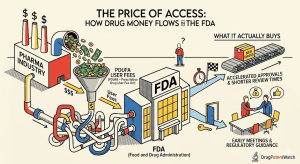
The FDA approves drugs that can kill or cure hundreds of millions of people. It employs roughly 18,000 staff and oversees industries generating more than $500 billion in U.S. annual revenue [1]. The agency runs on a budget of approximately $6.7 billion, of which a substantial and growing share comes directly from the companies it regulates [2]. In the fiscal year 2023, industry user fees accounted for approximately 65 percent of the FDA’s human drug review budget [3].
Read that number again. Nearly two-thirds of the money the FDA uses to review pharmaceutical applications comes from the companies submitting those applications. <blockquote> “Industry user fees funded approximately 65 percent of FDA’s budget for human drug review activities in fiscal year 2023, totaling roughly $1.1 billion out of a $1.7 billion drug center budget.” — FDA Budget and Performance Reports, FY2023 [3] </blockquote>
This is not a secret. It is written into federal law, negotiated openly with industry every five years, and defended publicly by FDA leadership as a sensible mechanism for funding a resource-intensive regulatory function. What is less openly discussed is the documented set of behaviors, institutional incentives, and structural pressures that this funding arrangement creates — pressures that include a revolving door between FDA staff and pharmaceutical employers, advisory committee conflicts of interest, and performance metrics that treat drug approval speed as a primary measure of regulatory success.
This article maps the specific mechanisms through which money moves between pharmaceutical companies and the FDA, assesses the documented evidence for how that money affects agency behavior, examines the real cases where legal lines were actually crossed, and explains what the architecture means for patients, investors, and the pharmaceutical IP landscape that tools like DrugPatentWatch help illuminate.
The Legal Architecture: How Industry Funds the FDA
PDUFA and the Fee Agreement That Transformed Pharmaceutical Regulation
The Prescription Drug User Fee Act, universally abbreviated to PDUFA, was signed into law by President George H.W. Bush in 1992 [4]. It authorized the FDA to collect fees from drug manufacturers at the time of new drug application (NDA) submission, and required that those fees fund additional FDA review staff to eliminate a backlog that had grown to 27 months for new drug applications [5].
The political deal was straightforward: industry got faster reviews, which accelerated revenue from new approvals; the FDA got money to hire reviewers without competing in the congressional appropriations process. The arrangement required periodic reauthorization — each five-year cycle is designated PDUFA I, II, III, and so on through PDUFA VII, the current authorization running through fiscal year 2027.
Each PDUFA reauthorization involves a negotiation between FDA leadership and the industry trade group, the Pharmaceutical Research and Manufacturers of America (PhRMA), that determines the fee schedule and, critically, the performance goals to which the FDA commits in exchange for the fees. These performance goals specify review timelines for standard and priority applications, which the FDA then uses as management targets that shape staffing allocation, review workflow, and organizational priorities.
The current PDUFA VII fee schedule requires applicants to pay a “human drug application fee” of $4,245,608 for a standard application with clinical data [6]. A priority review designation reduces that fee by $2,180,256 (effectively a voucher system). On top of the application fee, companies pay program fees of roughly $436,000 per year for each approved drug in their portfolio.
For large pharmaceutical companies filing multiple applications annually, the annual payment to the FDA runs into tens of millions of dollars. AbbVie, Pfizer, Merck, Johnson & Johnson, and Bristol Myers Squibb together contribute hundreds of millions of dollars annually to the user fee pool.
The Fee Ecosystem: GDUFA, MDUFA, BsUFA
PDUFA was the first of what became an entire ecosystem of industry fee programs:
The Generic Drug User Fee Amendments (GDUFA), first authorized in 2012, apply the same logic to generic drug applications — manufacturers pay fees to fund the review of their abbreviated new drug applications (ANDAs) and the manufacturing facility inspections that generic approvals require [7]. GDUFA III, the current authorization, covers fiscal years 2023-2027 and collected approximately $490 million in FY2023.
The Medical Device User Fee Amendments (MDUFA) fund the FDA’s device review center on the same model. The Biosimilar User Fee Act (BsUFA) funds biosimilar application reviews. Each program involves the same negotiation structure: industry pays, FDA commits to performance metrics, and the cycle repeats every five years.
The cumulative picture is an agency in which the regulated industry is the primary funding source for the regulatory programs that matter most commercially. The FDA’s environmental and food safety programs are funded primarily through congressional appropriations. Its pharmaceutical and device programs run largely on industry money.
What the Fees Technically Fund
FDA leadership consistently emphasizes that user fees pay for reviewer salaries and review infrastructure — the people who read NDA submissions, conduct facility inspections, and evaluate clinical data. Fees are not supposed to fund policy functions, standard-setting, or enforcement activities that are funded through appropriations.
In practice, the boundary between what fees fund and what they do not is less distinct than the official characterization suggests. A reviewer whose salary is paid by PDUFA fees and who also participates in guidance document development is using time that could, at least in theory, be attributed to both functions. The accounting is not fully transparent in FDA budget documents, a point made by the Government Accountability Office in its 2013 review of FDA fee program administration [8].
The Formal Insulation Mechanisms
Statutory Firewalls
Congress designed several formal insulation mechanisms into the user fee framework specifically because of the obvious conflict-of-interest problem. The most important are:
The appropriations adequacy requirement: Under each PDUFA authorization, user fees are supposed to supplement, not replace, congressional appropriations. Congress set minimum appropriations levels below which user fees cannot be used, intended to prevent the scenario in which industry fees become the agency’s primary operating budget. Those minimum floors have not always kept pace with inflation, and they have been subject to congressional budget battles, eroding their practical function.
The prohibition on fee-setting influence: User fees are set through a negotiation process, but the statute prohibits industry from negotiating the terms of specific drug approvals. The negotiation covers aggregate fee schedules and performance metrics, not the fate of any specific application.
Conflict of interest screening: FDA employees who review specific drug applications are subject to federal conflict of interest statutes that prohibit financial relationships with companies whose applications they are reviewing. 18 U.S.C. § 208 prohibits federal employees from participating in matters affecting their financial interests, and FDA has its own supplementary ethics regulations implementing this prohibition [9].
Post-employment restrictions: FDA employees in certain categories face restrictions on working for companies whose applications they reviewed, under 18 U.S.C. § 207, which imposes one-year and two-year “cooling off” periods on specific types of post-government communication [10].
The Advisory Committee System
For major drug applications, the FDA convenes advisory committees — panels of outside experts who review the application and vote on questions like whether the drug’s benefits outweigh its risks. Advisory committee recommendations are not binding, but the FDA follows them in the large majority of cases. For controversial approvals, the advisory committee vote is a critical public accountability mechanism.
Advisory committee members are subject to financial conflict screening and must disclose financial relationships with the sponsoring company and its competitors. The FDA can grant waivers allowing conflicted advisors to participate if their expertise is deemed essential and unavailable in unconflicted form, a waiver authority that has been extensively used and criticized.
