase III programs involve extensive study of pharmacokinetics, pharmacodynamics, dose-response relationships, sub-population effects, formulation behavior under real-world conditions, combination regimens, and biomarker correlates of response. Each data stream can generate patentable inventions — but only if the organization has the processes to identify and capture them.
A systematic approach to clinical IP mining embeds an IP attorney or patent agent into each major clinical program team, reviews clinical data packages for patentable features at defined protocol milestones, and requires clinical investigators to disclose unexpected findings through a formal invention disclosure process. The types of clinical-stage inventions most frequently missed include: optimal dosing regimens differing substantially from early-phase doses; patient sub-populations defined by biomarkers with disproportionate response; specific administration conditions that materially affect pharmacokinetics; unexpected secondary endpoints supporting a new indication claim; and combination regimens that show synergistic activity not predicted by either component’s prior data. Each of these, if patented with adequate specificity and data support, adds a distinct patent layer with its own expiration date extending beyond the primary composition of matter term.
Commercialization: Aligning Patent Estate with Launch Strategy
As a drug approaches FDA approval, the patent strategy must align with the commercial plan. A commercial strategy targeting a specific patient sub-population defined by a genotypic biomarker should have corresponding method-of-use patent claims covering that sub-population. A pricing strategy based on a specific formulation’s superior administration profile should be backed by formulation and device patents with sufficient remaining term to support that price premium. A partnering or out-licensing strategy requires a clean, well-documented patent estate that a partner’s diligence team can evaluate without finding holes. For pre-commercial biotechs, every patent prosecution decision affects future deal valuation. Narrowed claims, abandoned continuations, and missed foreign filings are permanent liabilities that surface in acquirer diligence.
Investment Strategy Note
In due diligence for a preclinical or Phase I asset, review not just the granted patents but the prosecution histories. A company that has repeatedly agreed to claim-narrowing amendments to overcome prior art has signaled that its core IP is vulnerable. A company with multiple continuation applications pending, each capturing different aspects of the clinical program, has built a dynamic estate that will continue generating new patents as data matures. The continuation pipeline is as valuable as the granted portfolio.
Key Takeaways — Section 3
Patent landscape analysis is a capital allocation tool, not a legal checkbox. It should precede significant R&D investment commitments and be updated at each development phase.
The patent filing date is a financial decision. Building an explicit model of the expected value of a filing delay — weighted by competitor preemption probability — produces a more defensible decision than instinct or legal conservatism alone.
Clinical trials are IP generation events. Organizations that treat them only as data generation events consistently leave patentable inventions undocumented and uncaptured.
For pre-commercial companies, every patent prosecution decision affects future deal valuation. Narrowed claims, abandoned continuations, and missed foreign filings are permanent liabilities that appear in acquirer diligence.
Section 4: Lifecycle Management and the Full Evergreening Technology Roadmap
Technical Evergreening Strategies: The Full Menu
Lifecycle management extends a drug product’s effective market exclusivity beyond the original composition of matter patent term by generating genuine or incremental technical innovations that qualify for new patent protection. The full menu of technical strategies is wider than most summaries acknowledge, and the most effective programs execute multiple strategies in parallel, with careful attention to the patent filing timeline for each.
Extended-Release and Modified-Release Formulations
The technical approaches to achieving modified release are numerous: matrix tablets using hydrophilic polymers (hydroxypropyl methylcellulose, carbomers, polyethylene oxide), reservoir systems with rate-controlling membranes (ethylcellulose, Eudragit polymer coatings), osmotic drug delivery systems (the OROS technology platform, originally developed by ALZA Corporation), and multiparticulate systems using coated pellets or microparticles in a capsule. Each of these approaches, and the specific formulation parameters within each, can be independently patented. The combination of polymer type, molecular weight, ratio, particle size distribution, coating thickness, and processing conditions that achieves a specific plasma concentration target represents a substantial design space, each corner of which is potentially patentable if it achieves a non-obvious result.
Eli Lilly’s development of once-weekly fluoxetine (Prozac Weekly) as the original twice-daily Prozac approached its composition of matter patent expiry is a textbook execution. The clinical rationale — improved patient adherence with weekly versus daily dosing — was real, the patent was legitimate, and the commercial strategy of switching the prescribing base to the new formulation before generic fluoxetine was available was well-timed. Bristol-Myers Squibb’s Glucophage XR (extended-release metformin) employed the same playbook with the additional benefit that the once-daily formulation reduced the gastrointestinal side effects that caused a significant percentage of patients to discontinue the immediate-release formulation — a genuine clinical improvement that also happened to extend effective exclusivity by years.
The durability of a modified-release formulation patent depends on the non-obviousness and clinical utility of the specific formulation design. Courts have found that simply applying a standard extended-release technology to a known drug compound, with predictable pharmacokinetic outcomes, does not clear the non-obviousness bar. Strong formulation patents rest on specific parameters that achieve a non-obvious clinical benefit — a particular polymer combination that maintains drug stability under GI pH variability in a way not predicted by any individual polymer’s prior art performance, or a particle size distribution that achieves an unexpected bioavailability profile.
New Routes of Administration
Reformulating an oral drug for intranasal, transdermal, subcutaneous, inhalation, or intrathecal delivery involves developing entirely new pharmaceutical compositions and delivery systems, each with its own patentable features. GlaxoSmithKline’s development of intranasal sumatriptan (Imitrex Nasal Spray) alongside the original oral and injectable formulations created multiple patent-protected product lines from a single API, each with distinct pharmacokinetic profiles and clinical utility in different patient scenarios. Route-of-administration changes carry a higher R&D cost than reformulation within the same route, but they also produce more clinically differentiated products that are harder for generics to replicate with a simple bioequivalence study. In some cases, different routes require separate NDA approvals, which can generate new periods of three-year new clinical investigation data exclusivity.
Fixed-Dose Combination Products
A fixed-dose combination (FDC) product combines two or more APIs into a single dosage form. If the combination is novel and offers a non-obvious clinical advantage over taking the components separately — improved efficacy, pharmacokinetic synergy, reduced adverse events, or improved adherence — it qualifies for a combination patent covering the specific drug ratios, the co-formulated product, or the method of treating a patient with the combination. HIV treatment is the paradigmatic example. Gilead Sciences’ development of tenofovir/emtricitabine (Truvada), and subsequent single-tablet regimens including Atripla, Complera, and Genvoya, each created new patent-protected franchise assets from combinations of existing antiretrovirals. Each combination carried its own patent term, its own period of three-year new clinical investigation data exclusivity, and prescribing differentiation in the market. The clinical justification — simplified dosing regimens that dramatically improved adherence and outcomes in HIV patients — was genuine and provided durable intellectual property protection against obviousness challenges.
