Every year, hundreds of millions of dollars shift hands based on a single question: who filed first?
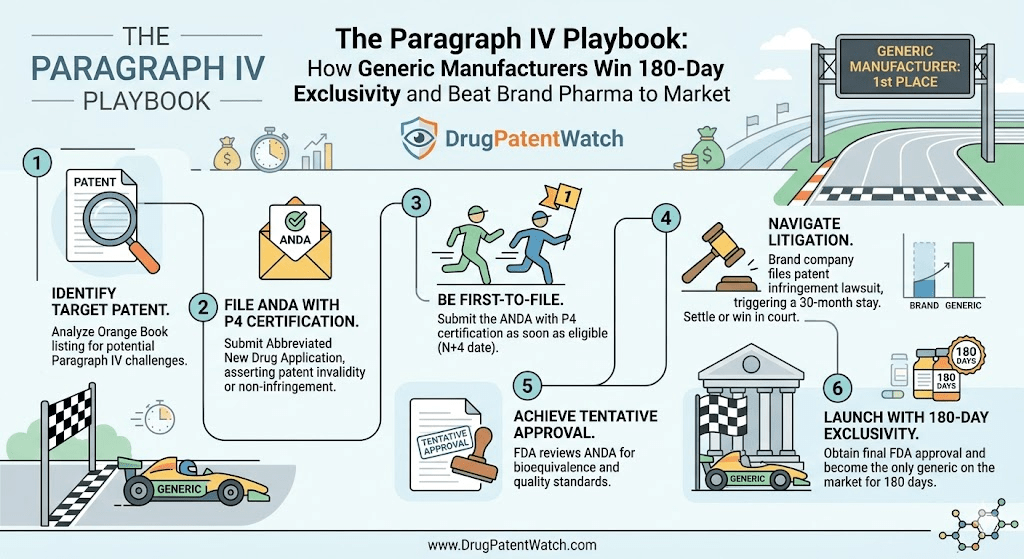
The Paragraph IV certification under the Hatch-Waxman Act is the most profitable legal maneuver available to a generic drug manufacturer in the United States. Get it right, and you control the first six months of a newly opened generic market — a window that, depending on the drug, can generate hundreds of millions in revenue while the rest of the industry watches from the sidelines. Miss the window, or botch the strategy, and you spend years in litigation only to launch into a market already crowded with competitors.
This is not a guide to the basics. You already know what a Paragraph IV certification is. This is an operational manual for the teams — legal, regulatory, and commercial — who need to win the race, protect the prize, and avoid the pitfalls that have killed otherwise solid exclusivity positions for well-resourced companies.
We will walk through every phase: pre-filing patent analysis, the mechanics of ANDA submission timing, the 30-month stay and how to use it, the six forfeiture triggers that can strip your exclusivity without warning, the antitrust constraints on settlement, the GSK v. Teva problem, and what the FTC’s 2023-2024 Orange Book campaign means for your target list. We will also look at how platforms like DrugPatentWatch give competitive intelligence teams the real-time visibility needed to move first when a brand’s patent wall starts to crack.
The prize is real. According to research published in the legal literature, generic companies typically generate 60% to 80% of their potential profit for a given product during the 180-day exclusivity window. [1] That concentration of revenue in six months is the reason this strategy commands the best attorneys, the largest pre-ANDA budgets, and the most intense internal scrutiny of any business decision a generic manufacturer makes.
1. The Foundation: What the Hatch-Waxman Act Actually Built
Congress passed the Drug Price Competition and Patent Term Restoration Act of 1984 — universally called the Hatch-Waxman Act — to solve a practical problem. Before the Act, generic manufacturers had to run full clinical trials to gain FDA approval, which made the generic business economically untenable. Brand manufacturers held de facto monopolies well beyond their patent terms because the economics of generic development simply did not work.
