Two federal statutes govern most of what happens when a pharmaceutical patent expires in the United States. One dates to 1984, the other to 2010. Between them, the Drug Price Competition and Patent Term Restoration Act (Hatch-Waxman) and the Biologics Price Competition and Innovation Act (BPCIA) define the battlefield on which brand-name drug companies and generic or biosimilar challengers fight for hundreds of billions of dollars in annual revenue.
The statutes share a common philosophy: give innovators a defined window of market exclusivity to recoup R&D investment, then open the door to competition. But the mechanics, durations, litigation procedures, and commercial implications diverge so sharply that treating them as analogous is a strategic error. The five-year exclusivity granted to a new chemical entity under Hatch-Waxman is a different instrument entirely from the twelve years granted to a reference biologic under BPCIA. So is the litigation dance that follows each.
This article breaks down both frameworks in full: the statutory architecture, the patent strategy layered on top of each, the court battles that have defined their scope, the FDA regulatory procedures that enforce them, and what all of this means for investment decisions, generic launch timing, and competitive positioning through the 2030s.
Patent intelligence platforms like DrugPatentWatch have become essential infrastructure for tracking Orange Book listings, ANDA filings, Paragraph IV certifications, and biosimilar patent dance timelines across both frameworks. We reference their data throughout this analysis.
The Statutory Architecture: What Hatch-Waxman and BPCIA Actually Say
What Is the Hatch-Waxman Act and Why Was It Created?
The Drug Price Competition and Patent Term Restoration Act of 1984, universally called Hatch-Waxman after its Senate and House sponsors, solved two problems simultaneously. Brand-name manufacturers had been losing effective patent life to FDA review delays, sometimes burning through five or more years of a twenty-year patent clock waiting for approval. Generic manufacturers, on the other hand, faced a statutory barrier: they could not begin clinical development or regulatory submission until after patent expiration without risking infringement. This created a period of de facto exclusivity that stretched years beyond any stated patent term.
Congress addressed both sides. Brand companies gained patent term extension under 35 U.S.C. § 156, which restores up to five years of patent life consumed by FDA review, subject to a cap of fourteen years of post-approval patent life. Generic companies gained the Abbreviated New Drug Application (ANDA) pathway under 21 U.S.C. § 505(j), which lets them reference the innovator’s safety and efficacy data without repeating clinical trials, provided they demonstrate bioequivalence.
The tradeoff mechanism is the Paragraph IV certification. When a generic applicant believes a listed Orange Book patent is invalid, unenforceable, or not infringed by its product, it files a Paragraph IV certification and notifies the brand. This triggers an automatic 30-month stay of ANDA approval, giving the brand time to litigate. The first ANDA filer with a Paragraph IV certification gets 180 days of generic market exclusivity upon launch, a prize worth fighting for.
What Is the BPCIA and How Does It Differ Structurally?
The Biologics Price Competition and Innovation Act, passed as part of the Affordable Care Act in 2010, created a parallel pathway for biosimilars under 42 U.S.C. § 262. It was modeled loosely on Hatch-Waxman but reflects the structural complexity of biologic products, which are large-molecule drugs manufactured in living cells rather than synthesized chemically.
The BPCIA created the 351(k) biosimilar application pathway and established two forms of market protection for reference product sponsors. The first is a twelve-year period of reference product exclusivity (RPE), during which no biosimilar application can be approved. The second is a four-year data exclusivity period, during which no biosimilar application can even be submitted using the reference biologic’s data.
In place of Orange Book patent listings, the BPCIA established the “patent dance,” a formal multi-step information exchange between biosimilar applicants and reference product sponsors designed to identify and resolve patent disputes before launch. The dance is largely optional after the Supreme Court’s 2017 decision in Sandoz Inc. v. Amgen Inc., but it still structures most litigation strategy in the biosimilar space.
Small Molecule vs. Biologic: The Core Legal Distinction That Drives Everything
The threshold question in any exclusivity analysis is whether a product is approved as a drug under the Federal Food, Drug, and Cosmetic Act (FD&C Act) or as a biologic under the Public Health Service Act (PHS Act). This determines which exclusivity framework applies.
Small molecules, defined chemically, are approved under the FD&C Act as NDAs. Biologics, which include proteins, monoclonal antibodies, recombinant enzymes, cell therapies, and gene therapies, are approved under the PHS Act as BLAs. Congress created a carve-out: as of March 23, 2020, the transition date mandated by the BPCIA, most protein-based drugs that had previously been approved as NDAs were deemed to have BLA status. This transition affected drugs like adalimumab (Humira), etanercept (Enbrel), and several insulin products, pulling them into the BPCIA framework even though they had existed for years under the prior regulatory regime.
The practical consequence of this distinction is stark. A new small molecule gets five years of NCE exclusivity after approval. A new biologic gets twelve. A generic challenging the small molecule can file an ANDA after four years and obtain approval after five. A biosimilar developer cannot even submit a 351(k) application until four years after the reference biologic’s approval date, and cannot receive approval until twelve years have elapsed.
The 5-Year NCE Exclusivity Timeline: A Step-by-Step Breakdown
What Triggers New Chemical Entity Exclusivity Under Hatch-Waxman?
New chemical entity (NCE) exclusivity is triggered when FDA approves an NDA for a drug containing an active moiety that has never previously been approved. The term “active moiety” is defined in 21 C.F.R. § 314.108 as the molecule or ion responsible for the drug’s physiological or pharmacological action, excluding appended portions that form salts, esters, or other noncovalent derivatives. A new salt of an existing drug does not generate NCE exclusivity. A new stereoisomer may, depending on whether FDA has previously approved the parent racemate.
NCE exclusivity runs for five years from the NDA approval date. During this window, FDA will not accept an ANDA that references the NCE. There is one exception: if a generic applicant files a Paragraph IV certification challenging an Orange Book patent, FDA will accept the ANDA after four years, not five. This four-year filing date sets up the 30-month stay, which if it runs its full course, still prevents approval until roughly 7.5 years after NDA approval. Aggressive generic filers bank on settlement or early litigation resolution to get in before the stay expires.
The Orange Book: What Gets Listed and Why It Matters for Generic Entry
The Orange Book, formally titled “Approved Drug Products with Therapeutic Equivalence Evaluations,” is FDA’s database of approved drug products and their associated patents and exclusivities. When an NDA sponsor submits a patent for listing, FDA does not verify the patent’s validity or relevance; it lists the patent as submitted, provided it meets the statutory criteria for patent types that may be listed (drug substance patents, drug product patents, and method-of-use patents).
Orange Book listings are commercially critical because listed patents generate the 30-month stay upon Paragraph IV certification. Non-listed patents do not. This creates an incentive to list as many patents as possible, and brand companies have become sophisticated at constructing patent portfolios specifically designed to generate multiple stays across overlapping ANDA filers.
FDA amended its Orange Book listing regulations in 2023 following court challenges by the FTC, which had argued that brand companies were improperly listing device patents (such as inhaler components) as drug patents to generate stays against generics. The revised regulations tightened the definition of listable patents, though litigation over specific listings continues.
Resources like DrugPatentWatch aggregate Orange Book data with ANDA filing histories, Paragraph IV notifications, and court docket information, letting analysts track the competitive landscape around any brand product in real time. This kind of integrated view is essential for forecasting LOE dates and planning generic entry strategy.
How the 30-Month Stay Works and When It Expires
When a brand company receives a Paragraph IV certification notice, it has 45 days to file an infringement lawsuit. If it does, FDA automatically stays ANDA approval for 30 months from the date the brand received the certification notice, or until the court issues a final judgment of invalidity or non-infringement, whichever comes first.
There is one stay per patent per ANDA filer. With multiple patents listed in the Orange Book, a brand can potentially generate overlapping or sequential stays. The Medicare Modernization Act of 2003 limited brands to one 30-month stay per ANDA, regardless of how many patents are challenged, but subsequent patent listings can still generate new stays for newly filed ANDAs.
The 30-month stay clock begins ticking from certification notice, not from the lawsuit filing date. If the brand is slow to receive notice, or if counsel delays formal receipt, this can compress the available litigation period. District court schedules, particularly in the District of Delaware and the District of New Jersey, the two most common venues for Hatch-Waxman litigation, typically produce trial dates within two to three years of filing, meaning many cases resolve before the stay expires.
What Happens at NCE Exclusivity Expiration: The Generic Entry Cascade
When NCE exclusivity expires and patent litigation resolves or is absent, the generic entry dynamic is well-documented. The first filer with 180-day exclusivity launches, typically pricing at 80 to 85 percent of brand price. Within six months, a second wave of generics enters at lower prices. Within twelve months, brand market share in units typically falls to below 20 percent in most therapeutic categories, while generics capture the volume.
Price erosion follows a predictable curve. With two to three generic entrants, prices settle at roughly 60 to 80 percent of brand. With six or more entrants, prices can fall to 20 percent or below. For oral solid dosage forms in large-volume therapeutic categories, generic price erosion can reach 90 percent within three years of loss of exclusivity.
Brand companies have deployed multiple strategies to extend revenue past NCE exclusivity, including authorized generic programs (launching their own generic at the same time as the first filer to split 180-day exclusivity), pay-for-delay settlements (which the Supreme Court scrutinized in FTC v. Actavis, 570 U.S. 136 (2013)), product switching to new formulations with independent exclusivities, and patient loyalty programs tied to branded products.
The 12-Year Reference Product Exclusivity Timeline: A Detailed BPCIA Analysis
How Does 12-Year Biologic Exclusivity Work Under BPCIA?
Reference product exclusivity (RPE) under the BPCIA runs for twelve years from the date the reference biologic was first approved under a BLA. No 351(k) biosimilar application can be approved during this window, and no biosimilar application can even be submitted during the first four years. This creates a hard floor under the biologic’s commercial exclusivity that does not depend on any patent.