Chiral Switch: Execution Mechanics
A chiral switch requires stereoselective synthesis of the single active enantiomer, characterization of its pharmacological and pharmacokinetic profile relative to the racemate, and a comparative clinical study demonstrating non-inferiority or superiority to support an NDA approval. The patent strategy involves a new composition of matter patent on the single enantiomer if not previously disclosed, method-of-use patents covering its therapeutic applications, and potentially formulation patents if the enantiomer requires a distinct formulation from the racemate. The critical IP vulnerability of a chiral switch strategy is obviousness. If prior art on the racemate teaches that one enantiomer is the more active stereoisomer, isolating and patenting that enantiomer faces a serious challenge. The most defensible chiral switch patents rest on unexpected advantages not predicted by prior art: a different metabolic profile that extends half-life, different tissue distribution that reduces a class-effect adverse event, or a superior therapeutic window not predictable from racemate data.
The Regulatory Toolkit: Stacking Exclusivities to Build the Full Protection Timeline
Technical patent strategies operate in parallel with regulatory exclusivity provisions that are distinct from patents, run on their own timelines, and can either extend or overlap with the patent protection period. The most sophisticated lifecycle management programs model the full stack of patent plus regulatory exclusivities to identify the true effective LOE date.
Patent Term Extension (PTE) Under Hatch-Waxman
Section 156 of Title 35 allows a patent holder to apply for a term extension compensating for time lost during FDA regulatory review. The extension applies to one patent per product and is calculated as one-half the time in IND clinical trials plus the full time in FDA review, reduced by any period of applicant non-diligence. The maximum is five years, subject to the condition that total post-approval remaining patent life cannot exceed 14 years. In practice, most drugs receive PTE extensions in the range of two to four years — often the difference between seven and eleven years of post-approval exclusivity. That difference, measured in billions of dollars for a major commercial product, is one of the highest-return activities in pharmaceutical legal strategy. The application must be filed within 60 days of FDA approval, and the specific patent to be extended must be chosen strategically — not necessarily the primary composition of matter patent, but whichever patent in the estate provides the greatest remaining exclusivity protection.
NCE Data Exclusivity and Three-Year Clinical Exclusivity
The five-year new chemical entity (NCE) data exclusivity provision prohibits the FDA from approving any ANDA relying on the brand’s safety and efficacy data for five years from NDA approval, running independently of the patent term. For a drug with a short remaining patent life, data exclusivity can be the primary barrier to generic entry during the five-year period. The three-year new clinical investigation exclusivity is available for supplemental NDAs supported by new clinical studies — new indications, dosing regimens, or formulations. For an active lifecycle management program, three-year exclusivity on each new indication or formulation generates a rolling series of exclusivity windows that can extend protection years beyond the original patent estate. Each supplemental NDA approval with qualifying new clinical studies triggers a fresh three-year window, independent of any patents covering the change.
Orphan Drug Exclusivity
Orphan Drug Designation under the Orphan Drug Act of 1983 provides seven years of market exclusivity from the date of FDA approval for drugs treating conditions affecting fewer than 200,000 people in the United States. Orphan drug exclusivity (ODE) prevents FDA approval of any identical drug for the same orphan indication for seven years, regardless of the designated drug’s patent status. This is a uniquely powerful provision because it operates as an absolute exclusivity bar, not just a data protection mechanism. A generic company cannot obtain approval for the same drug in the same indication during the seven-year period even if it develops its own clinical data independently. IQVIA data shows that orphan drugs now account for approximately 20 percent of total U.S. pharmaceutical spending despite treating a small fraction of the patient population, because the exclusivity and pricing environment for orphan products is uniquely favorable. ODE is also per-indication: a single drug can accumulate multiple ODE periods by obtaining designation and approval in multiple orphan conditions.
Pediatric Exclusivity
The six-month pediatric exclusivity extension, granted when a company completes FDA-requested pediatric studies under the Best Pharmaceuticals for Children Act, attaches to all of the drug’s existing patent and regulatory exclusivities simultaneously. It does not generate a new, separate period of exclusivity but extends every existing exclusivity by six months. For a drug with multiple overlapping patent expirations, a PTE, and an ODE period, six months of pediatric exclusivity can be worth hundreds of millions in revenue on a high-revenue product. The requirement is not that the pediatric studies show clinical benefit in children — only that they be completed in accordance with the FDA’s Written Request. The commercial incentive structure is therefore highly favorable for large-revenue products regardless of pediatric clinical outcome.
The Full Exclusivity Stack: Modeling the Effective LOE Date
Exclusivity Type
Statutory Duration
Trigger
Independent of Patent?
Stackable?
NCE Data Exclusivity
5 years
First NDA approval for a new molecular entity
Yes
Yes (runs concurrently with patent protection)
3-Year New Clinical Investigation
3 years per supplemental NDA
Supplemental NDA with new clinical studies
Yes
Yes; applies to each new indication or formulation independently
Patent Term Extension (PTE)
Up to 5 years; post-approval life capped at 14 yrs
Application within 60 days of FDA approval
No (extends a specific patent)
One patent per approved product
Orphan Drug Exclusivity (ODE)
7 years per indication
Orphan Drug Designation plus FDA approval
Yes
Yes; per-indication; multiple ODE periods possible for multiple orphan indications
Biologic Data Exclusivity (BPCIA)
12 years
First BLA approval for a reference biologic
Yes
Yes (runs concurrently with patent protection)
Pediatric Exclusivity
6 months (extends all existing exclusivities)
Completion of FDA-requested pediatric studies
No (extends existing exclusivities)
Yes; applies to all active patent and regulatory exclusivities simultaneously
Investment Strategy Note
When modeling pharmaceutical revenue, the statutory patent expiry date from an Orange Book lookup is consistently the wrong figure to use. Build the full exclusivity stack: add PTE years to the composition of matter patent, identify applicable orphan or pediatric exclusivity, model the probability of each secondary patent surviving IPR challenge, and generate a probability distribution of generic entry dates across the full scenario set. The midpoint of that distribution is closer to the true effective LOE date than primary patent expiry. Companies with well-constructed secondary patent estates and stacked regulatory exclusivities consistently have effective LOE dates two to seven years later than naive patent expiry analysis suggests.
Key Takeaways — Section 4
No single evergreening strategy is sufficient. The most defensible programs combine technical innovations (new formulations, chiral switches, combination products) with regulatory exclusivity tools (PTE, ODE, three-year clinical exclusivity) into a stacked timeline of overlapping protection.