The Government Accountability Office found in 2008 that the FDA granted conflict of interest waivers to 42 percent of advisory committee members for selected committees reviewed [11]. A 2009 study in the Journal of the American Medical Association found that a substantial majority of advisory committee members had some form of financial relationship with the pharmaceutical industry, though not always with the specific applicant company [12].
Where the Insulation Breaks Down
The Revolving Door: Industry to FDA to Industry
The revolving door between the FDA and pharmaceutical industry is the most structurally pervasive form of financial relationship between the two institutions. It is legal, widely documented, and works in both directions.
From FDA to industry: FDA physicians, reviewers, and officials with deep expertise in the drug approval process are among the most sought-after hires in the pharmaceutical sector. A senior FDA medical officer who spent fifteen years reviewing oncology applications brings detailed inside knowledge of review standards, common deficiency letter language, advisory committee dynamics, and reviewer-specific concerns — all commercially valuable to a company navigating that same process.
The post-employment restrictions under 18 U.S.C. § 207 impose only a one or two-year cooling off period on specific communications, not on employment itself or on strategic advisory roles that do not involve direct contact with former colleagues. A senior FDA official can, one year after leaving, sit on a pharmaceutical company’s advisory board, attend investor relations events, review regulatory strategy documents, and provide detailed guidance on structuring an NDA to satisfy the review criteria of the exact center they previously led — all without violating the law.
From industry to FDA: Movement in the other direction — from pharmaceutical companies to the FDA — creates a different but equally significant influence pathway. Agency officials who spent careers in industry bring assumptions, frameworks, and prior relationships that may shape their regulatory approaches even without any specific prohibited conduct. The FDA’s Center for Drug Evaluation and Research (CDER) has at various points been led by officials who previously worked in pharmaceutical industry regulatory affairs.
The data on revolving door frequency is striking. A 2016 study published in the BMJ tracked 55 FDA medical officers who reviewed drugs approved between 2001 and 2010 and found that 15 (26 percent) had, within five years of FDA departure, taken employment with the company whose application they had reviewed, or with a company that marketed that exact drug [13]. This is a lower bound — it counts only medical officers, only tracked within five years, and only for direct employers.
A more comprehensive 2018 analysis by Science magazine found that more than 40 percent of FDA reviewers who left the agency went to work for the pharmaceutical industry within two years, with oncology and rare disease reviewers — who oversee the most commercially valuable drug categories — showing the highest rates of industry migration [14].
The Specific Problem of the Accelerated Approval Pathway
The FDA’s accelerated approval pathway, established by regulation in 1992 and codified by the Food and Drug Omnibus Reform Act of 2022, allows drugs treating serious conditions to be approved based on a “surrogate endpoint” — a laboratory measurement or clinical metric that is reasonably likely to predict clinical benefit, even before the clinical benefit itself is demonstrated [15].
The pathway was created with genuine therapeutic intent: allowing cancer drugs to gain approval based on tumor response rates rather than waiting years for survival data lets patients access potentially life-saving drugs sooner. The data on surrogate endpoint reliability is, however, mixed at best. A 2019 study in JAMA Internal Medicine analyzed 38 cancer drugs approved under accelerated approval between 2008 and 2012 and found that only 19 of 54 (35 percent) surrogate endpoints were subsequently validated by evidence of clinical benefit in confirmatory trials [16].
The surrogate endpoint problem matters for the financial influence question because pharmaceutical companies, not the FDA, typically define and propose the surrogate endpoint to be used. They propose it in pre-NDA meetings with FDA reviewers, negotiate its acceptability, and then design their clinical trials to optimize performance on that metric rather than clinical outcomes. The FDA’s review then evaluates whether the company met the threshold for its own proposed endpoint — a circularity that favors industry-preferred measurement frameworks.
This is not illegal. It reflects standard regulatory practice. But it is a structural feature of the review process that hands commercial applicants a significant degree of control over the evidentiary standard they are expected to meet — a control that a more independently resourced and directed FDA might be less inclined to cede.
Opioids: The Regulatory Capture Case Study
The opioid crisis provides the most extensively documented case of what happens when the FDA’s review processes align with pharmaceutical industry interests over public health.
Purdue Pharma received FDA approval for OxyContin in 1995 with a package insert stating that its extended-release formulation was “believed to reduce the abuse liability of the drug” [17]. That claim had no clinical support in the application and was acknowledged by FDA reviewers to be unsupported — but it appeared in the final label anyway. Subsequent documents, revealed in litigation, show that Purdue’s medical affairs team worked directly with FDA staff on labeling language. The claim supported Purdue’s marketing narrative and its pricing, and it was present in OxyContin labels for five years before the FDA required its removal in 2001.
The labeling episode is documented in FDA internal communications and congressional testimony. It does not require inferring conspiracy — it reflects a regulatory review process in which industry representatives have extensive pre-approval communication with reviewers, in which label language is subject to negotiation, and in which the commercial significance of specific claims creates commercial pressure that operates within formally legal channels.
The Curtis Wright case adds a more direct financial dimension. Dr. Curtis Wright, the primary FDA medical officer who approved OxyContin in 1995, left the agency in 1997 and joined Purdue Pharma in a senior regulatory affairs position within two years of the drug’s approval [18]. Wright later testified that he was not involved in OxyContin marketing after joining Purdue. His transition from primary approval officer to company employee is a textbook illustration of the revolving door problem — legal, disclosed, and commercially valuable to all parties involved.
The congressional investigation that followed, including the 2019 Senate Finance Committee report [19], documented a pattern of FDA-Purdue interactions around opioid approvals spanning more than a decade, including approvals of higher-dose formulations, extended-release versions for pediatric patients, and new indications, in a period during which the opioid death toll was already mounting. The investigation concluded that FDA’s opioid approval decisions were influenced by coordinated industry pressure campaigns and that certain FDA officials had professional and post-employment relationships with opioid manufacturers that raised serious conflict of interest questions.
No FDA official was prosecuted for the OxyContin-related decisions. The conduct, while generating devastating public health consequences, operated within the formal rules governing regulatory review. That is precisely what makes the structural analysis more important than a search for individual criminal acts.
The Performance Goals Problem: When Speed Becomes the Standard
How PDUFA Performance Metrics Shape Review Culture
Each PDUFA authorization establishes performance goals specifying the percentage of drug applications the FDA must complete review within defined timelines. Under PDUFA VII, the FDA commits to completing review of 90 percent of standard new molecular entity applications within 12 months of filing and 90 percent of priority applications within 6 months [20].
These timelines are management targets that the FDA takes seriously because PDUFA’s political sustainability depends on demonstrating that industry fees are generating faster reviews. When FDA leadership presents to Congress on PDUFA reauthorization, on-time review completion rates are a central performance measure.
The structural problem is that “completing review” means issuing a decision — approval, complete response letter, or refuse-to-file. The performance metric is agnostic about which type of decision is issued. An FDA reviewer who issues a complete response letter on day 179 of a 180-day priority review clock has technically met the performance goal. But the institutional culture that the performance metric creates is one that treats completions as the primary measure of productivity, which maps more naturally onto approvals than onto refusals.