The Act did two things that changed the industry permanently. First, it created the Abbreviated New Drug Application (ANDA), which lets a generic manufacturer demonstrate bioequivalence to a reference listed drug (RLD) without replicating the innovator’s full clinical trial package. Second, it created a structured mechanism for challenging brand patents before they expire — the Paragraph IV certification — and attached a financial incentive to that challenge: 180 days of generic exclusivity for the first filer.
The quid pro quo for brand manufacturers was an extension of patent terms (up to five years) to compensate for regulatory review time, plus a 30-month automatic stay of ANDA approval if they chose to sue within 45 days of receiving notice of a Paragraph IV filing. The system was designed to accelerate generic entry while giving innovators a defined window to defend legitimate patents.
In practice, the system has produced enormous generic savings. Ninety percent of prescriptions filled in the United States are generic, yet generics account for roughly 13% of total drug spending. [2] That asymmetry traces directly to Hatch-Waxman mechanics. But the system has also produced decades of strategic gamesmanship — by both brand and generic manufacturers — that has forced continuous regulatory and judicial refinement.
Understanding the current state of the system requires knowing its history. The 2003 Medicare Modernization Act (MMA) significantly reformed the exclusivity provisions, adding six forfeiture triggers to prevent the “parking” of exclusivity by first filers who delayed commercial launch. The FTC v. Actavis decision in 2013 brought antitrust law directly into the settlement calculus. The GlaxoSmithKline v. Teva line of cases from 2020 through 2022 made Section viii skinny-label strategies significantly more dangerous. And the FTC’s 2023-2024 Orange Book challenges opened new offensive opportunities for generic manufacturers willing to use the regulatory process as a weapon.
All of those developments affect how you should structure a Paragraph IV campaign today.
2. Picking the Target: Patent Analysis Before You File Anything
The most consequential decision in any Paragraph IV strategy is target selection — and most teams make it too quickly. The commercial analysis (peak brand sales, remaining patent life, number of potential competitors) comes first, but it cannot be the only input. A drug generating $2 billion annually is worthless as a generic target if the compound patent has 12 years left and the active ingredient cannot be designed around.
2.1 Reading the Orange Book Correctly
The FDA’s Orange Book lists the patents that a brand manufacturer has self-certified as relevant to its approved drug product. The listings fall into three categories: drug substance (active ingredient) patents, drug product (formulation) patents, and method of use patents. Each category carries different strategic implications.
Compound patents covering the active ingredient are typically the hardest to challenge. A valid composition of matter patent gives the brand manufacturer a nearly impenetrable position — invalidating it requires prior art that predates the earliest priority date of the patent, and generic manufacturers have been searching for such art for decades on every major drug. The compound patents on genuinely novel molecules tend to survive litigation.
Formulation patents and manufacturing process patents are different. These are the targets where generic manufacturers have succeeded repeatedly. A brand company that reformulates an existing drug to add an extended-release mechanism, a specific excipient, or a new salt form and then lists those formulation patents in the Orange Book has created an attack surface. If the generic can demonstrate that the original formulation is prior art, that the patent claims are obvious, or that the generic product does not infringe the formulation claims, the path to early market entry opens.
Method of use patents listed in the Orange Book correspond to specific FDA-approved indications. These are relevant to Paragraph IV challenges that assert non-infringement, and also to the Section viii carve-out strategy discussed later. A generic manufacturer can potentially avoid a method of use patent entirely by labeling its product only for non-patented uses — though the GlaxoSmithKline v. Teva decision demonstrated that a carve-out label does not automatically insulate a manufacturer from induced infringement claims.
The first analytical step is building a complete, patent-by-patent inventory of every Orange Book listing for the target drug. Services like DrugPatentWatch provide structured access to Orange Book data, patent expiration timelines, and existing ANDA filing histories, allowing a team to determine quickly whether first-filer status is still available, whether same-day filings have already occurred, and which patents have been challenged before and survived. That historical record matters: a patent that has been litigated and upheld is not necessarily unassailable, but it tells you where prior arguments have failed and what new theory you need to bring.