The effective LOE date is a probability-weighted estimate, not a single date. It requires mapping all active patent and regulatory exclusivities, assessing each patent’s litigation vulnerability, and modeling the expected outcome of anticipated Paragraph IV challenges.
Orphan drug exclusivity is underutilized for drugs treating conditions that can be defined narrowly enough to qualify. The seven-year absolute exclusivity bar is more durable protection than most secondary patents in a thicket.
Pediatric exclusivity is the highest-return, lowest-effort extension strategy for high-revenue products. Six months of peak revenue for the cost of a pediatric clinical program is almost always a favorable investment, regardless of pediatric clinical outcome.
Section 5: Global Patent Strategy and PCT Filing Mechanics
Why a U.S.-Only Patent Strategy Is a Liability
The United States accounts for roughly 40 to 45 percent of global pharmaceutical revenue. A company protecting its drug only in the United States exposes roughly half its global commercial opportunity to immediate generic competition in every other jurisdiction from day one. The decision of where to seek protection outside the United States involves four considerations: market revenue potential (which countries represent the largest commercial opportunities for the specific drug), patent enforcement reliability (countries with functioning judicial systems that enforce patent rights reliably), manufacturing risk (whether the API could be manufactured in a country without patent protection and exported to a market where the patent exists), and competitor activity (where key competitors are based and likely to operate).
For most major therapeutic categories — oncology, immunology, metabolic disease, cardiovascular — the priority markets beyond the United States are the EU5 countries (Germany, France, Italy, Spain, and the United Kingdom), Japan, and China, followed by Canada, Australia, South Korea, Brazil, and a handful of other significant markets. Coverage in those twelve to fifteen markets accounts for approximately 85 percent of the global commercial revenue opportunity for most indications. Filing in all 158 PCT member states would be neither cost-effective nor strategically necessary; the objective is to align the patent filing map with the commercial commercialization plan.
PCT Filing: The Strategic Delay as a Capital Allocation Decision
The Patent Cooperation Treaty, administered by WIPO, creates a single-application pathway preserving the right to seek patent protection in any of its 158 member states for up to 30 to 31 months from the original priority date. The PCT system does not grant international patents — patents remain national rights granted by national offices. What the PCT provides is a centralized examination phase and a delay of up to 18 months beyond the 12-month Paris Convention deadline for national phase entry.
At 12 months from priority filing, a drug candidate is typically somewhere in Phase I clinical testing. The clinical data package is thin, the commercial market size estimates are speculative, and the probability of eventual approval is uncertain. Committing to national phase filings in the EPO, Japan, China, Canada, Australia, and Brazil at this point means paying potentially $200,000 to $500,000 or more in aggregate expenditure for an asset that may not survive Phase II. At 30 months from priority date, Phase I is complete. Early Phase II data may be available. The company has a much better basis for determining which markets are commercially relevant and whether the asset has survived the attrition that eliminates most drug candidates before significant external investment is justified. The PCT allows a company to make that more data-informed national phase entry decision with the same legal standing as if it had filed national applications at 12 months.
The PCT also provides an 18-month window during which an International Searching Authority (ISA) examines the application and issues an International Search Report (ISR) and a Written Opinion on the patentability of the claims. The Written Opinion is not binding — it does not grant or deny the patent — but it provides an early independent assessment of the claims against the prior art, identifying objections on grounds of novelty, non-obviousness, or written description. Applicants can file a response and request International Preliminary Examination, resulting in an International Preliminary Report on Patentability (IPRP). A favorable IPRP can accelerate examination in national offices and reduce prosecution costs. The ISR and Written Opinion allow the applicant to identify and address claim weaknesses before entering the national phase, when prosecution costs per jurisdiction are much higher and narrowing claims has more serious commercial consequences.
National Phase Entry: Building the International Portfolio
The European Patent Office (EPO) provides a regional filing pathway that, once a European patent is granted, can be validated in up to 44 EPC member states through a single validation process — dramatically reducing the cost of pan-European coverage compared to filing separately in each country. A practical priority framework for a drug targeting a broad autoimmune or oncology indication would include: EPO filing (covering the EU5 plus other European markets), the USPTO if not the priority jurisdiction, Japan, China, Canada, and possibly Brazil, Australia, India, South Korea, and other markets depending on the drug’s specific commercial profile. The total cost of this portfolio through prosecution and initial maintenance fees runs to several million dollars over the patent term — a meaningful but manageable cost against the drug’s potential revenue.
Key Takeaways — Section 5
A U.S.-only patent strategy forfeits coverage for more than half of global pharmaceutical revenue. The cost of PCT filing plus selective national phase entry is orders of magnitude smaller than the revenue at risk.
The PCT 18-month delay is a data-driven capital allocation decision. Companies should model what clinical and commercial data will be available at 30 months versus 12 months and price the delay accordingly.
The EPO regional patent is the most cost-efficient way to secure broad European protection. A single EPO prosecution produces a patent validatable across 44 jurisdictions.
The PCT Written Opinion provides early, no-additional-cost feedback on claim patentability. Companies that use it to refine claims before national phase entry enter prosecution with a stronger position and reduce the likelihood of costly prosecution delays in individual national offices.
Section 6: Competitive Intelligence, Freedom to Operate, and Litigation Risk
Reading Competitors Through Their Patent Filings
Patent applications are public documents, published 18 months after filing. Because drug development requires early filing to establish priority dates, those published applications reveal a competitor’s R&D program typically two to four years before any clinical data is disclosed, press release issued, or ClinicalTrials.gov entry posted. A systematic program of competitor patent monitoring is one of the most cost-effective intelligence tools available to a pharmaceutical R&D organization.
The most useful intelligence applications of competitor patent monitoring include: identifying new therapeutic targets or compound classes being explored by a competitor before any public disclosure; detecting a competitor’s entry into a new therapeutic area; tracking the evolution of a competitor’s formulation strategy for a drug approaching LOE; and identifying changes in a competitor’s manufacturing process that might indicate a product reformulation or production scale-up. Platforms such as DrugPatentWatch, Derwent Innovation, PatBase, and Orbit Intelligence aggregate pharmaceutical patent data with structured search and alerting tools that automate the monitoring process. A competitor filing 40 new patent applications in a twelve-month period for a drug approaching LOE is signaling an aggressive lifecycle management program — a signal worth tracking months or years before any public product announcement. Conversely, a competitor with declining patent application volume in a previously active area may be deprioritizing that program, creating a potential competitive opening.