The twelve-year period is absolute in the sense that it is not subject to PTAB challenge, patent invalidity, or litigation outcome. Even if every patent on a reference biologic is invalidated, the RPE still runs. This makes BPCIA exclusivity more durable than Hatch-Waxman exclusivity in one respect: NCE exclusivity can be undermined by Paragraph IV challenges to the underlying NDA approval (though this is rare), but RPE cannot be shortened by patent litigation.
The flip side is that RPE does not extend. If a biologic company receives approval for a new indication of its reference product, that approval does not restart the twelve-year clock. It may generate three years of new clinical investigation exclusivity for the new indication, but the RPE runs from the original approval date, not the new use approval.
The 4-Year Submission Bar: Why Biosimilar Development Timelines Are Front-Loaded
The four-year submission bar under BPCIA means biosimilar developers cannot even file a 351(k) application until four years after the reference product’s approval. This effectively means biosimilar developers must compress their entire analytical characterization, clinical pharmacology, and potentially clinical comparative work into a timeline that ends at year four, with submission at year four and approval only possible at year twelve.
In practice, sophisticated biosimilar developers begin their programs well before the four-year bar lifts. Companies like Pfizer (via its Hospira acquisition), Amgen, Samsung Bioepis, Sandoz, Mylan (now Viatris), and Celltrion have built dedicated biosimilar pipelines that run parallel programs against multiple reference products simultaneously. The analytical work, including reverse engineering the reference product’s molecular structure and glycosylation pattern, can take two to three years before any clinical program begins.
FDA’s biosimilar review process also takes twelve to eighteen months for most 351(k) applications, meaning biosimilar developers targeting a twelve-year RPE product must file by roughly month 10 of year eleven to hope for approval at the end of year twelve. The practical effect is that the earliest commercial biosimilar launches typically occur twelve to fourteen months after RPE expiration, not the day after.
What Is the BPCIA Patent Dance and How Does It Affect Launch Timing?
The patent dance is a statutory information exchange process under 42 U.S.C. § 262(l). After a biosimilar application is accepted for review by FDA, the biosimilar applicant must provide the reference product sponsor with a copy of the application and all related manufacturing information within twenty days. The reference product sponsor then has sixty days to identify patents it believes are infringed and would be willing to license. The applicant responds with its non-infringement and invalidity positions. The parties then negotiate a list of patents to litigate in a first wave, with remaining patents reserved for a second wave triggered by a notice of commercial marketing.
The Supreme Court’s 2017 decision in Sandoz Inc. v. Amgen Inc., 582 U.S. 1 (2017), established that the patent dance steps are largely optional from the biosimilar applicant’s perspective. An applicant that declines to engage in the dance cannot be compelled to do so through an injunction; the reference product sponsor’s only remedy is to bring an immediate declaratory judgment action. As a result, many biosimilar applicants now elect to skip parts of the dance, accepting the risk of earlier litigation in exchange for strategic flexibility.
The notice of commercial marketing, a 180-day pre-launch notice the biosimilar applicant must provide under 42 U.S.C. § 262(l)(8), was also addressed in Sandoz. The Court held that the notice can be given before FDA approval, which eliminated a potential six-month delay that brands had argued was mandatory.
Does the BPCIA 12-Year Exclusivity Apply to Interchangeable Biosimilars?
Interchangeable biosimilar designation under BPCIA is a separate status from standard biosimilarity. An interchangeable biosimilar has demonstrated, through additional clinical data, that it can be substituted for the reference product by a pharmacist without physician intervention, analogous to the AB-rated therapeutic equivalence designation under Hatch-Waxman. The first interchangeable biosimilar for any reference product receives one year of interchangeability exclusivity, during which FDA will not designate any other biosimilar for that reference product as interchangeable.
Boehringer Ingelheim’s Cyltezo (adalimumab-adbm) received the first interchangeable designation for a biosimilar to AbbVie’s Humira (adalimumab) in October 2021. When Humira’s patent and exclusivity situation allowed commercial launches beginning in January 2023, Cyltezo carried its interchangeable designation into a competitive market that already included multiple non-interchangeable adalimumab biosimilars. The interchangeability designation proved commercially valuable in payer and pharmacy benefit manager negotiations, though less determinative than initially expected because formulary placement depends more on rebate negotiations than on automatic substitution laws.
Side-by-Side Comparison: Hatch-Waxman vs. BPCIA Exclusivity Mechanics
Hatch-Waxman vs. BPCIA: A Structural Comparison Table
Feature
Hatch-Waxman (Small Molecule)
BPCIA (Biologic)
Statutory basis
21 U.S.C. § 505(j); 35 U.S.C. § 156
42 U.S.C. § 262; 21 U.S.C. § 355(c)
Application pathway
ANDA (Abbreviated New Drug Application)
351(k) biosimilar application
Reference data used
NDA data for brand drug
BLA data for reference biologic
Core exclusivity period
5 years (NCE) or 3 years (new clinical study)
12 years (reference product exclusivity)
Submission bar
4 years after NDA approval (if Paragraph IV)
4 years after BLA approval
Patent listing mechanism
Orange Book
No Orange Book; patent dance exchange
Litigation trigger
Paragraph IV certification notice
Patent dance or notice of commercial marketing
Automatic stay upon litigation
30-month stay on ANDA approval
No automatic stay; PI motion required
First-filer exclusivity
180 days for first Paragraph IV ANDA filer
1 year for first interchangeable biosimilar
Pediatric exclusivity add-on
6 months added to NCE or other exclusivity
6 months added to 12-year RPE
Orphan drug exclusivity
7 years (if designated)
7 years (if designated)
Patent term extension
Up to 5 years; 14-year post-approval cap
Up to 5 years; 14-year post-approval cap
Primary regulatory venue
FDA CDER
FDA CDER or CBER
When Does the 5-Year Clock Start vs. When Does the 12-Year Clock Start?
Both exclusivity periods begin on the approval date of the reference product. For NCE exclusivity, the trigger is FDA approval of an NDA for a drug whose active moiety has never been previously approved. For reference product exclusivity, the trigger is FDA approval of a BLA for a biological product.
One practical difference: the twelve-year RPE clock can also run from the date of a deemed BLA approval for products that transitioned from NDA to BLA status on March 23, 2020. FDA published guidance addressing transition products, and the twelve-year clock for those products began on their original BLA approval date, not the transition date. For insulin products like Eli Lilly’s Humalog (insulin lispro), which transitioned from NDA to BLA, this meant the twelve-year RPE was calculated from the original 1996 approval date, meaning it had already expired by the time of transition.
Can Exclusivity Periods Overlap With Patent Protection?
Exclusivity and patent protection are separate legal instruments. A brand drug can simultaneously benefit from NCE exclusivity and multiple Orange Book-listed patents. When NCE exclusivity expires, the patents remain in force. When the last patent expires, NCE exclusivity may have long since ended. The effective commercial protection period is usually the longer of the two, but the interaction is more nuanced.
Consider a brand drug approved in year zero with a composition-of-matter patent expiring in year fifteen. NCE exclusivity runs years zero through five, blocking ANDA submissions entirely. After year five (or year four for Paragraph IV filers), generic applicants can challenge the patent. If they succeed, they enter before year fifteen despite the patent. If they fail, they must wait until year fifteen regardless of exclusivity expiration. The exclusivity period thus matters most when the patent clock is shorter than the exclusivity period, which is rare for typical composition-of-matter patents but common for method-of-use or formulation patents.
For biologics, the same logic applies at longer timescales. Amgen’s etanercept (Enbrel) had dozens of patents covering it across its product and process, some extending into the 2030s. Its twelve-year RPE had long expired by the time those patents became the focal point of biosimilar challenges. The patents, not the RPE, were the dominant competitive barrier once RPE elapsed.
NCE Exclusivity in Practice: Real Cases and Launch Timelines
Lipitor (Atorvastatin) and the Pfizer LOE Playbook
Pfizer’s Lipitor (atorvastatin calcium) lost exclusivity in November 2011 after generating approximately $13 billion in annual U.S. sales at peak. The loss-of-exclusivity (LOE) event was one of the largest in pharmaceutical history. Pfizer’s response illustrates the full spectrum of Hatch-Waxman strategy.
Atorvastatin’s NCE exclusivity had expired years before the 2011 LOE date. The commercial protection in the final years came from a cluster of formulation and method-of-use patents, including U.S. Patent No. 4,681,893 (the original composition patent, which expired in 2010 with PTE), and process patents that Pfizer litigated aggressively against ANDA filers. Ranbaxy Laboratories received first-filer status for its Paragraph IV ANDA and launched an authorized generic simultaneously with Pfizer, a strategy Pfizer used to dilute the 180-day exclusivity value and depress prices during Ranbaxy’s exclusivity window.
Within six months of generic entry, Lipitor’s branded unit market share fell from near-total dominance to below 20 percent. The price of generic atorvastatin collapsed to roughly 10 to 15 percent of brand price within two years as twelve or more generic manufacturers entered the market. Pfizer’s revenues from atorvastatin fell from $9.6 billion globally in 2011 to under $2 billion by 2013.
Lyrica (Pregabalin) Patent Cluster Strategy and Extended NCE Defense
Pfizer’s Lyrica (pregabalin) illustrates how patent strategy can extend commercial protection well past NCE exclusivity expiration. Pregabalin received NCE exclusivity upon its 2004 FDA approval. That exclusivity expired in 2009. But Pfizer used a method-of-use patent covering pregabalin’s use in fibromyalgia (U.S. Patent No. 6,197,819) to block generic entry for an additional six years, until 2019 for that specific patent.
Generic manufacturers filed ANDAs seeking carve-out labels that excluded the fibromyalgia indication, relying on the doctrine that a skinny label approved for a different indication does not infringe a method-of-use patent. Pfizer successfully argued that pharmacists would nonetheless use the generic product for fibromyalgia under the skinny label, a theory of induced infringement that yielded mixed results across multiple courts.