2.2 The Prior Art Search
The strength of a Paragraph IV challenge correlates directly with the quality of the prior art search conducted before filing. This is not a task to delegate to a junior associate. The search must cover scientific literature, international patents, conference proceedings, regulatory submissions in other jurisdictions (the European Medicines Agency’s European Public Assessment Reports are particularly valuable), foreign pharmacopeias, unpublished doctoral theses, and any publicly disclosed research predating the priority date of the patents at issue.
For formulation patents, the search focuses on whether the claimed formulation was obvious based on the state of the art at the time of filing. The combination of previously known excipients in previously known ratios rarely survives an obviousness challenge if the generic manufacturer can demonstrate that a skilled formulation scientist would have been motivated to combine them and would have had a reasonable expectation of success. Courts have been willing to invalidate formulation patents on these grounds, particularly where the brand’s own prior publications disclose the components of the claimed formulation.
For compound patents on secondary salts or polymorphs, the analysis turns on whether the specific salt or polymorph was an obvious modification of a known compound. The Federal Circuit’s obviousness jurisprudence has made these patents increasingly vulnerable — but only if the prior art search surfaces the right disclosures.
The notice letter that accompanies the Paragraph IV certification must include a detailed statement of the factual and legal basis for each invalidity and non-infringement position. [3] Courts have found inadequate notice letters to be grounds for dismissal, and the arguments in the letter — while not binding in subsequent litigation — set the intellectual framework for discovery and trial strategy. Invest in the prior art search. The return on that investment compounds through every phase of what follows.
2.3 Freedom to Operate and Non-Infringement Analysis
A freedom-to-operate (FTO) analysis runs parallel to the prior art search. The question is whether the proposed generic product, in its actual formulation and using its actual manufacturing process, infringes the valid claims of any Orange Book-listed patent. This requires a claim-by-claim comparison against the generic product’s specifications — not against a hypothetical product, but against the product you plan to manufacture and sell.
If infringement is unavoidable on a formulation patent, the options are three: challenge validity (the invalidity route), design around the patent claims (the non-infringement route), or consider whether a Section viii carve-out is available for method of use patents (with full appreciation of the GSK v. Teva risk). Each path has different cost profiles, different risk profiles, and different implications for the notice letter that must accompany the filing.
The design-around approach deserves more emphasis than it gets in most strategic discussions. If a brand’s key formulation patent claims a specific polymer at a specific concentration range, and your formulation can achieve bioequivalence using a different polymer or a different concentration, you have a clean non-infringement position that is substantially stronger than an invalidity argument in litigation. Brand manufacturers spend enormous effort claiming their formulations broadly — but broad claims are often invalid for lack of written description or enablement, and narrow claims can frequently be designed around.
3. The ANDA Filing: Mechanics, Timing, and First-Filer Status
First-filer status determines whether you receive 180 days of exclusivity or spend that 180 days watching a competitor collect the revenue. The statute awards exclusivity to the applicant or applicants who file a substantially complete ANDA containing a Paragraph IV certification first. Under the MMA, if multiple companies submit on the same calendar day, all same-day filers share the exclusivity. [4]
3.1 What ‘Substantially Complete’ Means
A substantially complete ANDA contains all the sections required under 21 C.F.R. Part 314, Subpart C, sufficient for the FDA to begin substantive review. An ANDA that is missing critical sections — bioequivalence data, chemistry, manufacturing and controls (CMC) data, or labeling — is not substantially complete and will not be stamped as a first filing.
This creates a real tension. Generic manufacturers want to file as quickly as possible to secure first-filer status, but they cannot file a placeholder application. The FDA’s receipt date for a substantially complete application is the operative date for first-filer determination, and applications that arrive incomplete are not held — they are returned or received with a later date once the deficiencies are corrected.