Freedom to Operate: From Legal Clearance to Strategic Roadmap
Freedom to Operate (FTO) analysis determines whether a planned product or process can be developed, manufactured, and commercialized without infringing valid, in-force patent rights of third parties. It is not a single check at pre-launch but an iterative process conducted at multiple development points, with the scope and depth scaled to the stage of development and the magnitude of capital at risk.
The FTO process involves four sequential components. The scope definition phase establishes exactly what product features will be searched: the API’s structural identity, the intended formulation, the manufacturing process, the target indications, and the delivery system. Without a precise scope definition, the search will be either too broad to be actionable or too narrow to catch blocking patents in adjacent claim spaces. The search phase covers issued patents in the USPTO, EPO, and other target jurisdictions, along with pending published applications. The analysis phase requires qualified patent counsel to interpret the scope of identified claims relative to the product features. The action phase translates the risk assessment into strategic options: proceed, design around, seek a license, or challenge the blocking patent.
The most analytically valuable FTO reports treat the exercise as a strategic input rather than a compliance checkbox. An FTO analysis that concludes ‘high infringement risk due to Competitor X’s HPMC matrix tablet patent’ is a legal opinion. An FTO analysis that continues ‘however, no patents in the search cover osmotic delivery formulations for this compound class, and two Phase II trials in the literature have used osmotic delivery without adverse PK effects, suggesting this is a viable design-around path that also offers a differentiated once-daily release profile’ is a strategic asset. The data from a defensive FTO analysis should feed directly into the R&D design-around effort, the commercial differentiation strategy, and the next patent landscape analysis cycle.
Paragraph IV Certification and the 30-Month Stay
When a generic company files an Abbreviated New Drug Application (ANDA) for a drug with patents listed in the FDA’s Orange Book, it must make one of four patent certifications. A Paragraph IV certification is a declaration that the listed patent is invalid, unenforceable, or will not be infringed by the generic. It constitutes a statutory act of patent infringement under 35 U.S.C. section 271(e)(2), even though no product has been marketed. This triggers the brand company’s right to file a patent infringement suit within 45 days. If the brand company files suit within that window, the FDA is automatically stayed from approving the ANDA for 30 months — giving the brand company time to litigate before generic entry regardless of how strong the generic company’s case is. The 30-month stay creates an asymmetric procedural advantage for brand companies: filing suit is sufficient to trigger it, regardless of the ultimate merits of the litigation. The first generic company to file a successful Paragraph IV certification receives 180 days of generic market exclusivity, making the first-filer competition among generic companies intense and expensive.
PTAB Proceedings: IPR as a Challenger’s Primary Tool
The America Invents Act created Inter Partes Review (IPR) and Post-Grant Review (PGR) at the PTAB — trial-like post-grant proceedings that have become central to pharmaceutical patent strategy for both brand and generic companies. IPR allows a third party to challenge an issued patent’s validity based on prior art patents or printed publications, through a proceeding substantially faster and less expensive than district court litigation. IPR institution rates in pharmaceutical cases have historically been high, and final written decisions have invalidated a substantial fraction of challenged pharmaceutical patent claims. For brand companies, the threat of IPR creates a new dimension to lifecycle management strategy: secondary patents that would survive Paragraph IV litigation in district court under the clear and convincing evidence standard may not survive IPR review under the preponderance of evidence standard. Patent thicket strategy must account for the probability of IPR petition when assessing the durability of each patent layer.
Key Takeaways — Section 6
Competitor patent monitoring is one of the highest-return intelligence activities in pharmaceutical R&D. Published patent applications provide advance intelligence on competitor programs two to four years before public disclosure.
FTO analysis converts patent landscape data into strategic R&D guidance. Organizations that use FTO reports only for legal clearance miss the design-around and white-space opportunities that the same data reveals.
The 30-month stay creates asymmetric procedural advantage for brand companies. Filing suit is sufficient to trigger the stay regardless of the merits of the litigation.
IPR has lowered the durability of weak secondary patents, particularly polymorph and formulation patents without strong non-obviousness foundations. Patent thicket strategy must now account for the probability of IPR petition at each patent layer.
Section 7: Navigating the Patent Cliff
The Revenue Collapse: Understanding Loss of Exclusivity Dynamics
The pharmaceutical patent cliff is the rapid, sustained revenue decline that follows generic market entry. In the United States, with its highly competitive generic manufacturing sector, aggressive PBM formulary management, and mandatory generic substitution laws in most states, the revenue collapse following first generic entry is typically 60 to 80 percent within the first year and 85 to 95 percent within two years. The mechanism is formulary-level, not patient-level: once generic versions are available, PBMs remove the branded product from preferred formulary tiers and impose step therapy requirements or quantity limits. Patients move to generics through payer-driven substitution, not primarily through individual prescriber choice.
Pfizer’s experience with Lipitor (atorvastatin) following its November 2011 U.S. patent expiry illustrates the velocity. Atorvastatin had been the best-selling drug in the world with annual U.S. revenues exceeding $10 billion at peak. Within the first year of generic entry, Lipitor’s U.S. market share by prescription volume fell below 20 percent, and branded revenues declined by more than 70 percent. Looking ahead from March 2026, IQVIA analyses identify drugs with combined revenues of more than $300 billion at risk of losing exclusivity within the current decade, with 190 drugs including 69 blockbusters facing their own patent cliffs. Companies whose portfolio construction depends disproportionately on drugs approaching LOE without a next-generation pipeline asset in late-stage development are structurally exposed.
Pre-LOE Strategy: Maximizing Revenue Before the Fall
The 24 to 36 months before an expected LOE date are the period of maximum strategic leverage for a brand company. Price management in the pre-LOE period requires balancing two competing objectives. Gross price increases in the final years of exclusivity extract maximum net revenue per unit. But historical analysis suggests that price increases above approximately 9 percent net annualized trigger payer backlash that can accelerate formulary tier changes and reduce volume above the price increase, producing negative net revenue impact. The optimal pre-LOE pricing strategy requires modeling the price elasticity of the specific payer mix and the threshold above which volume losses outweigh price gains.
Rebate contract management in the pre-LOE period involves a deliberate phase-down. As LOE approaches, payers anticipate the switch to cheaper generics, reducing their leverage in formulary negotiations. Brand companies can begin reducing rebate commitments in the 12 to 18 months before LOE without proportionate loss of formulary access, improving net revenue per unit in the final exclusivity period. The commercial team should simultaneously be positioning the next-generation product — the extended-release formulation, combination product, or chiral switch — for formulary negotiations that establish it in preferred position before generic entry on the original product triggers payer re-evaluation.