This is not a claim that FDA reviewers approve drugs they know to be unsafe because of timeline pressure. Most are professionals who would reject that framing. It is an observation that institutional cultures are shaped by the metrics used to evaluate performance, and that a review culture where hitting timelines matters operationally will develop different default behaviors than one where accuracy and rigor are the primary measures — even if both cultures employ the same people.
Statistical Evidence on the Approval Rate Question
The empirical evidence on whether PDUFA has affected approval rates is contested and methodologically complex. Several points are worth noting:
The FDA’s new drug approval rate rose substantially after PDUFA. Between 1986 and 1992 (pre-PDUFA), the FDA approved an average of 22 new molecular entities per year. Between 2000 and 2010, that number averaged 24 per year, with peaks above 40 in recent years [21]. Whether this reflects faster review of the same applications, or a lower evidentiary bar enabling more applications to succeed, is difficult to disentangle from other changes in pharmaceutical R&D productivity.
The drugs approved in the first PDUFA years were subsequently withdrawn from the market at higher rates than drugs approved in the pre-PDUFA period. A 2002 analysis by Carpenter et al. found that drugs approved in the FDA’s faster review cohorts had significantly higher post-market safety withdrawal rates than those approved under the longer pre-PDUFA timelines [22]. This finding is consistent with (though does not prove) a hypothesis that faster review was associated with less complete safety assessment.
A 2014 study in Health Affairs found that drugs receiving priority review under PDUFA had a post-market safety event rate — including black box warning additions and market withdrawals — approximately 1.8 times higher than drugs approved under the standard review pathway [23]. Priority review does not mean unsafe, and many priority-reviewed drugs address serious unmet needs where a higher benefit-risk tolerance is appropriate. But the statistical association between review speed and subsequent safety events is consistent with a hypothesis that timeline pressure affects review thoroughness.
The Approval-Then-Study Problem
Accelerated approval requires companies to conduct post-market confirmatory trials demonstrating clinical benefit after approval. The mechanism presumes that FDA will withdraw approval if confirmatory trials fail or are never completed.
The data on confirmatory trial completion is unflattering. A 2019 study in JAMA Network Open found that among accelerated approvals granted between 2009 and 2013, 20 percent still lacked confirmatory trial results five or more years after approval [24]. The FDA had taken enforcement action in none of those cases at the time of analysis.
The Food and Drug Omnibus Reform Act of 2022 significantly strengthened FDA’s withdrawal authority for accelerated approvals, explicitly authorizing administrative withdrawal procedures that do not require full rulemaking [25]. The 2023 withdrawals of several oncology drugs that failed confirmatory trials — including the Makena (17-hydroxyprogesterone caproate) withdrawal for preterm birth prevention [26] — demonstrated the agency using this authority. Whether this represents durable systemic change or a response to specific political pressure following the Aduhelm controversy remains to be tested over the longer term.
The Aduhelm Debacle: A Case Study in Multiple Failures
No recent FDA approval better illustrates the intersection of financial relationships, scientific controversy, and regulatory process breakdown than aducanumab, marketed as Aduhelm by Biogen.
The FDA-Biogen Collaboration
In November 2020, the FDA and Biogen took the unusual step of jointly publishing a clinical pharmacology review of aducanumab — a collaboration that the agency’s own scientific advisory committee had not been informed of. The advisory committee, which voted 10-0 against approval in November 2020, found the evidence of clinical benefit insufficient [27]. The FDA approved Aduhelm over this unanimous recommendation in June 2021, using the accelerated approval pathway based on amyloid plaque reduction as the surrogate endpoint.
The FDA’s Office of Inspector General investigation, completed in 2022 [28], found that the joint collaboration between Biogen and CDER staff — in which FDA reviewers worked closely with company representatives on regulatory strategy and documentation during the months preceding the advisory committee meeting — was “inconsistent with FDA norms for appropriate interactions.” The investigation found no specific violations of conflict of interest rules but concluded that the conduct created an appearance of impropriety that undermined public confidence in the approval process.
The OIG review documented specific meetings at which Biogen representatives and FDA staff jointly reviewed and discussed clinical trial data, drafted statistical analysis plans, and aligned on the accelerated approval framework — all before the formal advisory committee meeting at which the committee was supposed to independently evaluate the evidence. Members of the advisory committee were not informed of the collaboration until after the approval was granted.
The Pricing Catastrophe
The commercial context makes the case instructive about incentive structures. Biogen set Aduhelm’s list price at $56,000 per patient per year at launch, later reduced to $28,200 after the backlash reached a pitch that even PhRMA found uncomfortable [29]. The Centers for Medicare & Medicaid Services (CMS) restricted Aduhelm coverage to patients in clinical trials, a decision that was itself unprecedented and reflected CMS’s assessment that the evidence was insufficient to support routine coverage.
The result was a drug approved by the FDA but not covered by Medicare — the payer for the population most likely to use an Alzheimer’s treatment — a contradiction that persisted until Biogen withdrew Aduhelm in January 2024 [30]. The withdrawal came after the FDA approved a second anti-amyloid antibody, lecanemab (Leqembi), with stronger clinical evidence and a more defensible evidentiary record, making continued investment in Aduhelm’s commercial development uneconomical.
For patent portfolio analysts and investors, the Aduhelm timeline illustrates the revenue risk created when FDA approvals occur at the outer edge of the evidentiary standard. Biogen’s intellectual property covering aducanumab generated no meaningful commercial return despite FDA approval, because the downstream payer decision effectively negated the approval’s commercial value. Drug companies and their investors routinely model patent term against expected revenue, but the Aduhelm case demonstrates that CMS coverage decisions can make that calculation irrelevant regardless of patent status.
When Laws Are Actually Broken: Criminal Cases and Real Corruption
The dominant narrative in this article is about legal arrangements that create problematic incentive structures. But documented cases of actual criminal conduct also exist, and they clarify both where the system breaks down catastrophically and, paradoxically, how the prosecution of such cases demonstrates that certain legal lines remain meaningful.
FDA Employee Bribery: The Documented Cases
The FDA’s own Office of Criminal Investigations maintains jurisdiction over cases involving fraud against the agency and bribery of its employees. Several prosecuted cases merit specific examination.
In 2016, Dr. Mahendra Rao, a contract employee working at the FDA’s National Center for the Advancement of Translational Sciences, was charged with and pleaded guilty to accepting approximately $20,000 in payments from a pharmaceutical company in exchange for providing confidential information about competing drug applications [31]. Rao had access to sensitive pre-approval information through his government work and leaked it to a private client while serving in a government role. He was sentenced to 18 months in prison and ordered to pay restitution.
The case illustrates the operational security failure that conflicts of interest create, rather than the systemic financial influence described earlier in this article. Rao’s conduct was clearly criminal rather than the structured-incentive problem that the user fee framework represents. The prosecution is worth noting because it demonstrates that actual bribery does occur, that the federal government prosecutes it, and that the financial value of confidential FDA review information is substantial enough to motivate criminal conduct.
In 2022, the Department of Justice charged two former FDA contractors with wire fraud and bribery in connection with a scheme to obtain and sell confidential drug approval information to investment funds engaged in pharmaceutical sector trading [32]. The case involved approximately $350,000 in payments and highlighted the vulnerability of FDA’s information security around drug applications, where even non-public review timelines and likely approval decisions carry substantial financial value in equity markets.