The practical implication: bioequivalence studies must be completed and the data must be in hand before the ANDA is filed. CMC sections must be finalized. Every section of the application that requires original data generation has a lead time measured in months. The competitive race to file first begins not when the brand’s patent wall starts to crack, but years earlier when the strategic decision is made to develop a specific generic product.
3.2 The Notice Letter: Your Opening Legal Brief
Within 20 days of receiving the FDA’s acknowledgment letter for the ANDA, the applicant must send formal notice of the Paragraph IV certification to both the NDA holder and each patent owner listed in the Orange Book. [5] The notice must be sent to the NDA holder even if the NDA holder and the patent owner are the same entity.
The notice letter has to include the ANDA application number, a detailed statement of the factual and legal basis for each patent’s invalidity or non-infringement, and — if the certification is based on non-infringement — an Offer of Confidential Access (OCA) allowing the brand manufacturer to examine the relevant sections of the ANDA before deciding whether to sue.
The OCA is not optional. Its absence can expose the generic manufacturer to a claim that it failed to provide adequate notice, potentially affecting the 45-day clock for the brand to file suit. The OCA typically includes confidentiality protections limiting who within the brand company can view the ANDA data and restricting use of that data solely to the patent litigation.
The notice letter is your first substantive communication to the brand about your legal theory. It sets the parameters of the dispute. If your invalidity arguments are based on anticipation by a specific prior art reference, that reference will anchor the litigation discovery on both sides. Crafting the notice letter with the trial in mind — not just as a compliance exercise — is the difference between teams that win Paragraph IV cases and teams that settle on unfavorable terms.
3.3 The 45-Day Decision and the 30-Month Stay
Once the brand manufacturer receives the notice letter, it has 45 days to decide whether to file a patent infringement lawsuit in federal district court. If it sues within 45 days, the FDA cannot grant effective approval of the ANDA until the earlier of: (a) the expiration of the 30-month stay, (b) a court decision finding the patents invalid or not infringed, or (c) a settlement permitting the generic to enter the market. [6]
Brand manufacturers sue approximately 75% of the time. [7] The 25% who do not sue have typically made a commercial or legal judgment that the patent cannot survive litigation and that filing suit would simply accelerate a loss while providing the generic manufacturer with a cleaner litigation posture. A decision not to sue is itself informative intelligence — if the brand passes on the 45-day window, the generic may be able to obtain a declaratory judgment that the patent is invalid, clearing the path to earlier launch.
The 30-month stay is not a litigation deadline. Patent cases in the District of Delaware and the District of New Jersey — where the overwhelming majority of ANDA cases are filed — routinely take longer than 30 months to reach trial. Many cases settle. The stay creates a defined floor: if the case is not resolved within 30 months and the ANDA is otherwise approvable, the FDA grants final approval regardless of whether litigation continues.
For the generic manufacturer, the 30-month stay period is not dead time. It is the period in which you must:
Complete any remaining ANDA review cycles and respond to FDA deficiency letters
Prepare manufacturing scale-up and commercial launch readiness
Secure supply chain agreements for active pharmaceutical ingredient (API) and finished dosage form
Monitor the litigation for early resolution opportunities
Tentative approval — granted when the ANDA is scientifically approvable but the 30-month stay has not expired — is a milestone that generic teams often underestimate. A tentative approval letter tells you that the FDA’s technical review is complete and that you will receive final approval when the stay expires or the litigation resolves. For a same-day filer competing for shared exclusivity, tentative approval also has implications for the failure-to-market forfeiture trigger, discussed below.
4. The 30-Month Period: Using the Stay Productively
Thirty months is not much time to litigate a complex pharmaceutical patent case to judgment. Most ANDA cases settle, and many of the settlements that do occur happen in the second half of the stay period when both parties have a clearer view of how trial will go and what the business economics of settlement look like.