Post-LOE Playbook: Authorized Generics, Product Hopping, and Patient Programs
An authorized generic is a generic version of a branded drug manufactured by the brand company or a contract partner and distributed under a non-branded label at generic prices. Because it does not require a separate FDA approval process, it can be launched on the first day of generic market entry. Brand companies use authorized generics to capture a share of the generic revenue stream and to compete directly with the first-filer generic in the 180-day exclusivity period, compressing first-filer margins and reducing the commercial value of Paragraph IV litigation. Pfizer’s launch of an authorized generic atorvastatin immediately after Lipitor’s patent expiry is a well-documented execution of this strategy.
Product hopping — transitioning the patient base from an older drug to a new, patent-protected version before the older product loses exclusivity — is the most effective long-term mitigation strategy when executed correctly. When Forest Laboratories introduced Namenda XR (once-daily extended-release memantine) and sought to withdraw the original twice-daily Namenda before generic entry on the IR formulation, the New York Attorney General obtained an injunction blocking the product discontinuation on antitrust grounds. The court found that the product hop was designed to eliminate the possibility of automatic generic substitution under state pharmacy laws rather than to provide genuine clinical benefit. Product hopping strategies must rest on genuine clinical differentiation and be structured to withstand antitrust scrutiny. The Namenda litigation established that restricting supply of the original product to force patient transition will be challenged by state attorneys general and federal courts.
For some drugs with high brand loyalty and patient populations for whom cost-sharing is a significant barrier, patient access programs — co-pay cards, financial assistance programs, and free drug programs for underinsured patients — can help retain a commercially insured patient base after generic entry. Novartis extended its co-pay assistance program for its multiple sclerosis drug Gilenya following its 2022 LOE to slow the erosion of the commercially insured segment. These programs do not affect payer-level formulary decisions, which drive the bulk of generic switch volume, but they can meaningfully slow the erosion of the residual branded segment.
Investment Strategy Note
The patent cliff is not a single-day event in financial modeling. It is a multi-year erosion curve with a shape determined by the competitive structure at LOE. The most adverse scenario for a brand company’s equity valuation is the combination of multiple generic ANDAs approved on LOE day one, no authorized generic strategy, no second-generation follow-on product in the market, and a payer landscape that has already negotiated aggressively in anticipation of generic entry. Companies in that scenario typically see 80 percent revenue declines in year one. Companies with one or more of those elements addressed see materially more gradual erosion curves.
Key Takeaways — Section 7
The patent cliff is a predictable event, typically visible ten or more years in advance. Companies without a follow-on product in late-stage development five years before LOE are too late to avoid a structural revenue discontinuity.
Pre-LOE price increases above approximately 9 percent net annualized have historically reduced total pre-LOE revenue relative to more moderate increases. Surge pricing in the final years of exclusivity is not an automatically profitable strategy.
The authorized generic strategy is the fastest post-LOE execution tactic but also the one that most directly cannibalizes branded revenue. It makes strategic sense for high-volume, low-brand-loyalty products where generic revenue capture outweighs the value of defending a shrinking branded base.
Product hopping requires genuine clinical differentiation and careful antitrust structuring. Supply restriction to force patient transition is not a viable tactic in the current regulatory environment.
Section 8: Case Studies — Humira, Enbrel, and Revlimid
Humira (adalimumab): The 247-Patent Fortress
AbbVie’s IP strategy around adalimumab (Humira) is the most extensively studied example of pharmaceutical patent thicket construction. Humira received FDA approval in December 2002 for rheumatoid arthritis. Its primary composition of matter patent on the adalimumab molecule expired in December 2016 — the date at which biosimilar competition would have begun in a simple patent-expiry model. Instead, biosimilars did not reach the U.S. market until January 2023, a seven-year gap created almost entirely by AbbVie’s secondary patent estate and the litigation and settlement strategy built on top of it.
I-MAK’s analysis of AbbVie’s Humira patent filings found 247 U.S. patent applications, of which 89 percent were filed after the drug received FDA approval. Post-approval patents covered formulation variations (including a high-concentration, citrate-free formulation that reduced injection site pain and was actively used to switch patients from the original version to the new, separately patented one), manufacturing process variations, and method-of-use claims covering each subsequently approved indication — Crohn’s disease, plaque psoriasis, ulcerative colitis, and juvenile idiopathic arthritis. A 2021 U.S. House Committee on Oversight and Reform investigation disclosed internal documents showing AbbVie worked with consultants to develop a plan to ‘broaden’ Humira’s patent estate specifically to minimize biosimilar competition, with the strategy formulated as early as 2010. The investigation found that this deliberate secondary patent accumulation strategy contributed to biosimilar entry delays of seven years in the U.S. compared to Europe, where biosimilars launched in 2018, at an estimated cost of over $14 billion to the U.S. healthcare system in excess drug spending.
AbbVie used its patent portfolio to sue every company that attempted to launch a U.S. biosimilar. Faced with the prospect of years of costly, high-risk litigation over more than 100 patents, every biosimilar competitor eventually settled, accepting a staggered U.S. launch schedule beginning January 2023. The settlement structure — allowing AbbVie to control when, and in what sequence, biosimilars entered the market — illustrates how a patent thicket can be used not to win litigation but to create a negotiating position producing settlements entirely on the patent holder’s terms. The commercial outcome was unambiguous. Seven additional years of U.S. monopoly sales, at peak Humira revenues above $20 billion annually, generated revenue that would not have been available without the thicket strategy.
Strategic Tactic
Description
Impact on Exclusivity
Post-Approval Patent Filings
AbbVie filed over 220 patent applications after Humira’s 2002 FDA approval
Created a dense thicket of secondary patents with expiry dates decades after the molecular patent
Formulation Patents
Patents on a high-concentration, citrate-free formulation reducing injection site pain
New patent barriers; used to switch patients to a newly protected version
Method-of-Use Patents per Indication
Separate patents for each approved indication: Crohn’s, psoriasis, ulcerative colitis, others
Each indication covered by its own patent set, expanding the thicket across disease areas
Aggressive Litigation
AbbVie sued every potential biosimilar entrant for infringement of dozens of patents
Created immense legal cost and uncertainty, delaying market entry for all biosimilar developers
Settlement Agreements
All biosimilar competitors settled, accepting a staggered launch schedule beginning January 2023
Secured AbbVie 7 additional years of U.S. monopoly sales beyond European biosimilar entry
Enbrel (etanercept): The 37-Year Timeline
Amgen’s experience with etanercept (Enbrel) illustrates that longevity in pharmaceutical patent protection can sometimes rest on two or three carefully constructed and well-defended patents rather than a large thicket. Enbrel received FDA approval in November 1998 for rheumatoid arthritis. As of early 2026, the earliest date at which a biosimilar can realistically launch in the United States remains 2029 — 31 years after approval, and 37 years after the earliest relevant patent application was filed.