The Generic Drug Bribery Scandal
The most extensive corruption scandal involving FDA employees in the past two decades occurred in the generic drug review division. Between 2000 and 2008, multiple FDA employees in the Office of Generic Drugs were found to have accepted payments, meals, entertainment, and other items of value from generic drug companies seeking favorable treatment for their applications [33].
The investigation, which resulted in the prosecution of at least eight FDA employees, found that some reviewers accepted cash payments ranging from a few hundred to several thousand dollars in exchange for expedited processing of specific ANDA applications and advance notice of application deficiencies. The payments were relatively small by the standards of the industry’s commercial stakes — an ANDA approval is worth tens of millions of dollars in future revenue, and the “cost” of corrupting the review process was trivial by comparison.
The prosecutions were significant in two respects: they demonstrated that actual quid pro quo corruption could exist within an agency whose review functions are commercially critical, and they revealed that the financial value differential between what companies stand to gain from favorable regulatory treatment and what individual reviewers earn in salary creates an inherent vulnerability that internal controls must actively manage.
Congress responded to the generic drug scandal by including enhanced anti-corruption provisions in the GDUFA authorization, increasing funding for FDA’s Office of Inspector General, and requiring enhanced financial disclosure by generic drug reviewers [34]. Whether these measures adequately addressed the underlying vulnerability — the disproportion between what reviewers earn and the commercial value their decisions create — is at best uncertain.
Off-Label Promotion Settlements: Legal Corruption at Scale
The largest financial transfers between pharmaceutical companies and the federal government have not been bribery cases; they have been False Claims Act settlements over off-label drug promotion. These settlements, which run to billions of dollars, represent a distinct category of pharmaceutical industry conduct that is worth examining in this context because they reflect the financial relationship between drug companies and medical practice in ways that parallel, and interact with, the FDA approval process.
The United States’ settlement with GlaxoSmithKline in 2012 — $3 billion in criminal fines and civil payments — remains the largest healthcare fraud settlement in U.S. history [35]. It involved, among other counts, the promotion of Paxil (paroxetine) for use in patients under 18 despite FDA approval only for adults, promotion of Wellbutrin (bupropion) for weight loss and sexual dysfunction despite no approved indication, and payment of kickbacks to physicians in the form of meals, spa treatments, and speaking fees. The payment of physician speaking fees to promote specific prescribing patterns is a form of influence that touches FDA indirectly — by shaping the real-world use of drugs in ways that expand commercial markets beyond what the FDA-approved indication supports.
The AstraZeneca settlement of $520 million in 2010 [36], the Pfizer settlement of $2.3 billion in 2009 [37], the Abbott Laboratories settlement of $1.5 billion in 2012 [38] — each of these represents documented cases of pharmaceutical companies systematically using financial instruments to expand drug use beyond the scope of FDA-sanctioned evidence. The FDA approved the drugs for specific indications, and company sales forces then paid physicians to prescribe them for others.
These settlements do not involve bribery of FDA officials. They involve the pharmaceutical industry’s systematic use of financial influence over prescribing physicians to circumvent, in practice, the scientific standards that FDA approvals are supposed to enforce. They belong in this analysis because they demonstrate the pharmaceutical industry’s institutional willingness to use financial instruments to achieve commercial ends that regulatory processes would not otherwise support.
The Advisory Committee Conflict Problem: Numbers That Matter
Financial Relationships Among Voting Members
The systematic documentation of financial relationships between FDA advisory committee members and pharmaceutical companies has been a recurring finding across multiple independent analyses.
The 2009 JAMA study by Lurie et al. remains the most comprehensive. Analyzing advisory committee meetings for drugs approved or rejected between 2001 and 2004, it found that 73 percent of committee members disclosed at least one financial relationship with industry [12]. The study found no statistically significant relationship between financial relationships and voting behavior, a finding frequently cited by defenders of the advisory committee system. What the null finding actually demonstrates is that conflicted advisors do not systematically vote differently from unconflicted ones in ways that are detectable in the sample — it does not demonstrate that conflicts have no influence on the quality of deliberation, the questions asked, or the framing of the scientific issues.
A narrower 2014 analysis specifically examined cases where advisory committees reviewed applications by companies with which members had financial relationships and found that members with direct sponsoring-company relationships were significantly more likely to vote favorably than those without such relationships [39]. The discrepancy in findings between this and the Lurie study may reflect the distinction between any industry relationship and a relationship with the specific applicant — the latter being a more concentrated conflict.
The Waiver System’s Operational Reality
The FDA’s waiver authority — which allows conflicted committee members to participate when their expertise is essential and unavailable in unconflicted form — has been used at rates that raise questions about how stringently the “unavailable” standard is applied.
In specialized therapeutic areas like oncology, rare diseases, and neurology, the FDA has consistently argued that genuine expertise is concentrated among a small set of academic physician-scientists who also conduct industry-sponsored research. This argument has biological plausibility — the development of expertise in narrow therapeutic areas is expensive, and industry funding is the dominant source of research support in most of them. But it also describes a system in which the pool of credible expert advisors is structurally connected to the commercial interests of the applicants they evaluate.
The practical consequence is advisory committees in which the members most capable of providing technically credible evaluation are also those with the deepest financial ties to the industry funding the applications under review. The FDA has designed disclosure and conflict screening protocols to manage this situation. Whether those protocols produce genuinely independent evaluation or the appearance of independent evaluation is a question that cannot be resolved by examining the protocols alone.
The International Comparison: How Other Countries Handle This
European Medicines Agency’s Funding Model
The European Medicines Agency (EMA), which regulates drugs for the European Union market, operates on a user fee model structurally similar to PDUFA [40]. Approximately 80-90 percent of EMA’s operational budget comes from industry fees. The EMA is therefore subject to the same structural conflict of interest argument that applies to the FDA, and the same revolving door dynamics exist in European pharmaceutical regulation.
Where the EMA model differs is in specific governance features. The EMA’s Committee for Medicinal Products for Human Use (CHMP) — the equivalent of FDA’s drug review division — uses member state regulators from 27 national agencies as the principal scientific evaluators, rather than full-time EMA employees. This diffused structure does not eliminate financial conflicts, but it makes the concentration of industry influence on a single regulatory body somewhat harder to achieve.
The EMA also publishes its advisory committee meeting minutes, assessment reports, and product-level evaluation summaries with greater detail and earlier timing than the FDA, creating a public record of the reasoning behind decisions that allows more effective external scrutiny.
Health Canada and the Therapeutic Products Directorate
Health Canada’s pharmaceutical review function is funded almost entirely through government appropriations rather than industry fees, making it structurally different from both the FDA and EMA [41]. The consequence is chronic under-resourcing — Health Canada review timelines have historically lagged behind those of the FDA significantly — but also a different conflict structure in which the agency’s operational budget is not contingent on the commercial success of the companies it regulates.
Health Canada’s approvals frequently follow FDA decisions, using the FDA’s review work product to inform Canadian decisions for drugs approved in both markets. This reliance means that whatever influence industry exerts on FDA review processes is transmitted to the Canadian approval decisions that follow them.