4.1 Claim Construction and Early Dispositive Motions
Claim construction — the court’s determination of the meaning of patent claim terms — is the pivot on which most ANDA cases turn. If the court adopts the generic manufacturer’s construction of key claim terms, the non-infringement argument typically becomes easier to sustain. If the court adopts the brand’s construction, the generic may need to rely more heavily on invalidity.
Generic manufacturers who have invested in strong prior art searches frequently file early motions for summary judgment of invalidity. Courts in the District of Delaware have been willing to grant these motions when the prior art is clear and the brand’s secondary considerations of non-obviousness (commercial success, long-felt need, failure of others) are weak. A successful summary judgment motion can resolve the case before the 30-month stay expires, granting the generic early commercial launch rights.
The invalidity arguments available include anticipation (a single prior art reference that discloses every element of the claimed invention), obviousness (the claimed invention would have been obvious to a person of ordinary skill at the time of filing), and the utility requirements of written description and enablement. Obviousness is the most frequently litigated ground in formulation patent cases and has produced the most generic-favorable outcomes. For process patents, prior use defenses under 35 U.S.C. § 273 can be relevant if the generic manufacturer or its supplier was using the claimed process before the brand’s priority date.
4.2 Inter Partes Review: The Parallel Track
The America Invents Act of 2011 created inter partes review (IPR) at the Patent Trial and Appeal Board (PTAB), and generic manufacturers have used IPR aggressively as a parallel challenge to Orange Book-listed patents. An IPR petition filed in coordination with an ANDA Paragraph IV challenge can create multiple points of pressure on the brand — district court litigation running alongside a PTAB proceeding on overlapping patents.
IPR has produced a higher rate of patent invalidation than district court litigation for many categories of pharmaceutical patents, particularly those relying on obviousness grounds. The PTAB applies a “preponderance of the evidence” standard for invalidity, which is more favorable to challengers than the “clear and convincing evidence” standard applied in district court. For strong prior art-based invalidity arguments, IPR can be a faster and less expensive path to invalidation than full trial.
The strategic interaction between IPR and ANDA litigation is complex. A PTAB final written decision finding a patent invalid has estoppel consequences in district court, but the timing of PTAB proceedings relative to ANDA litigation schedules requires careful management. District courts have discretion to stay ANDA litigation pending PTAB review — or to proceed on parallel tracks. The decision of whether to file an IPR petition, and when, requires coordinated analysis by the district court litigation team and the PTAB counsel.
One caution: filing an IPR petition after the one-year window from service of a district court complaint (the statutory IPR filing deadline) bars the IPR petition entirely. If IPR is part of the strategy, the petition must be filed within one year of the brand’s infringement complaint — which is typically filed within weeks of the 45-day window after the notice letter.
5. Winning the 180 Days: What Exclusivity Actually Covers
The 180-day exclusivity period blocks the FDA from granting effective approval to any other ANDA applicant for the same drug product. It does not block approval of an NDA-authorized generic — a brand manufacturer who launches a generic version of their own drug through their NDA can do so during the 180-day period without triggering the exclusivity bar.
5.1 The Authorized Generic Problem
The authorized generic (AG) is the most potent competitive response available to a brand manufacturer facing generic entry. An AG is a generic version of the brand drug sold under the brand’s NDA approval but labeled as a generic. Because it is approved under the NDA rather than an ANDA, it is not blocked by the 180-day exclusivity that shields the first-filer generic from other ANDA competitors.
The FTC has identified “no-AG commitments” as a significant component of pay-for-delay settlements. In the Endo/Lidoderm litigation, the FTC alleged that Endo paid the first-filing generic companies by committing not to launch an authorized generic during the 180-day exclusivity period — a form of reverse payment that preserved the value of the exclusivity window. [8] This structure came under antitrust scrutiny post-Actavis because the no-AG commitment has the same effect as a cash payment: it increases the value of the settlement to the generic by eliminating the AG competitive threat.