The key to Enbrel’s extended exclusivity is two patents acquired by Amgen from Roche: U.S. Patent Nos. 8,063,182 and 8,163,522, covering a TNFR:Fc fusion protein and the DNA encoding it. These patents were originally filed in the early 1990s, before a 1995 amendment to U.S. patent law changed the patent term from 17 years from grant to 20 years from filing. Because of a long examination delay at the USPTO, the patents were not issued until 2011 and 2012. Under the pre-1995 law applicable to applications pending at the time of the statutory change, these patents carry terms of 17 years from grant, meaning they do not expire until 2028 and 2029. Novartis’s Sandoz division received FDA approval for its Enbrel biosimilar, Erelzi, in August 2016. Sandoz challenged the two Roche-derived patents on grounds of obviousness-type double patenting. The Federal Circuit ultimately upheld both patents. The Enbrel case demonstrates that a few strategically critical and procedurally resilient patents can deliver more durable exclusivity than dozens of weaker secondary patents.
Revlimid (lenalidomide): The REMS Shield and Volume-Cap Settlement
Celgene (acquired by Bristol Myers Squibb in 2019) negotiated settlement agreements with all major generic ANDA filers for lenalidomide (Revlimid) that allowed generic entry beginning in 2022 but imposed volume restrictions — caps on the quantity of generic lenalidomide each generic company could sell in each calendar year. These volume caps will not be fully lifted until 2026. The volume cap structure was novel in pharmaceutical IP settlement history. By limiting generic quantity rather than delaying generic entry date, Celgene/BMS avoided a straightforward ‘pay for delay’ antitrust challenge while maintaining the effective scarcity of generic supply that preserved branded Revlimid pricing power for years after the nominal generic entry date. The FTC investigated the settlement structure, and the volume cap mechanism has since been cited in legislative discussions about anti-competitive pharmaceutical practices.
Celgene’s separate tactic of using its FDA-mandated REMS program (RevAssist) as a supply barrier also warrants examination. When generic companies attempted to purchase reference product samples to conduct the bioequivalence studies required for ANDA approval, Celgene repeatedly refused to sell them product, citing REMS restrictions. This tactic was the subject of antitrust litigation and a Congressional investigation. Courts have since ruled in multiple cases that brand manufacturers cannot refuse to sell REMS-covered product to ANDA filers for legitimate bioequivalence testing purposes, but the tactic successfully delayed generic development by several years while litigation was ongoing.
Key Takeaways — Section 8
Humira’s thicket strategy succeeded not because any individual secondary patent was strong but because the aggregate litigation cost of challenging 247 patents made settlement the economically rational choice for every biosimilar developer. Volume is a strategic weapon when the opponent’s budget is finite.
Enbrel’s long exclusivity rests on a small number of strategically critical, procedurally durable patents. In biologics, two or three well-structured fusion protein or manufacturing process patents can deliver more durable exclusivity than dozens of weak secondary patents.
Revlimid’s volume cap settlement was a novel mechanism for controlling post-LOE market dynamics that attracted FTC scrutiny. The model may not survive further regulatory or legislative intervention but demonstrates the continued expansion of post-LOE management creativity.
REMS programs cannot be used as supply barriers to deny biosimilar developers access to reference product samples for bioequivalence testing. Courts have established this clearly. The strategy carries substantial legal and reputational risk and is unlikely to remain viable for more than a short delay period.
Section 9: Biologics, Biosimilars, and the BPCIA Patent Dance
The Biologic IP Stack: Why the Thicket Is Denser
Biologics present a fundamentally different IP environment from small molecule drugs. A biologic is a large, complex macromolecule whose three-dimensional structure, post-translational modifications (glycosylation patterns, disulfide bond configurations), and higher-order assemblies are not fully determinable by any current analytical method and are critically sensitive to the specific manufacturing process used to produce them. A monoclonal antibody manufacturing process involves cell line selection and engineering, culture medium composition, bioreactor parameters (pH, dissolved oxygen, temperature, feeding strategy), purification train design (protein A capture, ion exchange, viral inactivation steps), and formulation processing conditions. Each of these elements can be independently patented, and because biosimilar developers must use their own manufacturing processes, process patents create substantial development barriers even after the primary sequence composition patents expire.
The BPCIA’s 12-year data exclusivity provision prevents FDA from approving a biosimilar application referencing the original BLA data for 12 years from the original biologic’s approval date, regardless of the biologic’s patent status. For a biologic approved at year five of its composition of matter patent’s 20-year term, data exclusivity covers the period from year 5 through year 17 — extending effective protection essentially through the full primary patent period. The 12-year BPCIA data exclusivity is therefore the dominant near-term barrier to biosimilar entry for recently approved biologics, and patent strategy for those products is primarily a post-data-exclusivity concern.
The BPCIA Patent Dance: Mechanics and Strategic Options
The BPCIA created a mandatory pre-litigation information exchange process between reference product sponsors and biosimilar applicants, defined in 42 U.S.C. section 262(l) and popularly called the ‘patent dance.’ Within 20 days of FDA accepting the biosimilar application, the applicant must provide the reference product sponsor with its complete aBLA application and manufacturing process information. The sponsor then has 60 days to provide a list of patents it believes could be asserted against the biosimilar. The applicant responds with its position on each listed patent. The two parties then have 15 days to agree on which patents will be litigated in the first wave and which reserved for a second wave after market entry. If they do not agree, the statute provides default rules for the selection. Litigation on first-wave patents must be filed within 30 days of completing the dance.
The Supreme Court’s decision in Sandoz v. Amgen (2017) established that a biosimilar applicant can forgo the patent dance entirely by not providing the aBLA information to the reference product sponsor — in which case the sponsor loses the right to bring a first-wave action but retains the right to sue for infringement after market entry. Biosimilar applicants with a strong invalidity case may prefer the dance to get a first-wave adjudication that clears the path before launch. Applicants that prefer to launch and litigate may forgo the dance to avoid revealing manufacturing process information to the reference product sponsor. The decision is a material strategic choice with significant litigation and commercial consequences, not just a procedural formality.