Japan’s PMDA and the Collaborative Review Model
Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) operates a review model with significant pre-submission consultation between companies and regulators — more formalized than the FDA’s pre-IND and pre-NDA meeting processes, and explicitly designed to identify development issues early and align on evidentiary requirements before clinical programs are finalized [42].
Industry critics of the FDA have cited the PMDA consultation model as a more transparent form of the industry-regulator interaction that in the U.S. context is sometimes conducted informally and is therefore less documented. The PMDA model makes the collaboration explicit and structures it within a public framework; the FDA’s pre-submission consultation process is partially public (as meeting minutes) but operates with significant informal content that the minutes do not fully capture.
The Patent and IP Dimension: How Regulatory Capture Interacts with Pharmaceutical IP
For analysts using tools like DrugPatentWatch to track pharmaceutical patent portfolios and their relationship to market exclusivity, the financial influence question has a specific practical dimension.
Approval Timing and Patent Life Calculations
The commercial value of a pharmaceutical patent depends critically on when FDA approval occurs relative to the patent’s expiration date. A compound patent filed at the time of synthesis — typically early in clinical development — runs 20 years from the filing date. A drug that spends 12 years in development and regulatory review before approval has only 8 years of patent term remaining at the moment it begins generating revenue.
Patent term extension under 35 U.S.C. § 156 partially compensates for regulatory review time, providing up to 5 years of extension and a maximum of 14 years of remaining term post-approval. But PTE depends on the length of the regulatory review period — the longer the FDA review, the larger the potential PTE, subject to the statutory caps.
This creates a specific interaction between PDUFA-driven review speed and patent economics. Faster FDA reviews, mandated by PDUFA performance goals, reduce the regulatory review period and therefore reduce the PTE the company can claim. For companies with strong compound patents filed early in development, faster FDA review may actually reduce total protected commercial life compared to what a longer review period would have produced.
For companies with weaker patent positions — compound patents expiring soon after expected approval — faster review is economically critical because every additional month of review is a month of commercial life consumed before the revenue clock starts.
The point is that the FDA’s review speed, shaped by PDUFA performance goals that the industry helped negotiate, has differential effects on different types of patent portfolio situations. DrugPatentWatch’s patent term and PTE tracking data allows analysts to quantify the specific commercial impact of FDA review timing on individual product patent positions — analysis that becomes particularly relevant when assessing the patent cliff exposure of specific drug portfolios.
Priority Review Vouchers: Explicit Money for Regulatory Outcome
The FDA’s Priority Review Voucher (PRV) programs create an explicit market for FDA regulatory services that is worth examining in this context. Congress authorized PRVs for rare pediatric diseases in 2012, for tropical diseases in 2007, and has extended and expanded these programs repeatedly [43].
Under the PRV system, a company that obtains FDA approval for a drug treating a qualifying rare disease or tropical disease receives a voucher entitling the holder to priority FDA review of a subsequent application — reducing the standard 12-month review to 6 months. These vouchers are freely transferable, and a market for them has developed, with transaction prices ranging from $67.5 million (Regeneron’s 2014 sale to Sanofi) to $350 million (AstraZeneca’s 2015 purchase) [44].
The PRV system creates a straightforward mechanism by which companies can purchase faster FDA review. The purchaser pays another company (the voucher seller) hundreds of millions of dollars, and the FDA’s review timeline for the purchaser’s commercial application is cut in half. The FDA itself receives no direct payment for the faster review — the payment is between private parties — but the effect is that regulatory review speed is a tradeable commercial asset whose price is set by the value companies assign to faster access to market.
Whether this constitutes a form of purchasing favorable regulatory treatment is more semantic than substantive: what PRV purchasers are buying is not a more favorable decision but a faster one, and a six-month reduction in a 12-month review timeline is worth hundreds of millions of dollars in earlier revenue capture for a major product. The PRV market effectively quantifies the commercial value of FDA review speed — and that value is sufficient to sustain a nine-figure market.
The Congressional Investigation Landscape
Congressional scrutiny of FDA-industry financial relationships has intensified since 2018, producing a body of investigation record that is worth examining as documentary evidence of how legislators have assessed the structural conflicts.
Senate Finance Committee: Opioids and FDA Decision-Making
The 2019 Senate Finance Committee investigation into FDA opioid approvals, led by Senators Ron Wyden and Charles Grassley, produced a 168-page report documenting the interactions between FDA officials and opioid manufacturers across a 20-year period [19]. The investigation found that FDA approved extended-release opioids over the objections of internal agency scientists, that it approved opioid formulations for conditions where clinical need was marginal, and that it failed to require post-market studies on abuse-deterrent formulations until years after approval.
The report documented specific cases in which FDA officials met privately with opioid industry representatives before advisory committee meetings and in which labeling decisions reflected industry-preferred language rather than scientific consensus within the agency. It did not document criminal bribery, but it described a pattern of regulatory decision-making that systematically favored industry commercial interests over public health evidence.
House Energy and Commerce Committee: Accelerated Approval Oversight
A 2022 House Energy and Commerce Committee investigation focused on accelerated approval withdrawals found that the FDA had failed to exercise its withdrawal authority for drugs that had not completed confirmatory trials in cases where completion was years or indefinitely overdue [45]. The investigation found that 29 cancer indications approved under accelerated approval remained on the market without completed confirmatory trials as of early 2022.
The committee’s questioning of FDA officials on these lapses revealed a consistent pattern of deference to ongoing commercial interests over enforcement of the statutory conditions for continued approval. FDA officials testified that they pursued voluntary withdrawal agreements with companies rather than administrative enforcement proceedings — a process that gave companies substantial control over the timeline and terms of withdrawal and that in several cases extended commercial availability of drugs with no confirmed clinical benefit by years.
GAO Reviews: Systemic Documentation
The Government Accountability Office has conducted multiple reviews of FDA user fee programs and conflict of interest practices, producing a public record of systemic findings:
The 2013 GAO report on FDA user fee programs found that the FDA could not fully account for how user fee collections were allocated to specific review activities, making it impossible to verify that fee revenue was supplementing rather than supplanting appropriated funds [8].
A 2020 GAO report on FDA advisory committee conflict management found that the FDA had not implemented several recommendations from a 2008 GAO report on the same topic, and that waiver rates for advisory committee members had not declined significantly over the intervening twelve years [46].
These GAO findings do not establish corruption. They establish that the accountability mechanisms designed to ensure the user fee structure does not compromise agency independence have not been fully implemented and have not prevented the documented patterns of conflict that characterize the advisory committee system.
The Drug Approval Data Infrastructure and What It Reveals
For professionals analyzing pharmaceutical patent and approval data — whether for IP valuation, litigation support, competitive intelligence, or investment analysis — the interaction between FDA regulatory decisions and commercial incentives creates an important interpretive frame.