The Lyrica litigation eventually reached the Federal Circuit, which in Warner-Lambert Co. v. Apotex Corp., 316 F.3d 1348 (Fed. Cir. 2003), had previously addressed the limits of method-of-use patents against skinny label ANDAs. The ongoing tension between brand companies defending late-cycle patents and generic companies seeking skinny labels continues to generate significant litigation, most recently around GSK’s Coreg (carvedilol), where the Federal Circuit’s GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc. decisions in 2020 and 2021 established a more permissive induced infringement standard that benefited brand companies.
Sovaldi (Sofosbuvir) and the Hepatitis C Patent Estate
Gilead’s Sovaldi (sofosbuvir) and Harvoni (ledipasvir/sofosbuvir) represent the most commercially successful patent estate in small-molecule drug history. Sofosbuvir generated $10.3 billion in U.S. revenues in 2014 alone, its first full year on the market. Gilead’s exclusivity strategy was a model of defensive patent layering.
The core sofosbuvir composition patent, U.S. Patent No. 7,964,580, was listed in the Orange Book and challenged by multiple generic companies including Merck and Gilead’s own licensees. Gilead survived a challenge before the Patent Trial and Appeal Board (PTAB) that attempted to invalidate the ‘580 patent on obviousness grounds. The IPR proceedings, which became a key battleground in pharmaceutical patent strategy after the America Invents Act created PTAB in 2012, tested whether sofosbuvir’s prodrug chemistry was obvious in light of prior art.
PTAB ultimately confirmed the ‘580 patent’s validity in proceedings that cost Gilead significant legal resources. The outcome validated Gilead’s prosecution strategy and preserved what was at that point the most profitable pharmaceutical patent in the world.
“Generic entry within twelve months of patent or exclusivity expiration has been documented in more than 80 percent of small-molecule brand drugs with annual U.S. revenues above $250 million, according to IQVIA data published in 2022, underscoring how reliably the Hatch-Waxman system functions as a commercial cliff.”
BPCIA 12-Year Exclusivity in Practice: Biologics LOE Case Studies
Humira (Adalimumab) Biosimilar Launches: The Most Watched LOE in Biologic History
AbbVie’s Humira (adalimumab) generated over $20 billion in annual global revenues at peak, making it the best-selling drug in history by cumulative revenue. Its U.S. LOE situation became the industry’s most closely watched exclusivity case study because it combined expiring BPCIA exclusivity with one of the most elaborate patent thickets ever constructed.
Adalimumab’s original BLA approval in December 2002 set the twelve-year RPE clock. By 2014, that exclusivity had expired, and FDA had received multiple 351(k) applications. But AbbVie had constructed a portfolio of over 250 patents covering adalimumab, including manufacturing process patents, formulation patents (notably the citrate-free formulation), device patents covering the autoinjector, and method-of-use patents covering its approved indications.
AbbVie entered into settlements with all U.S. biosimilar developers that delayed U.S. launch dates until January 2023, while European biosimilar launches proceeded from 2018 onward. The settlements included license agreements granting the biosimilar manufacturers rights to launch in January 2023, with AbbVie collecting royalties on biosimilar sales. These settlements were structured to comply with the Actavis rule by tying the settlement to the remaining IP term rather than paying biosimilar developers to stay out.
When January 2023 arrived, nine adalimumab biosimilars launched: AbbVie’s own authorized biosimilar Hadlima (through a Samsung Bioepis partnership), Amgen’s Amjevita, Pfizer’s Abrilada, Boehringer Ingelheim’s Cyltezo (as the first interchangeable), Coherus’s Yusimry, Fresenius Kabi’s Idacio, Mylan’s Hadlima, Organon’s Hadlima, and Sandoz’s Hyrimoz. The immediate price competition produced high-concentration and low-concentration product variants at varying rebate rates, complicating formulary decisions for PBMs.
The Humira situation validated a concern regulators and academics had raised since BPCIA passage: that the twelve-year RPE period, combined with aggressive patent strategy and settlement agreements, could effectively extend commercial exclusivity far beyond the statutory window. The FTC has investigated AbbVie’s conduct, and Senator Elizabeth Warren has introduced legislation to limit biologic exclusivity periods.
Remicade (Infliximab) and the First Major U.S. Biosimilar Launch
Janssen’s Remicade (infliximab) faced the first significant U.S. biosimilar competition when Pfizer/Celltrion’s Inflectra (infliximab-dyyb) launched in November 2016. Remicade’s twelve-year RPE had expired in 2010 (based on its 1998 approval), and its key patent, U.S. Patent No. 7,598,083, was challenged in both PTAB IPR proceedings and district court litigation.
The infliximab biosimilar story illustrated an early BPCIA tension: despite RPE expiration and favorable patent outcomes for biosimilar developers, hospital and payer adoption was slow. Remicade held over 95 percent of its U.S. market share through 2017, two years after European biosimilar competition had eroded Janssen’s EU market substantially. The reason was contracting: Janssen’s bundled contracting practices locked in hospital systems under all-or-nothing formulary terms, a practice that triggered antitrust litigation.
Pfizer sued Janssen in the Eastern District of Pennsylvania in 2017, alleging anticompetitive bundling in violation of the Sherman Act. The case, Pfizer Inc. v. Johnson & Johnson, settled in 2018 on undisclosed terms, but prompted Janssen to adjust its contracting practices. By 2019, infliximab biosimilar market penetration began climbing toward 20 percent, still far below European levels but representing genuine commercial progress.
Enbrel (Etanercept) and the Impact of Patent Thickets on Biosimilar Entry
Amgen and Pfizer’s Enbrel (etanercept) presents the starkest example of patent exclusivity extending commercial protection far beyond BPCIA’s twelve-year RPE. Etanercept’s original approval in 1998 set an RPE clock that expired in 2010. Sandoz received FDA approval for its etanercept biosimilar Erelzi in August 2016. But Erelzi had not launched in the U.S. as of early 2024 because Amgen’s patent portfolio covering etanercept extended into the 2030s.
Amgen’s etanercept patents include process patents covering the CHO cell-based manufacturing process, purification process patents, and formulation patents. Sandoz’s attempt to design around these patents resulted in litigation in the Northern District of California that produced settlements constraining launch dates. The effective commercial exclusivity of etanercept in the United States has extended more than twenty-five years beyond the drug’s approval, not because of RPE but because of patent law.
Immunex, the original developer, had filed process patents in the early 1990s that covered methods for producing etanercept in mammalian cell culture. These patents received patent term extensions under Hatch-Waxman’s § 156 provisions and were subsequently acquired by Amgen as part of its Immunex acquisition. The combination of original process patents and later manufacturing improvement patents created a thicket that even well-resourced biosimilar developers struggled to penetrate.
Patent Term Extension: How Brands Add Time to Both Frameworks
How Does Patent Term Extension Work Under 35 U.S.C. § 156?
Patent term extension (PTE) under 35 U.S.C. § 156 compensates brand drug companies for patent life consumed during FDA regulatory review. The extension equals the sum of the regulatory review period (from IND filing to NDA/BLA filing) plus half the clinical investigation period, minus any time the applicant failed to act with due diligence, subject to a maximum extension of five years and a cap of fourteen years of post-approval patent life.
PTE applies to both Hatch-Waxman and BPCIA products. Only one patent per approved product can receive PTE, which means the brand must choose strategically which patent to extend. The standard choice is the composition-of-matter patent with the latest expiration date, as this provides the longest effective commercial protection.
The fourteen-year post-approval cap is a binding constraint for drugs that receive approval late in their patent life. A drug approved in year sixteen of a twenty-year patent with a five-year review period would be entitled to five years of extension but is capped at fourteen years post-approval, meaning the extended patent expires fourteen years after approval rather than at year twenty-five.
Pediatric Exclusivity: The 6-Month Add-On and Its Commercial Value
Pediatric exclusivity under 21 U.S.C. § 505A (Best Pharmaceuticals for Children Act) grants a six-month extension to any existing patent or exclusivity period for a drug product if the sponsor conducts FDA-requested pediatric studies. This extension applies regardless of whether the pediatric studies show clinical benefit, as long as they are conducted per FDA’s Written Request and the data are submitted.
Pediatric exclusivity is notable for applying simultaneously to all listed Orange Book patents and exclusivities for the product. A drug with multiple Orange Book patents, all expiring on different dates, gets six months added to each. For a product with a long patent estate and high annual revenues, the commercial value of pediatric exclusivity can exceed $1 billion. Pfizer has been among the most consistent users of pediatric exclusivity for its portfolio products.
For biologics, pediatric exclusivity under 42 U.S.C. § 262(m) similarly adds six months to the twelve-year RPE, creating a 12.5-year effective exclusivity period for reference products that conduct FDA-requested pediatric studies. Few biologics have actually received this extension because the pediatric study requirements for complex biologics are technically demanding, but as the biologics market matures and FDA pediatric drug development expectations increase, this provision will likely see more use.
Orphan Drug Exclusivity: 7 Years That Can Override Both NCE and RPE
Orphan drug exclusivity under 21 U.S.C. § 360cc grants seven years of market exclusivity for a drug designated to treat a disease affecting fewer than 200,000 Americans. This exclusivity prohibits FDA from approving the same drug for the same orphan indication from a different sponsor during the seven-year period.
Orphan exclusivity operates independently of both Hatch-Waxman NCE exclusivity and BPCIA RPE. A small molecule with NCE exclusivity and orphan exclusivity benefits from both periods running concurrently or, if the orphan indication approval comes after NCE exclusivity begins, from an effective protection period that extends to the later of the two expiration dates. For biologics, orphan exclusivity can add three to seven years beyond RPE expiration for specific orphan indications.