A first-filer that does not receive a no-AG commitment must model the revenue impact of AG competition during the exclusivity window. Historical data shows that an authorized generic typically captures 40% to 60% of the generic market within weeks of launch. The 180-day exclusivity still has value in this scenario — no other ANDA generics enter the market — but the duopoly between the first-filer generic and the authorized generic produces substantially lower prices and lower revenue than a true monopoly exclusivity period would.
5.2 Triggering the Clock: Commercial Marketing
The 180-day exclusivity clock starts running on the date of first commercial marketing by the first-filer generic. [9] It does not start on the date of FDA approval. A first-filer who receives final approval but delays commercial launch is extending the period during which subsequent ANDA applicants cannot receive effective approval — which is precisely the “parking” behavior that Congress targeted with the MMA’s forfeiture provisions.
Once commercial marketing begins, the 180-day clock runs regardless of what else happens. The first-filer must notify the FDA of the date of first commercial marketing within 30 days. If the notification is not timely, the FDA deems the first commercial marketing date to be the date of ANDA approval — a constructive notice provision that can dramatically compress the effective exclusivity window.
The practical implication for commercial operations: launch readiness cannot be a parallel track to regulatory and legal work. It must be integrated from the beginning. Inventory build, distribution agreements, wholesaler access, and commercial launch operations need to be ready on the day that final approval or 30-month stay expiration creates the legal right to sell. Every day between final approval and first commercial sale is a day that the FDA’s approval block on subsequent ANDAs remains in place without generating revenue for the first filer — and depending on forfeiture timing, a day that competitors may use to trigger forfeiture.
6. The Six Forfeiture Triggers: Know Them Before You File
The MMA added six grounds for forfeiture of 180-day exclusivity. A first filer who forfeits exclusivity loses it entirely — the FDA grants effective approval to subsequent ANDA applicants without any exclusivity block. Understanding each trigger is not optional strategy; it is risk management for an asset worth hundreds of millions of dollars.
6.1 Failure to Market
The most commonly litigated forfeiture trigger is the failure to market provision. Under Section 505(j)(5)(D)(i)(I) of the FD&C Act, the first filer forfeits exclusivity if it fails to commercially market its drug by the later of: (a) 75 days after the date on which final approval is made effective, or (b) 30 months after the ANDA’s submission date. [10]
There is a secondary trigger. The FDA can also find forfeiture if a subsequent ANDA applicant obtains tentative approval and then: the court enters a final judgment of patent invalidity or non-infringement against the first filer’s challenged patent, or the first filer’s 30-month stay expires — and 75 days pass without commercial marketing.
The FDA clarified in its July 2018 letter that a subsequent applicant’s tentative approval can occur at any time prior to or after the triggering event — it does not need to precede the event. [11] This interpretation expands the circumstances under which a competitor can trigger the first filer’s forfeiture.
6.2 Withdrawal of Application
A first filer who voluntarily withdraws its ANDA forfeits exclusivity. This is rarely done strategically — withdrawal signals that the ANDA cannot be approved — but it can be triggered by manufacturing failures, supply chain problems with the API, or commercial decisions that make the product economically unviable.
6.3 Amendment of Certification
If the first filer amends its Paragraph IV certification to a later certification — for example, converting from a Paragraph IV certification to a Paragraph III certification acknowledging patent validity and agreeing to wait for patent expiration — it forfeits exclusivity. This amendment-based forfeiture prevents a generic manufacturer from using the Paragraph IV filing solely to secure first-filer status and then effectively abandoning the challenge.
6.4 Failure to Obtain Tentative Approval
If the first filer fails to obtain tentative approval within 30 months of ANDA submission and that failure is caused by the first filer’s conduct, the exclusivity is forfeited. This provision targets manufacturers who delay the FDA review process — by providing inadequate responses to Complete Response Letters, by failing to address manufacturing deficiencies, or by holding the ANDA in incomplete status for extended periods.