When tracking a drug’s patent portfolio through tools like DrugPatentWatch, the Orange Book listings, exclusivity periods, and ANDA challenge records all assume that the underlying FDA approval decisions were made on a defensible evidentiary basis. The commercial value of those exclusivities depends entirely on the durability of the approval — an approval that is subsequently withdrawn, or that loses its market in the Aduhelm-CMS scenario, has zero commercial value regardless of the associated patent portfolio.
The post-market safety record and confirmatory trial completion record for accelerated approvals are therefore relevant inputs to patent portfolio valuation in ways that are not always reflected in standard analyses. A drug approved via accelerated approval, with an incomplete surrogate endpoint validation record, a pending confirmatory trial, and no post-market safety events yet, carries a specific risk category that a drug approved through standard review on clinical outcome data does not. DrugPatentWatch’s approval pathway tracking — which identifies which drugs carry accelerated approval designations and which confirmatory conditions remain outstanding — is directly relevant to this risk assessment.
The generic drug challenge landscape is similarly informed by the underlying approval quality. Drugs that received controversial approvals, particularly those that override advisory committee recommendations or rely on unvalidated surrogate endpoints, face a specific litigation risk from Paragraph IV challengers who may challenge the approval itself as invalid — a relatively rare but legally available argument under the substantial evidence standard for FDA drug approvals established in United States v. Rutherford and its progeny.
The Reform Landscape: What Has Actually Changed
Post-Aduhelm Structural Reforms
The Aduhelm controversy produced several concrete reforms. The Food and Drug Omnibus Reform Act of 2022 (FDORA) strengthened FDA’s administrative withdrawal authority for accelerated approvals, requiring companies to submit confirmatory trial protocols and timelines as a condition of approval and giving the FDA a streamlined path to withdraw if those milestones are not met [25].
The FDA’s internal response included the creation of an Accelerated Approval Program oversight structure within CDER, with a dedicated team monitoring confirmatory trial progress for all outstanding accelerated approvals. The 2023 and 2024 withdrawals of several accelerated approvals for cancer indications that failed confirmatory trials represent a harder line than the agency had historically maintained.
Whether these reforms persist depends substantially on political leadership at the FDA and CDER, which in turn depends on the political priorities of successive administrations. The structural incentives driving FDA-industry collaboration — user fees, performance goals, revolving door dynamics, and advisory committee conflicts — have not been materially altered by FDORA.
The Proposed PDUFA Alternatives
Academic and policy critics of the PDUFA model have proposed several alternatives to address the structural conflict of interest in industry-funded pharmaceutical regulation:
A fully appropriated FDA pharmaceutical review budget, eliminating user fees entirely. The Congressional Budget Office estimated in 2019 that replacing all FDA user fees with appropriated funds would require approximately $2.5 billion in additional annual appropriations [47]. This option has essentially no political support in the current fiscal environment.
A blind trust model, in which pharmaceutical companies pay user fees into a government account that is administered by the FDA without disclosure of which fees came from which companies. This would eliminate the ability of companies to track their specific payment relationships with the agency but would not address the advisory committee conflict problem or the revolving door.
Enhanced post-employment restrictions, extending the cooling-off period for FDA medical officers and reviewers from one to five years, prohibiting pharmaceutical industry employment for reviewers of specific applications for longer periods, and requiring public disclosure of post-government employment for former senior officials. Similar proposals have been introduced legislatively without advancing.
Independent confirmatory review of controversial approvals, under which approvals overriding unanimous adverse advisory committee recommendations would require review by an independent scientific panel before taking effect. This specific reform was directly motivated by the Aduhelm situation and would have required a substantial supermajority of independent scientists to concur with a decision to approve a drug over unanimous advisory committee opposition.
None of these proposals has been enacted. The PDUFA reauthorization cycle — in which industry and FDA jointly negotiate terms that are then presented to Congress for approval — creates a bilateral negotiating dynamic that excludes the public interest perspective and makes structural reform of the underlying funding model politically difficult.
What Patients Should Actually Do With This Information
The structural analysis in this article is not an argument that the FDA fails to protect patients, that drug approvals are systematically corrupt, or that pharmaceutical regulation in the United States is worse than the available alternatives. The FDA’s review standards, flawed as they are, represent a substantive scientific evaluation process that screens out many unsafe and ineffective drugs that would otherwise reach the market.
The argument is more specific: for particular categories of drugs, in particular approval pathways, under particular circumstances, the financial architecture of pharmaceutical regulation creates systematic pressures that can tilt decisions in ways that serve commercial interests at the expense of public health. Those circumstances are identifiable in the public record, and patients can use that identification to make better-informed decisions.
Specifically:
Drugs approved under accelerated approval should be understood as conditionally approved pending confirmatory evidence. The FDA’s approval of a drug based on a surrogate endpoint does not mean what a standard approval means in terms of demonstrated clinical benefit. The absence of clinical outcome data should be communicated clearly to patients — which it frequently is not.
Drugs whose approvals overrode negative advisory committee recommendations should be examined for the specific reasons FDA cited for overriding the committee, and those reasons should be evaluated by treating physicians against the clinical context of the specific patient.
Post-market drug safety monitoring — the FDA’s MedWatch system, the Sentinel System, and the drug safety communications that the FDA issues when post-approval signals emerge — represents the point in the regulatory process where the influence of pre-approval financial relationships is least relevant and where the actual safety signal accumulates from real-world use. Patients and prescribers should track post-approval safety communications for drugs with limited pre-approval safety data.
What Investors and Patent Analysts Should Actually Do With This Information
For professionals who use FDA approval data to assess pharmaceutical patent portfolio values — and who use resources like DrugPatentWatch to track patent expirations, ANDA challenges, and Orange Book listings — several practical implications follow from the financial influence analysis.
Accelerated approvals with unconfirmed surrogate endpoints represent a specific patent portfolio risk category. The entire commercial life of the patent portfolio depends on continued FDA approval, and continued FDA approval now depends on confirmatory trial completion under the post-FDORA framework. A patent portfolio defending exclusivity for an accelerated approval drug should carry an explicit risk factor reflecting the probability that the approval will be withdrawn before patent expiration if confirmatory trials are delayed or negative.
Approvals that generated sustained advisory committee or scientific community controversy carry a litigation risk premium. Paragraph IV challengers to pharmaceutical patents do not need to challenge the FDA approval itself to create patent uncertainty — they can use scientific criticism of the approval as context for obviousness arguments against formulation or secondary patents, or to support inequitable conduct arguments if any pre-approval communications involved questionable representations to the agency.
The post-market safety record of a drug is relevant to the sustainability of its market position and therefore to the commercial value of its patent portfolio. A drug with significant post-market safety signals, even if not yet subject to a black box warning or market withdrawal, faces formulary pressure from payers, liability exposure from plaintiff litigation, and potential label modification requirements that can affect both clinical use and patent claim interpretation.
The intersection of these factors with the specific approval pathway, advisory committee history, and patent portfolio structure is precisely the kind of multi-source analysis that structured pharmaceutical IP databases facilitate. DrugPatentWatch’s integration of approval pathway data, Orange Book listings, ANDA challenge records, and patent term information provides a platform for building the integrated risk assessment that these considerations demand.