Alexion’s Soliris (eculizumab), approved for paroxysmal nocturnal hemoglobinuria in 2007 and for atypical hemolytic uremic syndrome in 2011, benefited from multiple orphan drug exclusivities. Combined with its twelve-year RPE (running from the 2007 approval) and a strong patent portfolio, Soliris maintained commercial exclusivity in its primary indications into the early 2020s. When biosimilar eculizumab finally launched, the market had already been partially disrupted by Alexion’s own successor product, Ultomiris (ravulizumab), a longer-acting C5 inhibitor that Alexion designed to capture patients switching from Soliris.
Paragraph IV Litigation vs. BPCIA Patent Dance: A Tactical Comparison
Paragraph IV Certification Strategy: How Generic Companies Plan Challenges
A Paragraph IV certification is a legal assertion, filed under penalty of perjury, that an Orange Book patent is invalid, unenforceable, or will not be infringed by the proposed generic product. Filing a Paragraph IV certification is an act of technical patent infringement under 35 U.S.C. § 271(e)(2), which is why it triggers the brand’s right to sue and the 30-month stay.
Generic companies select Paragraph IV challenges based on several factors: the strength of invalidity arguments over the patent (prior art, obviousness, written description, enablement, double patenting), the economics of the 180-day exclusivity prize, the competitive landscape among ANDA filers, and the litigation venue’s historical record on pharmaceutical patents. Mylan, Teva, Apotex, Sun Pharmaceuticals, and Dr. Reddy’s Laboratories have historically been the most active Paragraph IV filers, building legal teams specifically designed for Hatch-Waxman litigation.
The Southern District of New York, the District of Delaware, and the District of New Jersey are the three most common venues for Hatch-Waxman patent litigation. The District of Delaware, where the Court of Chancery and federal district courts have developed deep pharmaceutical patent expertise, handles the largest volume of complex cases.
PTAB IPR Proceedings: How They Changed Pharmaceutical Patent Strategy After AIA
The America Invents Act of 2011 created inter partes review (IPR) at the Patent Trial and Appeal Board as an alternative patent challenge mechanism. IPR allows any party, including generic and biosimilar manufacturers, to challenge patent validity at PTAB using patents and printed publications as prior art. The PTAB proceeding is faster and cheaper than district court litigation, typically resolving in twelve to eighteen months.
IPR has changed pharmaceutical patent strategy substantially. Generic companies now file IPR petitions against Orange Book patents as a supplement to or substitute for district court Paragraph IV litigation. Brands have responded by designing patents with broader claim scopes backed by robust prosecution histories intended to withstand IPR scrutiny. The Federal Circuit’s willingness to affirm PTAB invalidity findings at a high rate has made IPR a credible threat to even well-prosecuted pharmaceutical patents.
The Supreme Court addressed the constitutionality of IPR in Oil States Energy Services v. Greene’s Energy Group, 584 U.S. 325 (2018), upholding it as a valid exercise of Congress’s authority over public rights in patent grants. The decision removed the constitutional cloud over IPR and cemented its role in pharmaceutical patent strategy.
For biologics, IPR has been used extensively against monoclonal antibody patents. Coherus BioSciences petitioned PTAB challenging several of AbbVie’s adalimumab formulation patents before the 2023 Humira biosimilar launches. While those IPR proceedings did not fully resolve before the agreed launch dates under AbbVie’s settlement agreements, they informed the settlement negotiations and established Coherus’s litigation leverage.
Why the BPCIA Patent Dance Produces Different Litigation Dynamics Than Paragraph IV
The most important structural difference between Paragraph IV litigation and BPCIA patent dance litigation is the absence of an automatic stay in the biosimilar context. Under Hatch-Waxman, a patent suit triggers an automatic 30-month delay in ANDA approval. Under BPCIA, there is no equivalent. A reference product sponsor that wants to stop a biosimilar from launching must seek a preliminary injunction from a district court.
Preliminary injunctions in patent cases require the movant to show likelihood of success on the merits, irreparable harm absent an injunction, balance of hardships in the movant’s favor, and that public interest supports an injunction. In pharmaceutical patent cases, courts have historically been willing to grant preliminary injunctions on strong composition patents, but have been reluctant to do so on manufacturing process patents or method-of-use patents where the balance of hardships weighs against blocking a competitive product.
Amgen’s attempt to obtain a preliminary injunction against Sandoz’s filgrastim biosimilar Zarxio in 2015 illustrates the dynamic. Amgen sought to enjoin Zarxio’s launch pending litigation over its Neupogen (filgrastim) patents. The Northern District of California denied the injunction, and Zarxio launched in September 2015 as the first FDA-approved biosimilar in the United States. The Federal Circuit affirmed the denial in part, and Zarxio went on to capture significant market share before the underlying patent litigation resolved.
The Commercial Cliff: Forecasting LOE Impact for Small Molecules vs. Biologics
Why Biologic LOE Events Hit Slower Than Small-Molecule LOE Events
Generic drugs capture market share faster than biosimilars for several structural reasons. First, generics are therapeutically equivalent to their reference products as AB-rated products under the Orange Book, enabling automatic substitution at the pharmacy. Biosimilars, unless interchangeable-designated, require physician prescribing decisions or patient consent to switch, introducing friction that slows market penetration.
Second, biologic manufacturing complexity means biosimilar supply chains are more constrained. A generic oral solid can be manufactured in any number of qualified facilities using standard chemical synthesis. A biosimilar monoclonal antibody requires specialized bioreactor capacity, complex purification processes, and validated comparability programs that take years to establish.
Third, biosimilar pricing discounts are structurally smaller than generic discounts. While oral generic prices can fall to 5 to 10 percent of brand price within three years of LOE, biosimilar prices at launch typically come in at 10 to 30 percent below brand list price. Net price discounts after rebates can be larger, but WAC-based comparisons, which drive formulary decisions for many PBMs, show more modest biosimilar discounts.
IQVIA data from 2023 shows that biosimilar market penetration in the United States averages 35 to 45 percent of units within three years of first biosimilar launch, compared to 80 to 90 percent for small-molecule generics within the same window. The gap is narrowing as payers become more sophisticated and biosimilar prescribing habits mature among physicians, but it remains substantial.
Revenue Erosion Timelines: What the Data Shows for Both Drug Types
Brand revenue erosion after generic entry follows a well-documented pattern for small molecules. In the first year after NCE or final patent expiration with generic entry, brand revenue typically falls 60 to 80 percent in unit terms but only 30 to 40 percent in dollar terms because brand price often increases simultaneously with generic entry, a practice sometimes called the “last dance” pricing strategy.
For biologics, the revenue erosion timeline is slower and more variable. Reference product sponsors have responded to biosimilar competition with a range of strategies: launching authorized biosimilars, reducing list prices for rebate-eligible accounts, introducing next-generation products, and defending market share through long-term supply agreements with health systems.
AbbVie’s revenue experience after Humira biosimilar entry in January 2023 illustrates the biologic pattern. Global Humira revenues fell from $21.2 billion in 2022 to $14.4 billion in 2023, a 32 percent decline. U.S. revenues declined from $17.0 billion to $12.2 billion, while international revenues, already biosimilar-competitive, declined modestly. AbbVie had partially offset this by growing its successor immunology products Skyrizi (risankizumab) and Rinvoq (upadacitinib), which together generated $14.3 billion in 2023 revenues.
What Does LOE Mean for a Drug’s Supply Chain and Manufacturing Strategy?
Loss of exclusivity triggers manufacturing strategy changes on both sides. Brand companies often reduce manufacturing capacity or divest manufacturing assets for LOE products, shifting production to lower-cost contract manufacturers. Generic companies scale up manufacturing capacity in anticipation of first-to-market opportunities, often investing in ANDA-specific manufacturing lines years before expected approval.
For biosimilars, the manufacturing investment is far larger. Building or qualifying bioreactor capacity for a monoclonal antibody biosimilar can cost $200 to $500 million and take five to seven years. Companies like Samsung Bioepis, which operates massive biomanufacturing capacity in South Korea, have built their business models around serving as the manufacturing backbone for multiple biosimilar programs simultaneously.
Supply chain considerations extend to API sourcing for small molecules. When a brand drug loses exclusivity and multiple generic manufacturers enter the market, API prices typically fall as new sources enter the market. This has created quality control challenges documented by FDA’s inspection system: generic API manufactured in cost-competitive facilities has in several cases failed quality standards, generating shortages and recalls that temporarily disrupted the post-LOE market. The metformin nitrosamine contamination issue in 2020, which affected multiple manufacturers and stemmed from a manufacturing process that generated N-nitrosodimethylamine (NDMA) as a byproduct, illustrated the supply chain risks that accompany mass generic entry.
FDA Exclusivity Designations Beyond NCE and RPE: The Complete Map
3-Year New Clinical Investigation Exclusivity: When It Applies and When It Doesn’t
New clinical investigation exclusivity under 21 U.S.C. § 505(c)(3)(E)(iii) protects approved drug products for three years when the approval was based on new clinical investigations essential to the approval. This is distinct from NCE exclusivity: it applies to new formulations, new indications, new routes of administration, or new dosage regimens of already-approved active moieties, not to new chemical entities.
The three-year exclusivity period means FDA will not approve an application for the same condition of use relying on the same new clinical data for three years. Importantly, it does not prevent ANDA submissions for other conditions of use or ANDA submissions with Paragraph IV challenges; it only blocks a specific type of marketing approval. Its commercial value is significant for lifecycle management products like extended-release formulations, combination products, and new indication approvals.
Allergan used three-year exclusivity extensively in its Restasis (cyclosporine ophthalmic emulsion) lifecycle management strategy, obtaining multiple three-year exclusivities for new indications and formulation changes while concurrently litigating the core Orange Book patents. The Restasis situation also generated one of the most controversial patent strategy maneuvers in recent pharmaceutical history: Allergan’s 2017 assignment of Restasis patents to the Saint Regis Mohawk Tribe, which claimed sovereign immunity from PTAB IPR proceedings. The Federal Circuit ultimately rejected tribal sovereign immunity as a defense in PTAB proceedings in Saint Regis Mohawk Tribe v. Mylan Pharmaceuticals Inc., 896 F.3d 1322 (Fed. Cir. 2018).