6.5 Agreement with Brand or Other Applicants
An agreement between the first filer and the brand manufacturer, the NDA holder, or another ANDA applicant that causes a court to determine that antitrust laws have been violated triggers forfeiture. This is the provision that connects pay-for-delay antitrust liability to exclusivity forfeiture. A finding by a court that a reverse payment settlement violated the antitrust laws — applying the Actavis rule-of-reason standard — can strip the first filer of the exclusivity it received as consideration for staying out of the market.
6.6 Expiration of All Challenged Patents
If every patent that the first filer challenged under Paragraph IV expires before the exclusivity window runs, forfeiture follows. The exclusivity exists to reward patent challenges — once the patents have expired, there is nothing to reward and no competitive barrier to justify the exclusivity block.
“In general, most generic companies estimate that 60% to 80% of their potential profit for any one product is made during this exclusivity period.” — Coughlin & Dede, Hatch-Waxman Game-Playing from a Generic Manufacturer Perspective, 25 Biotech. L. Rep. 525 (2006). [1]
7. The Antitrust Constraint: Life After FTC v. Actavis
Before June 17, 2013, most circuits applied a “scope of the patent” test to reverse payment settlements: as long as the generic’s agreed entry date did not extend beyond the patent’s expiration, the settlement was presumptively legal. The FTC v. Actavis decision by the Supreme Court destroyed that framework. [12]
7.1 The Rule-of-Reason Standard
Actavis held that reverse payment settlements — where the brand pays the generic to delay market entry — are subject to antitrust scrutiny under the rule of reason. The Court did not rule that all such settlements are per se illegal. Instead, it held that a large, unexplained payment from brand to generic is itself evidence of anticompetitive harm, because a company does not pay to resolve a lawsuit it expects to win on the merits.
The rule-of-reason standard requires courts to weigh the anticompetitive effects of delayed generic entry against any procompetitive justifications for the payment. In the Actavis case itself — the AndroGel litigation involving Solvay Pharmaceuticals — the brand had paid $19-30 million annually to Actavis for nine years, plus additional payments to other generic challengers, in exchange for agreements not to enter the market until 65 months before Solvay’s patent expired. [13] The Supreme Court found that this payment structure warranted antitrust examination.
7.2 What Settlements Are Still Permissible
The Actavis decision does not prohibit all branded-generic settlements in Paragraph IV litigation. It specifically noted that settlements permitting the generic to enter before patent expiration, without payment to stay out prior to that agreed date, do not present the same antitrust concerns. Settlements structured around an agreed entry date — where the brand grants a license to the generic and both parties avoid the uncertainty of trial — are permissible as long as the payment flowing from brand to generic is proportionate to legitimate services rendered and is not simply compensation for delayed entry.
The practical effect has been a restructuring of settlement economics. Cash payments from brand to generic, which were the original currency of pay-for-delay, have been partially replaced by no-AG commitments (the FTC’s targeted equivalent), co-promotion agreements, API supply arrangements, and other commercial terms whose valuation is more ambiguous and harder to quantify as “large and unexplained.” Each of these non-cash compensation structures carries its own antitrust risk profile and requires careful valuation analysis before execution.
7.3 The No-AG Commitment as Reverse Payment
The FTC has consistently taken the position that a no-AG commitment is a form of reverse payment because it eliminates the authorized generic competitive threat during the 180-day exclusivity window, thereby increasing the value of the exclusivity to the first-filer generic. In the Endo/Lidoderm case, the FTC alleged that Endo’s no-AG commitment to Watson and Allergan was worth hundreds of millions of dollars in economic value, making it functionally equivalent to a cash payment. [8]
Generic manufacturers negotiating settlement terms need to understand that the no-AG commitment is not a safe harbor from Actavis. If the total value transferred from brand to generic — including the value of eliminating the AG — is “large and unexplained,” the settlement faces antitrust challenge. Structuring settlement around an early entry date, without a no-AG commitment, is the path most likely to survive scrutiny.