Biosimilar Interchangeability: The Regulatory Distinction That Drives Market Dynamics
Under the BPCIA, a biosimilar can receive one of two designations: ‘biosimilar’ (highly similar to the reference product with no clinically meaningful differences) or ‘interchangeable’ (meeting the additional standard that the product can be expected to produce the same clinical result in any given patient with no greater risk from switching than from using the reference product without switching). Interchangeability is a higher regulatory bar requiring additional switching studies, typically three-period switching pharmacokinetic studies. Biosimilar interchangeability matters commercially because most U.S. state pharmacy laws permit pharmacists to automatically substitute an interchangeable biosimilar for the reference biologic without a new prescription. Biosimilars without interchangeability designation require active prescriber switching — a much slower and more expensive market penetration process. For biosimilar companies seeking to capture significant market share quickly, biosimilar interchangeability designation is a material commercial asset, and the cost of the required switching studies (typically $20 to $50 million) is often justified by the accelerated market access it enables.
Investment Strategy Note
When evaluating biologic franchise assets, the correct effective LOE date model requires mapping both the patent estate and the 12-year BPCIA data exclusivity independently, then identifying which provides longer protection. For most major biologics approved in the 2010s, data exclusivity either equals or exceeds the remaining primary patent term, meaning data exclusivity — not patents — is the primary near-term barrier to biosimilar entry. Secondary patents in the biologic’s estate only become the dominant exclusivity mechanism once the data exclusivity barrier clears.
Key Takeaways — Section 9
The biologic IP environment produces denser and more durable patent thickets than small molecules because the manufacturing process, cell line, formulation, and delivery system each generate independent patentable subject matter in addition to the sequence composition itself.
BPCIA’s 12-year data exclusivity is the dominant near-term barrier to biosimilar entry for recently approved biologics. Patent strategy for biologics is primarily a post-data-exclusivity concern.
Biosimilar interchangeability designation drives market penetration rates substantially faster than those achievable through prescription-level switching alone. The cost of switching studies is typically justified by the commercial acceleration for high-volume products.
The BPCIA patent dance is a strategic negotiation. The decision to participate, which patents to include in the first wave, and whether to use the Sandoz forfeiture option are all material choices with significant litigation and commercial consequences.
Section 10: AI-Discovered Drugs and Personalized Medicine — The New IP Frontier
Who Owns the AI Invention? Inventorship Under USPTO 2024 Guidance
AI platforms are now capable of generating novel candidate molecules, identifying new drug targets through protein structure prediction, and proposing synthetic routes for complex APIs — in some cases with speed and accuracy that compress early-stage discovery timelines from years to months. Insilico Medicine’s development of INS018_055 for idiopathic pulmonary fibrosis, with a preclinical phase completed in approximately 18 months using AI-driven discovery, represents a documented case of AI-assisted drug development reaching clinical testing. That timeline matters because the traditional preclinical phase for a small molecule typically runs five to six years. If AI-assisted discovery becomes the industry standard, the implications for patent filing timing, claim scope strategy, and the effective patent term calculation change substantially.
U.S. patent law requires that every named inventor be a natural person who made a ‘significant contribution’ to the conception of at least one claim in the patent. In the 2022 case Thaler v. Vidal, the Federal Circuit affirmed that an AI system cannot be named as an inventor. The Supreme Court declined to hear the case. An AI-designed drug molecule, if the AI’s contribution to its conception was not accompanied by a sufficiently significant human contribution, faces the risk of having no valid inventor — which means the patent application cannot be granted.
The USPTO’s February 2024 guidance on AI-assisted inventions clarifies that AI-assisted inventions are patentable, provided that a human inventor made a ‘significant contribution’ to the conception of each claimed element. Contributions that may qualify include: designing and training the AI for a specific problem domain; curating the training dataset; selecting a candidate from among the AI’s outputs based on criteria requiring specialized scientific judgment; and experimentally validating or structurally modifying the AI’s output in ways requiring inventive skill beyond routine testing. Contributions that likely do not qualify include: operating an AI tool according to standard instructions, accepting its output without further analysis, and recognizing that the AI output might have commercial value without applying additional scientific judgment. The practical implication is documentation. The inventorship determination and the supporting documentation for ‘significant human contribution’ must be prepared contemporaneously with the discovery work, not reconstructed at filing.
The Obviousness Problem: AI Augments the PHOSITA Standard
Beyond inventorship, AI creates a subtler challenge to the non-obviousness requirement. The non-obviousness standard is assessed relative to a person having ordinary skill in the art (PHOSITA) at the time of the invention. As generative AI tools become widely available, the capabilities of this hypothetical PHOSITA are effectively augmented — a skilled medicinal chemist with access to a state-of-the-art generative chemistry AI can explore chemical space and identify active scaffolds far more efficiently than a chemist without such tools. The practical consequence is that the category of ‘obvious’ innovations will expand as AI tools become more capable and widely adopted. A molecule that a skilled chemist without AI would not have been expected to synthesize and test might be obvious for a skilled chemist operating with an AI platform specifically trained on the relevant compound class.
The strategic response is twofold. File applications on AI-assisted discoveries quickly, before competitors with similar AI tools can reduce them to practice and file first. Build patent specifications that go beyond the AI’s output — including experimental data, mechanistic insights, or structural modifications that could not have been generated by the AI alone and that support a non-obviousness argument based on unexpected results. A specification that says ‘from among the AI’s suggested class, we identified a specific compound with an unexpected pharmacokinetic profile resulting from a metabolic pathway not predicted by the AI’s training data, and we determined the structural basis for that unexpected advantage’ is substantially stronger than one that says the AI suggested a molecule and it was active in vitro.
Personalized Medicine and the Section 101 Minefield
Personalized medicine innovations that rest on natural biological correlations face Section 101 ineligibility under current U.S. law. In Mayo Collaborative Services v. Prometheus Laboratories (2012), the Supreme Court held that a method claim covering the observation that thiopurine drug metabolite levels correlate with therapeutic efficacy or toxicity, and using that correlation to adjust dosing, was not patent-eligible because it amounted to claiming a natural law. The additional steps in the method (administering the drug, measuring metabolite levels) were conventional. In Association for Molecular Pathology v. Myriad Genetics (2013), the Court held that isolated natural DNA sequences were products of nature and not patentable, while finding that synthetic cDNA sequences could be patent-eligible.
Since Mayo and Myriad, companies developing personalized medicine innovations have focused patent claims on practical applications of natural correlations rather than the correlations themselves. A patent on ‘a diagnostic kit comprising probe X and reagent Y for detecting mutation Z in a patient sample, using the following novel hybridization method’ is more likely to survive Section 101 analysis than a patent on ‘a method of treating patients with mutation Z, wherein patients with mutation Z respond to drug W.’ The distinction is between a claim to a natural correlation (not patentable) and a claim to a specific technical implementation of that correlation (potentially patentable if the implementation is itself non-obvious). The Federal Circuit’s Alice/Mayo two-step framework requires that additional claim elements involve a genuine inventive concept — not merely routine or conventional techniques applied to a natural phenomenon.