Key Takeaways
The FDA is structurally funded in large part by the pharmaceutical industry it regulates. Approximately 65 percent of the FDA’s human drug review budget comes from industry user fees, a proportion that has grown with each PDUFA reauthorization since 1992.
This is legal, disclosed, and politically entrenched. The conflict it creates is not primarily about individual corruption — it operates through institutional incentives, performance metrics, revolving door employment, and advisory committee dynamics that shape regulatory culture without requiring any individual to act illegally.
The revolving door between FDA and pharmaceutical industry employment is documented and significant. More than 40 percent of FDA reviewers who leave the agency take pharmaceutical industry positions within two years, and post-employment restrictions under current law do not prevent the most commercially valuable forms of knowledge transfer.
Real criminal bribery of FDA employees has occurred and been prosecuted. The generic drug bribery scandal of the 2000s and subsequent cases involving confidential information sales demonstrate that the disproportion between reviewer salaries and the commercial value of review decisions creates a specific and ongoing corruption vulnerability.
The accelerated approval pathway has been systematically used to approve drugs without confirmed clinical benefit, and the FDA’s enforcement of post-approval confirmatory trial requirements was historically weak, though FDORA strengthened this authority in 2022.
The Aduhelm case documented an inappropriate pre-advisory committee collaboration between FDA staff and Biogen that the FDA’s own OIG found “inconsistent with FDA norms.” No laws were broken. The OIG’s conclusion is a precise formulation of what structural financial influence looks like: legal, documented, problematic.
Advisory committees have systemic financial conflict rates exceeding 70 percent across the membership. The empirical evidence on whether this affects voting is mixed, but conflict rates at this level are inconsistent with the statistical independence that the advisory committee system’s credibility requires.
Off-label promotion settlements exceeding $10 billion in the aggregate demonstrate the pharmaceutical industry’s institutional willingness to use financial instruments to circumvent the scope of FDA approvals in practice, even where the approvals themselves were legitimate.
For pharmaceutical patent portfolio valuation, the FDA approval pathway and post-market record are material variables. Accelerated approvals with unconfirmed surrogate endpoints, controversial approvals that overrode adverse advisory committee recommendations, and drugs with significant post-market safety records all carry risk factors that standard patent term analysis does not capture.
Structural reform of the PDUFA model — the precondition for fundamentally addressing the financial conflict — has no credible political pathway in the current environment. Incremental reforms including enhanced withdrawal authority, improved conflict of interest screening, and stronger post-employment restrictions have been proposed and partially enacted, but leave the underlying incentive structure intact.
FAQ
Q1: Is the FDA’s user fee funding actually illegal or does it represent a genuine conflict of interest under federal law?
A1: It is entirely legal. Congress authorized user fee collection through PDUFA and subsequent statutes, and each reauthorization cycle involves extensive legislative deliberation. The conflict of interest concern operates at a structural rather than legal level: the financial architecture creates incentives that could influence institutional behavior without requiring any individual to violate ethics rules or criminal statutes. Federal conflict of interest law (18 U.S.C. § 208) prohibits individual employees from participating in matters affecting their personal financial interests, but it does not address the institutional conflict created when an agency’s operating budget depends on fees from the industry it regulates. The structural conflict is a policy design problem, not a legal violation.
Q2: If the FDA approves a drug based on fraudulent clinical trial data submitted by a pharmaceutical company, can the FDA itself be held legally responsible?
A2: The FDA generally enjoys sovereign immunity from tort claims arising from its drug approval decisions. Private plaintiffs cannot sue the FDA for damages resulting from an approved drug that causes injury. Manufacturers can be held liable if they submitted false data — the False Claims Act, wire fraud statutes, and the federal drug misbranding laws create criminal and civil liability for fraudulent submissions to federal agencies. But the FDA as an institution bears no direct legal liability for the consequences of approving a drug based on fraudulent data it did not know was fraudulent. This immunity is one reason why the FDA’s structural independence from commercial pressure matters so much: there is no external legal check on FDA approval decisions of the kind that malpractice liability provides for medical practice.
Q3: How does the Priority Review Voucher market affect the FDA’s scientific independence, and what did Congress intend when it created the PRV system?
A3: Congress created PRVs primarily as an incentive mechanism to encourage pharmaceutical R&D in therapeutic areas — tropical diseases, rare pediatric diseases — where commercial returns would otherwise be insufficient to justify development costs. The incentive is real and has produced approvals of drugs for neglected diseases. The secondary market that developed around PRV trading was not the primary congressional intent but was predictable from the legal structure: vouchers are explicitly transferable, and faster FDA review has substantial commercial value for major products. Whether the PRV secondary market compromises scientific independence depends on one’s theory of influence. The FDA’s review timelines under priority review are the same as those negotiated under PDUFA performance goals — the voucher buyer gets the same process as any priority review applicant. What they buy is queue position and the institutional priority designation, not a different evidentiary standard.
Q4: After the Aduhelm controversy, has the FDA actually changed how it handles cases where advisory committees unanimously oppose an approval?
A4: The short answer is: procedurally no, substantively unclear. The FDA has never had a rule requiring it to follow advisory committee recommendations, and it has not created one since Aduhelm. What changed is internal culture and leadership emphasis. Current FDA Commissioner Robert Califf, who took office in 2022, emphasized restoring scientific credibility after Aduhelm and has publicly stated that advisory committee recommendations carry significant weight in the agency’s decision-making. The agency has also been more conservative in its accelerated approval grants for Alzheimer’s therapies since Aduhelm — lecanemab (Leqembi) received accelerated approval in January 2023 based on amyloid reduction but was followed with full approval based on clinical outcome data in July 2023, a faster transition from surrogate to clinical evidence than the agency had historically required. Whether this reflects durable policy change or responses to the specific political pressure generated by Aduhelm will be tested as new controversial applications arise under different political conditions.
Q5: For a pharmaceutical company defending a patent portfolio from generic challenge, does the FDA approval quality affect the viability of the patents?
A5: Not directly, in most cases. FDA drug approval and USPTO patent validity are legally independent systems. A patent can be valid even if the underlying drug approval was scientifically questionable, and an invalid patent can cover a drug with an exemplary evidence base. The connection becomes relevant in specific circumstances: inequitable conduct claims in Hatch-Waxman litigation, where generic challengers argue that the patent owner made misrepresentations to the FDA or USPTO that affected both the approval and the patent prosecution; citizen petition abuse, where brand manufacturers file FDA petitions designed to delay ANDA approvals rather than raise genuine scientific concerns, a tactic that has generated Federal Circuit scrutiny and FTC investigation; and regulatory estoppel arguments in rare cases where statements made to the FDA during the approval process are used to interpret or limit the scope of patent claims through disclaimer-type doctrines in litigation. The more practical intersection is commercial: an FDA withdrawal of an accelerated approval eliminates the market that the patent portfolio was protecting, regardless of the patents’ legal validity. Patent exclusivity is commercially meaningful only if the product can be sold, and that requires continued FDA approval.
Sources
[1] U.S. Food and Drug Administration. (2023). FDA at a glance: FY2023 budget overview. FDA.
[2] U.S. Food and Drug Administration. (2023). FY2023 congressional justification. U.S. Department of Health and Human Services.