Breakthrough Therapy, Fast Track, and Accelerated Approval: Do They Create Exclusivity?
FDA’s expedited development and review programs, Breakthrough Therapy Designation, Fast Track Designation, Priority Review, and Accelerated Approval, do not themselves create additional exclusivity periods. What they do is accelerate approval, which can interact with exclusivity in commercially significant ways.
Accelerated Approval under 21 C.F.R. § 601.41 (for biologics) and 21 C.F.R. § 314.500 (for drugs) permits approval based on a surrogate endpoint reasonably likely to predict clinical benefit, with confirmatory trials required post-approval. A drug approved under Accelerated Approval receives the same exclusivity it would receive based on its product type and approval characteristics. The commercial significance is that Accelerated Approval often enables earlier market entry, starting the exclusivity clock sooner while confirmatory data accumulates. If the drug is ultimately withdrawn under Accelerated Approval for failure to confirm clinical benefit, as happened with several oncology drugs after the FDA’s 2021 oncology portfolio review, the exclusivity period is moot.
Breakthrough Therapy Designation specifically accelerates the development program through intensive FDA guidance, which can compress clinical development time. For a biologic with a twelve-year RPE, earlier BLA approval means the twelve-year clock starts earlier, but it also means the biosimilar submission window opens earlier (at year four) and approval occurs earlier (at year twelve). A biologic approved two years earlier through Breakthrough Therapy thus faces biosimilar competition two years earlier in absolute calendar terms, though with two additional years of commercial exclusivity as a tradeoff.
Qualified Infectious Disease Product Exclusivity: The QIDP Add-On
The Generating Antibiotic Incentives Now (GAIN) Act of 2012 created the Qualified Infectious Disease Product (QIDP) designation, which grants an additional five years of exclusivity for antibacterial and antifungal drugs treating serious or life-threatening infections. This add-on applies to whatever exclusivity the drug would otherwise receive: NCE exclusivity becomes ten years, three-year exclusivity becomes eight years, and orphan exclusivity becomes twelve years.
QIDP exclusivity has been granted to several recently approved antibiotics, including Merck’s Sivextro (tedizolid), The Medicines Company’s (acquired by Novartis) Vabomere (meropenem/vaborbactam), and Melinta’s Baxdela (delafloxacin). For drugs in the antibacterial space, where clinical trial economics are challenging and off-patent generic competition is severe, QIDP exclusivity was designed to make development economically viable.
Competitive Generic Therapy Designation: The FDA Program That Accelerates Generic Entry
Competitive Generic Therapy (CGT) designation, created by the FDA Reauthorization Act of 2017, provides expedited review for generic drugs that face inadequate generic competition. A drug with fewer than three approved generic manufacturers qualifies for CGT designation, which triggers priority review for the ANDA and the possibility of a 180-day period of exclusivity for the first CGT applicant.
CGT represents a rare example of FDA policy designed to accelerate generic entry rather than protect brand exclusivity. It has been particularly significant for drugs with manufacturing challenges, specialty drugs, and drugs with limited markets that did not attract multiple generic entrants under the standard Paragraph IV pathway. The program has resulted in meaningful price competition in several therapeutic categories where chronic supply constraints had maintained artificially high prices.
Biosimilar vs. Generic: What Do Payers, Hospitals, and PBMs Actually Do With LOE?
How PBMs Handle Formulary Transitions After Small-Molecule LOE
Pharmacy benefit managers handle small-molecule LOE systematically because the AB-rating system automates substitution. When a generic product receives an AB rating from FDA, indicating therapeutic equivalence to the brand, most state pharmacy practice laws permit or require pharmacists to substitute the generic unless the prescriber writes “dispense as written.” PBM formularies move generics to the lowest cost-sharing tier immediately upon generic launch, generating rapid adoption through economic pressure on patients.
Express Scripts, CVS Caremark, and OptumRx, the three largest U.S. PBMs, have refined their generic transition processes to maximize capture rates. They combine automatic generic substitution policies with prescriber outreach programs for physicians still prescribing branded products after generic availability, and they exclude brand drugs from formulary coverage (or move them to non-preferred tiers with high cost-sharing) shortly after generic entry.
The result is the well-documented generic capture rate: over 90 percent of prescriptions for products with multiple generic entrants are filled with a generic within twelve months of generic launch in most therapeutic categories. Exceptions occur in narrow therapeutic categories where physicians prefer branded products for dosing precision reasons (levothyroxine is the canonical example) or where patient assistance programs and co-pay cards reduce patient out-of-pocket costs to match generic prices.
How Health Systems Manage Biosimilar Adoption and What Slows It Down
Hospital formulary decisions for biologics and biosimilars involve clinical pharmacy committees, medical staff input, contract pricing negotiations, and supply security considerations that collectively make the transition slower and more variable than small-molecule generic transitions. Unlike automatic pharmacy substitution, biosimilar adoption requires active prescribing decisions or formulary-level restrictions that physicians must actively comply with.
Kaiser Permanente, a vertically integrated health system with its own formulary, has achieved biosimilar adoption rates above 80 percent for several biologics, including infliximab and adalimumab, by implementing formulary exclusions for branded products and creating clinical equivalency policies that enable pharmacists to dispense biosimilars for new and transitioning patients. Kaiser’s experience has become a reference case for biosimilar adoption best practices.
Community-based rheumatology and gastroenterology practices, which administer the majority of infliximab and adalimumab doses through buy-and-bill arrangements, face different incentive structures. Under buy-and-bill, physicians purchase biologics at contracted prices and are reimbursed by Medicare or commercial payers at a percentage above average sales price. Because biosimilars and reference products may have different ASPs and rebate structures, physician practice economics can influence prescribing decisions in ways that do not directly align with formulary recommendations.
Pricing Strategy After LOE: Brand Playbook and Biosimilar Response
Reference product sponsors have developed several pricing responses to biosimilar competition. The most common is the split-list strategy: maintain a high list price for payers who rely on rebates for their PBM contracts while offering deep net price discounts through rebate agreements to lock in formulary exclusivity. AbbVie executed this strategy explicitly with Humira after 2023, offering payers a choice between high-list-price/high-rebate Humira and lower-list-price Humira with reduced rebates, creating complexity in formulary management that biosimilar competitors found difficult to match.
Biosimilar developers counter with transparent pricing strategies: lower list prices, lower or no rebates, and value-based contracting models. Coherus BioSciences launched its adalimumab biosimilar Yusimry at $1,000 per carton list price, an approximately 85 percent discount to Humira’s list price, as a direct price competition strategy. The approach has limited adoption in the rebate-driven commercial market but has gained traction in Medicare Part D contexts where the Inflation Reduction Act’s drug price negotiation and out-of-pocket cap provisions alter the rebate economics.
Regulatory and Legislative Risks: What Could Change the Exclusivity Landscape
Proposed BPCIA Reforms: Should Biologic Exclusivity Be Shortened to 7 Years?
Debate over reducing biologic reference product exclusivity from twelve to seven years has been active since at least 2015. The Obama administration’s fiscal year 2016 budget proposed reducing RPE to seven years, citing the large and growing cost of biologic drugs to public payers. Subsequent administrations have made similar proposals.
The pharmaceutical industry, led by PhRMA and the Biotechnology Innovation Organization (BIO), argues that twelve-year RPE is necessary to sustain the investment required for biologic development, which typically exceeds $1 billion per approved product with a development timeline of ten to fifteen years. Proponents of reducing RPE point to the fact that most other OECD countries provide eight or fewer years of data exclusivity for biologics without measurable impact on biologic R&D output in those markets.
The academic literature is mixed. A 2021 study in the Journal of the American Medical Association found that most approved biologics had recouped development costs well within the twelve-year RPE window, often within seven years for blockbuster products. A competing analysis published in Health Affairs argued that average revenue calculations obscure the distribution of returns, where a small number of blockbusters cross-subsidize a larger number of less successful biologics, and that reducing RPE would make the economics of developing the next blockbuster uncertain enough to chill investment.
The Inflation Reduction Act and Its Effect on Biologic Patent Strategy
The Inflation Reduction Act of 2022 (IRA) introduced direct Medicare drug price negotiation, rebate reform, and out-of-pocket cap provisions that are reshaping pharmaceutical commercial strategy. For small molecules, drugs are eligible for negotiation after nine years post-approval. For biologics, the negotiation eligibility period is thirteen years post-BLA approval, one year longer than the twelve-year RPE period.
This thirteen-year threshold was designed to ensure that biologics do not become subject to government price negotiation while still under RPE protection. The effect is to maintain BPCIA’s twelve-year framework intact while adding a post-RPE negotiation risk that brands must incorporate into their pricing and launch strategies.
For brands, the IRA creates an incentive to extract maximum revenue during the pre-negotiation window, which may paradoxically increase list prices in the years before negotiation eligibility. For biosimilar developers, the IRA’s out-of-pocket cap for Medicare Part D beneficiaries reduces the financial benefit of biosimilar price discounts in that segment because patients’ out-of-pocket costs are capped regardless of biosimilar discount levels, reducing patient-level financial motivation to switch.
FTC Enforcement Against Pay-for-Delay Settlements: Post-Actavis Landscape
The Supreme Court’s 2013 decision in FTC v. Actavis, 570 U.S. 136 (2013), held that reverse payment settlements between brand and generic companies, where the brand pays the generic to stay off the market, are subject to antitrust rule-of-reason scrutiny rather than per se legality. The FTC had challenged Solvay Pharmaceuticals’ settlement with Watson and Paddock Laboratories over the testosterone gel AndroGel (testosterone) as anticompetitive.
The post-Actavis landscape has produced a series of significant FTC enforcement actions. The agency has pursued cases against Shire (for amphetamine salts), AbbVie (for AndroGel in the Third Circuit), and K-Pharm (for ADHD medications). Courts applying the rule-of-reason standard have generally required plaintiffs to show that the settlement payment was large enough relative to the reference product’s profits to suggest the brand was paying to avoid competition rather than resolving legitimate patent uncertainty.