Key Takeaways — Section 10
AI-assisted drug discovery does not eliminate the need for a human inventor with a ‘significant contribution’ to each claimed element. Documentation of specific human scientific contributions at each step of an AI-assisted discovery program is now a core IP risk management requirement.
The non-obviousness bar for AI-generated molecules will rise as AI tools become routine in the field. Patent specifications must demonstrate unexpected results or non-AI-predictable advantages to survive an obviousness challenge from a PHOSITA operating with state-of-the-art AI tools.
Personalized medicine innovations resting on natural biological correlations face Section 101 ineligibility under Mayo and Myriad. Claim strategy should focus on novel technological implementations of those correlations, not on the correlations themselves.
Third-party AI platform licensing agreements must explicitly address IP ownership of drug candidates generated using the platform. Without a contractual assignment of all output IP to the user company, the platform developer may assert co-inventorship or co-ownership claims over resulting patents.
Conclusion: The Integrated Pharmaceutical Patent Strategy Framework
The pharmaceutical patent system is, at its core, a revenue management system built on a legal infrastructure. The 20-year patent term, the Hatch-Waxman exclusivity toolkit, the BPCIA’s 12-year data exclusivity, the PCT delay mechanism — each is a financial instrument with a specific expected payoff that must be modeled explicitly. Companies that treat them as legal compliance requirements consistently underperform the companies that treat them as the primary levers of revenue and enterprise value.
Five operating principles frame the integrated strategy. The patent estate is a capital asset, not a legal service line. Patent strategy is a continuous process, not a project with a start and end date. The effective LOE date is a probability distribution, not a date, and every financial model that uses a single LOE date is wrong by construction. The data gathered for defensive FTO analysis is equally valuable as offensive competitive intelligence — the same landscape that shows where competitors’ patents block your path also shows where their coverage is absent, and absence means opportunity. The new frontiers of AI-assisted discovery, biologic complexity, and personalized medicine diagnostics require proactive engagement, not passive waiting for settled doctrine.
The pharmaceutical industry is a race against a clock that starts at patent filing and ends at generic entry. The companies that run that race best do not just discover better drugs. They build better IP architecture around those drugs — more durable claims, more strategically timed filings, more complete global coverage, more carefully stacked regulatory exclusivities, and more precise litigation and settlement strategy. That architecture is what converts scientific innovation into sustained enterprise value.
Frequently Asked Questions
What is the single biggest strategic mistake companies make in early-stage patent planning?
Filing the composition of matter patent too early. Companies that file as soon as a novel molecule is identified are unnecessarily sacrificing potentially two to three years of peak-commercial exclusivity. The correct approach is to model the expected value of delay explicitly, accounting for the probability that a competitor files first during the delay period, and to file at the latest point where that probability-adjusted expected value calculation supports filing. In most cases, that point is later than instinct or legal conservatism suggests.
How should a small biotech with limited capital prioritize its international patent filing strategy?
File a U.S. provisional application immediately to secure the priority date, then file a PCT application within 12 months. Use the PCT’s 30-month national phase entry window to defer major international filing costs until Phase II data is available. At 30 months, prioritize national phase entry in the EPO (covering 44 countries via a single prosecution), the USPTO if not the priority jurisdiction, and Japan. Add China, Canada, Australia, South Korea, and Brazil based on specific commercial opportunity assessment. For most drugs targeting major therapeutic categories, EPO plus USPTO plus Japan covers the majority of global pharmaceutical revenue for a combined prosecution cost well within a clinical-stage biotech’s IP budget.
Does evergreening always block access to affordable medicines, or can it produce genuine clinical benefit?
Both outcomes are real, and the key variable is the clinical differentiation of the new product. A chiral switch that isolates a single enantiomer with meaningfully better tolerability, lower dose requirements, and a cleaner drug interaction profile — backed by prospective head-to-head clinical data — produces genuine patient benefit and deserves patent protection. A polymorph patent on a form of an API with no clinically measurable advantage produces no patient benefit and exists primarily as a litigation deterrent. Both are legal under current law. Lifecycle management strategies resting on genuine clinical differentiation are more durable legally, more defensible commercially, and less likely to attract regulatory or antitrust intervention than those that do not.
My company is using a licensed AI platform for drug discovery. How do I structure the IP ownership terms?
This negotiation must happen before any research work begins. The licensing agreement must explicitly assign to your company all right, title, and interest in any patent-eligible inventions generated using the platform, including any patents the platform developer might claim based on contributions by their employees who designed or trained the model. Insist on a representation that the platform developer will not assert co-inventorship claims based on the platform’s role in discovery. Require that the agreement addresses the USPTO’s ‘significant human contribution’ standard and confirms that the platform developer’s activities in developing and training the AI do not constitute inventive contribution to any specific drug candidates generated using the platform. Have IP counsel review both the licensing agreement and the AI platform’s technical documentation before signing.
How do I build a ‘patent-aware’ scientific culture in an R&D organization?
Four mechanisms work in combination. Embed patent attorneys or patent agents into R&D program teams as standing members, not as external consultants called in at the end of a project. Real-time patent awareness requires real-time IP participation in scientific work. Implement a formal invention disclosure process with financial incentives — cash awards for submitted disclosures and additional recognition for patents that are granted or licensed. Scientists who have a direct financial stake in the outcome of the IP process pay more attention to it. Require patent landscape reviews as a mandatory checkpoint before each major project stage gate decision. When the IP landscape analysis is an explicit input into the investment decision for advancing a program, the entire organization learns that patent strategy affects resource allocation. Finally, run quarterly IP training sessions specifically covering what constitutes a public disclosure, how the grace period and first-to-file rules interact, and what information should be withheld from conference presentations and preprints until a patent application is filed.
What does the transition to AI-augmented PHOSITA mean for my existing patent portfolio?
Conduct an IPR vulnerability audit of your secondary patent estate specifically through the lens of AI-augmented obviousness. For each secondary patent — particularly formulation, polymorph, and method-of-use patents filed before the widespread availability of generative AI tools in medicinal chemistry — assess whether the claimed invention would be considered obvious to a PHOSITA now operating with current-generation AI platforms. Patents that would be vulnerable under an AI-augmented obviousness analysis are structurally weaker than they appeared at the time of filing. If they are critical to your effective LOE timeline, consider whether continuation applications or new divisional filings can add stronger, unexpected-results-based claims to the portfolio before a challenger tests them at PTAB.