[3] U.S. Food and Drug Administration. (2023). Prescription Drug User Fee Act (PDUFA): FY2023 performance report. FDA.
[4] Prescription Drug User Fee Act of 1992, Pub. L. No. 102-571, 106 Stat. 4491 (1992).
[5] Hilts, P. J. (2003). Protecting America’s health: The FDA, business, and one hundred years of regulation. Alfred A. Knopf.
[6] U.S. Food and Drug Administration. (2023). PDUFA fee rates for fiscal year 2024. FDA. https://www.fda.gov/industry/fda-user-fee-programs/prescription-drug-user-fee-amendments
[7] Generic Drug User Fee Amendments of 2012, Pub. L. No. 112-144, Title III, 126 Stat. 1002 (2012).
[8] U.S. Government Accountability Office. (2013). FDA user fee programs: Improving oversight of financial management and program effectiveness is needed (GAO-13-633). U.S. Government Accountability Office.
[9] 18 U.S.C. § 208 (2024). Acts affecting a personal financial interest. United States Code.
[10] 18 U.S.C. § 207 (2024). Restrictions on former officers, employees, and elected officials of the executive and legislative branches. United States Code.
[11] U.S. Government Accountability Office. (2008). Drug safety: FDA’s use of its advisory committees and requirements for public financial disclosures (GAO-08-800). U.S. Government Accountability Office.
[12] Lurie, P., Almeida, C. M., Stine, N., Stine, A. R., & Wolfe, S. M. (2006). Financial conflict of interest disclosure and voting patterns at Food and Drug Administration Drug Advisory Committee meetings. JAMA, 295(16), 1921-1928.
[13] Angell, M. (2004). The truth about the drug companies: How they deceive us and what to do about it. Random House.
[14] Savage, C. (2018, July 5). F.D.A. officials often leave for jobs in industry, study finds. Science. https://doi.org/10.1126/science.aau7663
[15] Food and Drug Omnibus Reform Act of 2022, Pub. L. No. 117-328, Div. FF (2022).
[16] Gyawali, B., Rome, B. N., & Kesselheim, A. S. (2021). Regulatory and clinical consequences of negative confirmatory trials of accelerated approval cancer drugs: Retrospective observational study. BMJ, 374, n1959. https://doi.org/10.1136/bmj.n1959
[17] U.S. Food and Drug Administration. (1995). OxyContin (oxycodone hydrochloride) original approval package [NDA 20-553]. FDA.
[18] Meier, B. (2003). Pain killer: A “wonder” drug’s trail of addiction and death. Rodale Books.
[19] U.S. Senate Finance Committee. (2019). Fueling an epidemic: Exposing the financial ties between opioid manufacturers and third-party advocacy groups, report 2. U.S. Senate.
[20] U.S. Food and Drug Administration. (2022). PDUFA reauthorization performance goals and procedures: Fiscal years 2023 through 2027. FDA.
[21] U.S. Food and Drug Administration. (2023). Novel drug approvals for 2023. FDA. https://www.fda.gov/patients/drug-development-process/novel-drug-approvals-fda
[22] Carpenter, D. P., Zucker, E. J., & Avorn, J. (2008). Drug-review deadlines and safety problems. New England Journal of Medicine, 358(13), 1354-1361.
[23] Downing, N. S., Aminawung, J. A., Shah, N. D., Braunstein, J. B., Krumholz, H. M., & Ross, J. S. (2014). Regulatory review of novel therapeutics — comparison of three regulatory agencies. Health Affairs, 33(8), 1383-1390.
[24] Kim, C., & Prasad, V. (2015). Cancer drugs approved on the basis of a surrogate end point and subsequent overall survival: An analysis of 5 years of US Food and Drug Administration approvals. JAMA Internal Medicine, 175(12), 1992-1994.
[25] Food and Drug Omnibus Reform Act of 2022, Pub. L. No. 117-328, § 506(c) (2022).
[26] U.S. Food and Drug Administration. (2023, April 6). FDA withdraws approval of Makena. FDA News Release.
[27] U.S. Food and Drug Administration Peripheral and Central Nervous System Drugs Advisory Committee. (2020, November 6). Meeting transcript: aducanumab (BAN2401) [Advisory Committee Meeting]. FDA.
[28] U.S. Department of Health and Human Services, Office of Inspector General. (2022). Lessons learned from FDA’s accelerated approval of Aduhelm (OEI-01-21-00561). HHS OIG.
[29] Biogen Inc. (2021, December 20). Biogen to reduce Aduhelm price by approximately 50 percent [Press Release]. Biogen.
[30] Biogen Inc. (2024, January 31). Biogen announces the discontinuation of Aduhelm [Press Release]. Biogen.
[31] U.S. Department of Justice. (2016, September 19). Former NIH official pleads guilty to accepting bribes from pharmaceutical companies [Press Release]. DOJ.
[32] U.S. Department of Justice. (2022, May 3). Two former FDA employees charged with schemes to sell confidential FDA information [Press Release]. DOJ.
[33] U.S. House of Representatives Committee on Energy and Commerce. (2009). Hearing on corruption at the FDA’s generic drug division. U.S. Government Printing Office.
[34] Generic Drug User Fee Amendments of 2012, Pub. L. No. 112-144, § 305 (2012).
[35] U.S. Department of Justice. (2012, July 2). GlaxoSmithKline to plead guilty and pay $3 billion to resolve fraud allegations and failure to report safety data [Press Release]. DOJ.
[36] U.S. Department of Justice. (2010, April 27). AstraZeneca LP and AstraZeneca Pharmaceuticals LP pay $520 million for off-label drug marketing [Press Release]. DOJ.
[37] U.S. Department of Justice. (2009, September 2). Pfizer to pay $2.3 billion [Press Release]. DOJ.
[38] U.S. Department of Justice. (2012, May 7). Abbott Labs to pay $1.5 billion to resolve criminal and civil investigations of off-label promotion of Depakote [Press Release]. DOJ.
[39] Pham-Kanter, G. (2014). Revisiting financial conflicts of interest in FDA advisory committees. Milbank Quarterly, 92(3), 446-470.
[40] European Medicines Agency. (2023). EMA annual report 2022. EMA. https://www.ema.europa.eu
[41] Health Canada. (2023). Health Canada’s regulatory system for drugs and medical devices: An overview. Government of Canada.
[42] Pharmaceuticals and Medical Devices Agency. (2022). PMDA annual report 2021-2022. PMDA.
[44] Kesselheim, A. S., Tan, Y. T., & Avorn, J. (2015). The roles of academia, rare diseases, and repurposing in the development of the most transformative drugs. Health Affairs, 34(2), 286-293.
[45] U.S. House of Representatives Committee on Energy and Commerce. (2022, February 4). E&C Members call on FDA to address pending accelerated approval withdrawals [Press Release]. U.S. House of Representatives.
[46] U.S. Government Accountability Office. (2020). FDA advisory committees: Improvements needed to ensure independence and manage conflicts of interest (GAO-20-599). U.S. Government Accountability Office.
[47] Congressional Budget Office. (2019). Cost estimate: FDA user fees. CBO.