For biosimilar settlements, which involve longer exclusivity periods and higher settlement values, the Actavis framework raises similar concerns. The FTC has stated that it applies the same antitrust standards to biosimilar settlement agreements, though the economic analysis is more complex given the longer development timelines, higher capital requirements, and different market structure of the biosimilar industry relative to the generic drug industry.
Patent Strategy Layer: How Brands Build Moats Above the Exclusivity Floor
The Orange Book Patent Thicket: Filing, Listing, and Defending Multiple Patents
Brand pharmaceutical companies construct patent portfolios around their drug products with a view to generating the maximum cumulative exclusivity period through overlapping layers of IP protection. A typical brand strategy around a new small molecule might include: a composition-of-matter patent covering the active moiety (primary defense, typically expiring fifteen to twenty years from first filing); a formulation patent covering a specific dosage form (secondary defense); a method-of-use patent covering specific indications or dosing regimens (tertiary defense, supporting a skinny label defense strategy); and a process patent covering the synthesis or purification process (manufacturing barrier).
Each of these can be listed in the Orange Book if it meets the listing criteria. The Orange Book listing subjects each patent to potential Paragraph IV challenge, but also provides the benefit of a 30-month stay per challenged patent per ANDA filer. Brand companies have used this architecture to construct litigation defense timelines that extend commercial protection substantially beyond NCE exclusivity expiration.
DrugPatentWatch tracks Orange Book patent listings for all marketed drug products, providing patent expiration dates, PTE applications, and Paragraph IV certification histories. For any brand drug approaching LOE, this data provides the critical input for competitive intelligence analysis and generic entry forecasting.
Biologic Patent Thickets: Why Manufacturing and Formulation Patents Are Different
Biologic patent thickets differ from small-molecule patent thickets in several structural ways. First, because the BPCIA does not have an Orange Book equivalent for biologic patents, the thicket is not as directly tied to an automatic stay mechanism. Brand companies must litigate through the patent dance or PI motion route, which requires active court engagement rather than passive stay enforcement.
Second, biologic manufacturing process patents are uniquely valuable because the manufacturing process defines the product. For small molecules, a generic company can often use a different synthesis route and avoid process patents entirely. For biologics, the critical quality attributes of the product, including glycosylation patterns, aggregation levels, and post-translational modifications, are determined in large part by the manufacturing process. A biosimilar using a substantially different process may produce a product with different quality attributes that fails FDA’s comparability standards, creating an indirect barrier that patent protection alone does not fully capture.
Third, the capital barrier to challenging biologic patents through IPR is higher than for small molecules. IPR petitions for complex biological process patents require expert declarations from qualified biochemists and bioprocess engineers that are expensive to prepare and difficult to find given the specialized nature of biologic manufacturing expertise.
Secondary Patents and Lifecycle Management: Do Brand Companies Game the System?
The phrase “gaming the system” is used loosely in public discourse about pharmaceutical patent strategy. The more accurate framing is that brand companies rationally exploit every available IP and regulatory tool to extend the revenue-generating period of their products, and Congress designed those tools to create exactly those incentives. The question is whether the incentives are calibrated correctly.
Critics point to specific practices: listing device patents (autoinjectors, inhalers) as Orange Book drug patents to generate stays; obtaining method-of-use patents on obvious dosing regimens to delay generic skinny labels; and prosecuting continuation patent applications timed to issue just before the NCE exclusivity or major patent expiration, generating new Orange Book entries that trigger new 30-month stays. The FTC’s 2024 study on Orange Book patent listings, which identified hundreds of potentially improper patent listings in inhaler products, is the most recent regulatory intervention targeting these practices.
For biologics, AbbVie’s patent prosecution strategy around adalimumab, which produced over 250 U.S. patents covering the product, process, and delivery system, represents the most aggressive documented example of patent layering in the biologic space. The strategy was successful in delaying U.S. biosimilar competition by roughly five years beyond European competition while RPE had already expired.
Financial Impact Analysis: What Exclusivity Expiration Means for Revenue Modeling
How Analysts Model LOE Risk for Pharmaceutical Companies
Sell-side pharmaceutical analysts model LOE risk as a revenue cliff event, applying generic penetration curves to projected brand revenues to arrive at post-LOE revenue estimates. The standard model applies product-specific generic capture curves based on the drug’s therapeutic category, the number of expected generic entrants, the presence of authorized generic competition, and the timing of first generic launch relative to the expected market date.
For small molecules, LOE modeling is relatively standardized. Generic penetration typically reaches 80 to 90 percent within twelve to eighteen months of first generic entry in standard therapeutic categories. The residual brand revenue, from patients on patient assistance programs, brand-loyal physicians, or specialty pharmacy channels, stabilizes at a low level.
For biologics, LOE modeling is more complex and variable. Analysts typically model biosimilar penetration as a function of interchangeability status, PBM formulary decisions, reference product sponsor contracting strategy, and therapeutic category dynamics. The range of analyst biosimilar penetration estimates for a given product can span from 20 to 60 percent within three years of biosimilar launch, reflecting genuine uncertainty about the pace of adoption.
What Is the Revenue Impact of a Successful Paragraph IV Challenge?
A successful Paragraph IV challenge, where a generic company obtains a court ruling of patent invalidity or non-infringement and launches at-risk or under a consent judgment, can advance generic entry by years. The commercial value of 180-day exclusivity for a successful first filer against a high-revenue brand drug is correspondingly enormous.
Teva’s successful Paragraph IV challenge against Pfizer’s Protonix (pantoprazole) patents resulted in an at-risk launch in 2007 that generated over $1 billion in Teva revenues during the 180-day exclusivity period before Pfizer’s other Orange Book patents were resolved. The Protonix litigation, which involved a complex sequence of patent challenges, authorized generic launches, and damage calculations, ultimately cost Pfizer several hundred million dollars in damages to Teva for improperly blocking the launch.
The calculation of first-filer exclusivity value has become sufficiently precise that generic companies run detailed financial models comparing the expected value of 180-day exclusivity (discounted by litigation risk and settlement probability) against the cost of the Paragraph IV campaign. For drugs with annual U.S. revenues above $500 million and weak Orange Book patents, the net present value of a successful first-filer challenge can exceed $500 million.
Biosimilar ROI: Does the 12-Year Wait Justify the Development Cost?
The economics of biosimilar development are harder to justify than small-molecule generic development because the investment is higher and the return is less certain. A typical small-molecule generic ANDA costs $1 to $5 million to prepare and file. A typical biosimilar 351(k) application requires $100 to $300 million in development costs for analytical characterization, manufacturing process development, clinical pharmacology studies, and potentially clinical comparative effectiveness studies.
The return on that investment depends on the biosimilar’s commercial success, which is far less predictable than generic drug commercial success. Small-molecule generics capturing 80 percent of units generates predictable revenue. Biosimilars capturing 30 to 40 percent of a market with smaller price discounts generate revenues that may not fully recover development costs for less successful products.
Samsung Bioepis has built a business model based on manufacturing biosimilars at scale across a diversified portfolio, spreading the fixed costs of bioreactor capacity across multiple programs and accepting below-average margins on individual products in exchange for portfolio-level returns. Amgen, by contrast, treats biosimilar development as a strategic capability for its own pipeline and as a competitive intelligence investment that informs its understanding of the biologic product class.
Recent Litigation and Regulatory Developments: 2022 to 2024
Key Federal Circuit Decisions That Affect Both Hatch-Waxman and BPCIA Strategy
The Federal Circuit, which has exclusive appellate jurisdiction over patent cases arising from district courts, continues to refine the law that governs Hatch-Waxman and BPCIA litigation. Several recent decisions merit attention.
In Amgen Inc. v. Sanofi, 598 U.S. 594 (2023), the Supreme Court, not the Federal Circuit, unanimously held that Amgen’s claims to a functional class of PCSK9 antibodies defined by their binding region rather than their specific structure were not adequately enabled under 35 U.S.C. § 112. The Court affirmed the Federal Circuit’s judgment invalidating claims that purported to cover the entire class of antibodies capable of binding to specific PCSK9 residues, including antibodies not yet discovered at the time of filing. This decision has significant implications for biologic patent strategy, as many monoclonal antibody patents were drafted using similar functional claiming approaches.
Post-Amgen v. Sanofi, brand biologic companies have been reviewing their antibody patent portfolios for enablement vulnerability, and biosimilar developers have been evaluating whether to file IPR petitions targeting functionally claimed antibody patents on enablement grounds. PTAB has accepted several such petitions, indicating that the enablement standard the Supreme Court articulated will be actively applied in inter partes review proceedings.
In the Hatch-Waxman context, the Federal Circuit’s 2023 decision in Teva Pharmaceuticals USA, Inc. v. Corcept Therapeutics Inc., addressing method-of-use patent protection for mifepristone (Korlym) in Cushing’s syndrome, continued to develop the induced infringement doctrine governing skinny label ANDAs. The court applied a detailed analysis of whether the generic label, read in full, would inevitably induce physicians to prescribe the generic for the patented indication. The ongoing evolution of skinny label law directly affects generic entry timing for dozens of drugs with method-of-use patents expiring after NCE exclusivity.
FTC Orange Book Delisting Actions: What Changed in 2023 and 2024
The FTC took an active role in challenging Orange Book patent listings beginning in 2023. Under Section 21 CFR 314.53(f)(1), any person may request FDA to remove an improperly listed Orange Book patent. The FTC exercised this authority for the first time in September 2023, challenging patent listings for AstraZeneca’s Symbicort (budesonide/formoterol), GlaxoSmithKline’s Advair Diskus (fluticasone/salmeterol), and Boehringer Ingelheim’s Spiriva Handihaler (tiotropium), among others.
The FTC’s theory was that inhaler device patents covering the mechanical autoinjector or powder dispenser do not qualify as Orange Book-listable “drug product” patents because they claim the container closure system rather than the drug substance or drug product itself. If correct, the listed device patents had been generating improper 30-month stays against ANDA filers for years.
FDA’s responses to the FTC’s delisting requests were mixed. For some products, FDA agreed with the FTC and initiated delisting proceedings. For others, FDA found the listed patents to be within the current listing regulations. The litigation over these delisting decisions will likely take years to resolve, but the FTC’s intervention has already changed the cost-benefit calculus for brand companies deciding whether to list device or process patents in the Orange Book.
Stelara (Ustekinumab) Biosimilars: A Current Real-Time BPCIA Case Study
Johnson & Johnson’s Stelara (ustekinumab), approved in 2009 for plaque psoriasis and later for Crohn’s disease and ulcerative colitis, had annual U.S. revenues of approximately $6 billion in 2022. Its twelve-year RPE expired in September 2021 (based on the 2009 approval date, plus four years for the indications that triggered new exclusivity calculations, depending on the formulation).
Multiple companies received FDA approval for ustekinumab biosimilars in 2023 and began negotiating launch agreements with J&J. Amgen’s Wezlana (ustekinumab-auub) was the first FDA-approved ustekinumab biosimilar, approved in October 2023 with an interchangeability designation. Commercial launches began in 2024 under agreements with J&J that included patent licenses.
The Stelara biosimilar situation illustrates post-Humira biosimilar market dynamics. J&J entered settlement agreements with biosimilar developers that included license start dates tied to resolved patent disputes, similar to the AbbVie/Humira model. The settlement structure reflected both parties’ interest in certainty: biosimilar developers wanted a guaranteed launch date; J&J wanted royalty revenue and controlled competitive dynamics. The result was a managed LOE rather than a litigated cliff.
What This Means for Strategic Planning in Pharma IP
What This Means for Small-Molecule Brand Companies Approaching LOE
Brand companies with small-molecule drugs approaching NCE exclusivity expiration face several decision points. First, they must assess the strength of the remaining patent portfolio: which patents are most vulnerable to Paragraph IV challenge, which method-of-use claims can be defended against skinny labels, and which formulation patents offer genuine non-obviousness arguments. Second, they must evaluate lifecycle management options: does a new formulation, indication, or combination product generate independent exclusivity that justifies investment? Third, they must decide on authorized generic strategy: should they launch their own authorized generic to dilute the 180-day exclusivity value for the first ANDA filer, and if so, at what price point?
Tracking ANDA filings and Paragraph IV certifications through resources like DrugPatentWatch gives brand companies early warning of competitive activity. A brand company that sees multiple ANDA filings and Paragraph IV certifications being filed four years into NCE exclusivity knows it faces a contested loss-of-exclusivity and can allocate litigation resources accordingly.
What This Means for Biosimilar Developers Choosing Reference Product Targets
Biosimilar developers choose target reference products based on commercial opportunity (peak sales, patent/exclusivity expiration timeline), technical feasibility (manufacturing complexity, analytical comparability), and competitive landscape (how many other biosimilar programs are under development for the same reference product). All three factors must align for a biosimilar program to generate adequate returns on the development investment.
The reference product exclusivity expiration date is the starting point for any timeline analysis. From RPE expiration, subtract twelve to eighteen months for FDA biosimilar review, subtract two to three years for the clinical program, and subtract two to three more years for analytical characterization and process development: a biosimilar program targeting a reference product approved in 2013 (RPE expiring in 2025) needed to begin analytical work no later than 2018 or 2019 to have a realistic path to a 2025-2026 launch.
The second critical variable is the patent landscape. Using DrugPatentWatch and PTAB filings to map the patent estate around the reference product, biosimilar developers assess which patents are vulnerable to IPR and which must be designed around or licensed. A reference product with a thin patent estate is far more commercially attractive than one with a 250-patent thicket, even if the commercial opportunity is identical.
What This Means for Generic Companies Planning First-Filer Campaigns
First-filer 180-day exclusivity remains one of the most valuable prizes in the generic industry, but competition for first-filer status has intensified as generic companies have become more sophisticated at identifying targets and executing Paragraph IV campaigns simultaneously. Multiple companies often file Paragraph IV certifications on the same day against the same Orange Book patent, sharing first-filer status and splitting the 180-day exclusivity period.
The strategy of filing the earliest possible ANDA with a Paragraph IV certification on the strongest available invalidity ground remains the core playbook. Generic companies that can identify prior art that escaped USPTO examination during brand prosecution, or that can construct a compelling obviousness case based on post-grant developments in the relevant scientific field, are most likely to succeed in challenging composition-of-matter patents.
For process patents and method-of-use patents, the carve-out and design-around strategies require careful label drafting and non-infringement analysis. The post-GlaxoSmithKline v. Teva induced infringement standard creates real risk even for generics that attempt skinny label strategies, and conservative generic companies sometimes wait for patent expiration rather than risk an at-risk launch that could expose them to damages.
International Comparisons: How U.S. Exclusivity Frameworks Stack Up Globally
EU Data Exclusivity vs. U.S. NCE Exclusivity: The 8-Year Rule and Market Protection
The European Union’s pharmaceutical exclusivity framework under Directive 2004/27/EC provides eight years of data exclusivity plus two years of market protection (the “8+2” system) for centrally authorized products, which can extend to eleven years for new therapeutic indications approved within the first eight years. This differs from the U.S. five-year NCE exclusivity in both duration and structure.
Under the EU system, generic applicants can submit their marketing authorization application after eight years of data exclusivity but cannot launch until ten years have elapsed. The two-year marketing protection period thus functions analogously to the U.S. exclusivity period that prevents approval, but it runs after an eight-year data-only exclusivity period rather than concurrently with it. The overall effective exclusivity for a new chemical entity approved in both jurisdictions is typically longer in the EU than the U.S. if no patent protection exists.
For biologics, the EU provides eight years of data exclusivity and ten years of market protection under the European Medicines Agency’s biosimilar guidelines, compared to the U.S.’s twelve-year RPE. This ten-year European exclusivity has not visibly depressed biologic R&D investment in the EU relative to the U.S., a fact that supports arguments for reducing the U.S. twelve-year period.
Japan, Canada, and Emerging Markets: How Non-U.S. Exclusivities Affect Global Patent Strategy
Japan provides eight years of data exclusivity for new drugs (reduced from ten years in regulatory reforms), with separate exclusivity provisions for orphan drugs and drugs receiving priority review. Canadian data protection provides eight years for new active substances and six years for new indications or formulations under the Data Protection Regulations.
Emerging market exclusivities, particularly in Brazil, China, and India, vary widely. China’s regulatory reform under the Amended Drug Administration Law of 2019 introduced a new data exclusivity system providing six years for innovative drugs approved simultaneously in China and abroad. India provides no dedicated pharmaceutical data exclusivity period under its Patents Act, though process patents (India does not recognize product patents on new chemical forms without enhanced efficacy) can protect some pharmaceutical products.
Global patent strategy for a new biologic launched in the U.S. must account for these varying exclusivity periods when making territory-specific launch, licensing, and commercialization decisions. The global intellectual property management of a multiproduct biologics portfolio is a full-time function at major pharmaceutical companies, involving dozens of regulatory counsel, patent attorneys, and commercial strategists coordinating across jurisdictions.
Key Takeaways
The five-year NCE exclusivity under Hatch-Waxman and the twelve-year reference product exclusivity under BPCIA are separate, non-overlapping statutory frameworks tied to product type (small molecule vs. biologic) and regulatory approval pathway (NDA vs. BLA).
Both exclusivity periods run from the date of FDA approval of the reference product, regardless of patent status, and cannot be shortened by patent invalidity findings.
The Hatch-Waxman 30-month stay is an automatic right triggered by a patent lawsuit. The BPCIA provides no equivalent; reference product sponsors must seek preliminary injunctions through courts.
Patent protection layered above exclusivity is often the dominant commercial barrier, particularly for biologics where RPE may expire years before a complex patent portfolio.
The biosimilar market penetration rate is structurally lower and slower than the generic market penetration rate due to the absence of automatic pharmacy substitution, higher manufacturing complexity, and smaller price discounts.
Pediatric exclusivity adds six months to any listed patent or exclusivity for both small molecules and biologics; orphan drug exclusivity provides seven years for qualifying indications in both frameworks.
The IRA’s thirteen-year threshold for biologic Medicare price negotiation eligibility preserves the twelve-year RPE structure while adding a post-exclusivity pricing risk that must enter brand companies’ commercial models.
FTC actions challenging Orange Book patent listings and biosimilar reverse payment settlements are reshaping the cost-benefit calculus of both listing decisions and settlement strategy in 2023 and 2024.
DrugPatentWatch provides the most comprehensive available aggregation of Orange Book data, ANDA filing histories, Paragraph IV certifications, and biosimilar patent dance timelines for competitive intelligence purposes.
The twelve-year BPCIA period is under sustained legislative pressure to be reduced to seven years; the commercial and R&D implications of such a change would reshape the biologics industry more fundamentally than any regulatory development since the BPCIA itself.
Frequently Asked Questions
FAQ 1: Can a biologic drug receive both BPCIA 12-year exclusivity and Hatch-Waxman NCE exclusivity?
No. NCE exclusivity applies only to drugs approved under the FD&C Act’s NDA pathway. Biologics approved under the PHS Act’s BLA pathway receive reference product exclusivity under BPCIA, not NCE exclusivity. Before the March 2020 BLA transition, some protein products were approved as NDAs and could have generated NCE exclusivity, but post-transition those products are governed by BPCIA.
FAQ 2: What happens to NCE exclusivity if FDA approves a new salt form of the same drug?
A new salt of a previously approved active moiety does not receive NCE exclusivity because the active moiety itself was previously approved. The new salt form may receive three-year new clinical study exclusivity if the approval was based on new clinical investigations essential to the approval of the new salt form, but it will not receive a full five-year NCE period.
FAQ 3: How does the first biosimilar interchangeable designation affect competition from subsequent biosimilars?
The first biosimilar to receive interchangeable designation for a given reference product receives one year of interchangeability exclusivity, during which FDA will not designate a second biosimilar as interchangeable for the same reference product for the same conditions. After one year, additional biosimilars can seek and receive interchangeable designation. Interchangeability designation enables automatic pharmacy substitution in states whose laws permit it, which can generate faster adoption rates in those markets.
FAQ 4: Does orphan drug exclusivity prevent FDA from approving a biosimilar for a non-orphan indication of the same drug?
No. Orphan drug exclusivity under 21 U.S.C. § 360cc is indication-specific. It prevents FDA from approving the same drug for the same orphan indication from a different sponsor for seven years. A biosimilar (or generic) seeking approval for a different indication is not blocked by orphan exclusivity covering only the orphan use. The applicant would need to obtain approval for the non-orphan indication through a separate pathway, which may still be subject to BPCIA RPE if the reference product is still within its twelve-year window.
FAQ 5: What is the “patent dance” and can biosimilar applicants opt out of it?
The patent dance is the multi-step information exchange and negotiation process under 42 U.S.C. § 262(l) that begins when a biosimilar applicant provides its application to the reference product sponsor. After the Supreme Court’s 2017 decision in Sandoz v. Amgen, biosimilar applicants can decline to participate in the dance without triggering injunctive remedies, though declination accelerates the timeline for patent litigation as the reference product sponsor can immediately seek a declaratory judgment. Most applicants participate in at least some parts of the dance because it provides strategic information and structures the litigation timeline.
FAQ 6: How does patent term extension interact with NCE exclusivity?
Patent term extension under 35 U.S.C. § 156 and NCE exclusivity under Hatch-Waxman are independent protections that run simultaneously. A drug may have a composition-of-matter patent that, after PTE, expires at year seventeen post-approval, while NCE exclusivity expires at year five. The effective commercial protection period is the latter of the two, here year seventeen, but the five-year NCE exclusivity period provides absolute protection from ANDA submissions (or four years for Paragraph IV filers) regardless of patent status during those first years.
FAQ 7: Can a generic company challenge a patent using IPR while also pursuing a Paragraph IV ANDA?
Yes, and many generic companies do exactly this. Parallel tracks of ANDA/Paragraph IV litigation in district court and IPR petition at PTAB allow generic companies to pursue invalidity on multiple fronts simultaneously. PTAB proceedings are typically faster and cheaper for invalidity challenges based on patents and printed publications. District court litigation can address non-infringement arguments that PTAB cannot and also provides a venue for claim construction that affects both patent and exclusivity analysis. Courts have developed procedures for managing overlapping proceedings, including stays of district court cases pending PTAB resolution.
FAQ 8: What happens to a biologic’s exclusivity if the reference product sponsor files for bankruptcy?
Reference product exclusivity is a statutory right that runs with the approved BLA, not with the company. If a reference product sponsor files for bankruptcy and the BLA is sold or licensed to another entity, the twelve-year RPE continues running from the original approval date. The biosimilar applicant’s 351(k) application remains viable against the new BLA holder. The practical complication is in patent litigation: a bankruptcy sale may separate the BLA from the patent portfolio, creating complex questions about who has standing to enforce which patents against biosimilar applicants.
FAQ 9: Does FDA track and publish biosimilar 351(k) application dates and patent dance timelines?
FDA publishes the Purple Book, which lists all FDA-approved biological products and their biosimilars, including approval dates, interchangeability designations, and reference product exclusivity expiration dates. Unlike the Orange Book, the Purple Book does not list patents. Patent dance information is largely confidential between the parties, though litigation filings in district court cases and PTAB proceedings provide public access to some patent dance communications that have been submitted as evidence.
FAQ 10: What is the practical difference between “biosimilar” and “interchangeable biosimilar” from a market access perspective?
A standard biosimilar requires a new prescription specifically for the biosimilar, or at minimum a prescriber decision to switch a patient. An interchangeable biosimilar can be substituted for the reference product by a pharmacist without prescriber intervention in states that have enacted automatic substitution laws for interchangeable biologics. As of 2024, most states have enacted or are considering such laws, modeled on the Model State Substitution Legislation developed by the National Alliance of State Pharmacy Associations. Interchangeable status is commercially valuable for retail pharmacy channels and increasingly for specialty pharmacy channels, though the difference in adoption rates between interchangeable and non-interchangeable biosimilars has been smaller than originally predicted because formulary decisions by PBMs often override individual substitution decisions.
References
Drug Price Competition and Patent Term Restoration Act of 1984, Pub. L. No. 98-417, 98 Stat. 1585 (codified at 21 U.S.C. § 505(j) and 35 U.S.C. § 156).
Biologics Price Competition and Innovation Act of 2009, Pub. L. No. 111-148, 124 Stat. 119 (codified at 42 U.S.C. § 262).
FTC v. Actavis, Inc., 570 U.S. 136 (2013).
Sandoz Inc. v. Amgen Inc., 582 U.S. 1 (2017).
Amgen Inc. v. Sanofi, 598 U.S. 594 (2023).
Saint Regis Mohawk Tribe v. Mylan Pharmaceuticals Inc., 896 F.3d 1322 (Fed. Cir. 2018).
GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc., 7 F.4th 1320 (Fed. Cir. 2021).
Oil States Energy Services, LLC v. Greene’s Energy Group, LLC, 584 U.S. 325 (2018).
Federal Trade Commission. (2023). FTC Sends Warning Letters on Improper Orange Book Listings. FTC.gov.
IQVIA Institute for Human Data Science. (2022). The Use of Medicines in the U.S. 2022: Usage and Spending Trends and Outlook to 2026. IQVIA.
Feldman, R., & Wang, C. T. (2021). A New Look at Pay-for-Delay: A Study of Pharmaceutical Settlements. UC Hastings Research Paper.
Kesselheim, A. S., & Avorn, J. (2021). New “Esther”–Type Rules for Biologic Exclusivity. JAMA, 325(3), 239-240.
FDA. (2023). Purple Book: Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations. U.S. Food and Drug Administration.
FDA. (2023). Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations (43rd ed.). U.S. Food and Drug Administration.
Grabowski, H., Long, G., Mortimer, R., & Boyo, A. (2014). Recent Trends in Brand-Name and Generic Drug Competition. Journal of Medical Economics, 17(3), 207-214.
Lichtenberg, F. R., & Philipson, T. J. (2002). The Dual Effects of Intellectual Property Regulations: Within- and Between-Patent Competition in the U.S. Pharmaceuticals Industry. Journal of Law and Economics, 45(S2), 643-672.
AbbVie Inc. (2023). Annual Report on Form 10-K for the Year Ended December 31, 2023. SEC EDGAR.
DrugPatentWatch. (2024). Patent and Exclusivity Analysis for Pharmaceutical Products. DrugPatentWatch.com.
America Invents Act, Pub. L. No. 112-29, 125 Stat. 284 (2011) (codified at 35 U.S.C. §§ 311-319).
Inflation Reduction Act of 2022, Pub. L. No. 117-169, 136 Stat. 1818 (codified in scattered sections of 26 U.S.C. and 42 U.S.C.).
Best Pharmaceuticals for Children Act, Pub. L. No. 107-109, 115 Stat. 1408 (2002) (codified at 21 U.S.C. § 505A).
Generating Antibiotic Incentives Now Act, Pub. L. No. 112-144, Title VIII, 126 Stat. 1002 (2012).
Council Directive 2004/27/EC, amending Directive 2001/83/EC on the Community code relating to medicinal products for human use. Official Journal of the European Union, L 136, 34-57 (2004).
Wouters, O. J., McKee, M., & Luyten, J. (2020). Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA, 323(9), 844-853.
Cohen, J. (2021). Biosimilar Uptake in the US: What Are the Barriers and How Can They Be Overcome? Drugs, 81(15), 1673-1680.
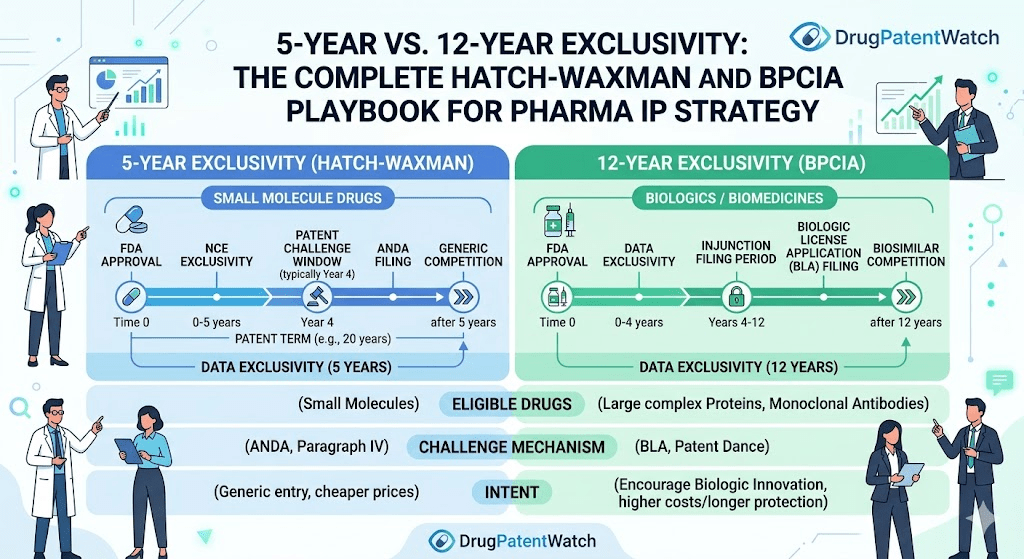













")