8. The Section viii Skinny Label: Opportunity and Risk After GSK v. Teva
Section viii of the Hatch-Waxman Act allows a generic manufacturer to avoid challenging a method of use patent by carving out the patented indication from its label. The generic markets its product only for non-patented uses and files a Section viii statement instead of a Paragraph IV certification for the method of use patent. The theory is that the generic cannot infringe a method patent for an indication it does not claim.
8.1 What the GSK v. Teva Decision Changed
The Federal Circuit’s August 5, 2021 decision in GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc. confirmed that Teva had induced infringement of GSK’s patent on a specific method of use for Coreg (carvedilol), even during a period when Teva’s label carried out the patented heart failure indication. [14]
The facts matter here. Teva launched with a skinny label in 2007, carving out the CHF indication covered by GSK’s patent. But Teva’s marketing materials, catalogs, and press releases — and testimony from Teva’s own witnesses — showed that Teva actively encouraged carvedilol sales for the carved-out CHF indication. The court held that this conduct, not the skinny label itself, established induced infringement.
The Federal Circuit stressed that its holding was fact-specific: a skinny label standing alone does not automatically generate inducement liability, and an AB-rated generic (interchangeable with the branded product) is not itself evidence of intent to induce infringement. [15] But the decision drew a clear line: if your commercial conduct — marketing materials, sales force messaging, physician communications — directs prescribers to use the generic for a carved-out indication, the skinny label provides no protection.
8.2 Practical Risk Management for Section viii Strategies
The Section viii carve-out remains a viable strategy, but it requires rigorous control of commercial execution post-launch. Generic manufacturers who deploy a skinny label must:
Train sales forces explicitly to not promote for carved-out indications
Review all marketing materials, press releases, and public communications for references that could be construed as directing use for patented methods
Document the scope of the carve-out and the product’s approved indications in all commercial contracts
Monitor physician prescribing patterns to understand what proportion of use is for carved-out indications
The risk does not disappear with careful commercial management — physicians who receive no information from the generic manufacturer may still prescribe the generic for the carved-out indication because the drug is listed as therapeutically equivalent to the brand. But eliminating the brand’s ability to point to the generic manufacturer’s own conduct as evidence of intent to induce is a substantial reduction in exposure.
For drugs where the primary commercial indication is covered by a method of use patent and the secondary indications are small markets, the Section viii strategy may not generate sufficient revenue to justify the litigation risk. Patent-by-patent analysis of the commercial value of carved-out versus patented indications should drive the Section viii versus Paragraph IV decision, not a blanket preference for one strategy over the other.
9. The FTC’s Orange Book Campaign: New Opportunities for Generic Challengers
The FTC’s 2023-2024 challenge campaign against Orange Book patent listings has created a category of offensive opportunity that generic manufacturers have historically underutilized.
9.1 The Scale of the Challenge
In November 2023, the FTC challenged more than 100 patent listings in the Orange Book using the FDA’s regulatory dispute process, asserting that the patents were improperly listed under the Hatch-Waxman listing criteria. In April 2024, it followed with challenges to over 300 additional listings, some of which were aimed at facilitating generic alternatives to drugs like Ozempic (semaglutide) and Victoza (liraglutide). [16]
The FTC’s theory was straightforward: the Hatch-Waxman Act requires brand manufacturers to list only patents that claim the approved drug product — its active ingredient, formulation, or a method of using it. Device patents covering an autoinjector or inhaler used to deliver the drug, and patents covering packaging or ancillary delivery mechanisms, do not meet the statutory listing criteria. By listing these patents, brand manufacturers trigger 30-month stays on ANDA applications that challenge them, even though the patents may not be validly listable under the Act.
FTC Chair Lina Khan’s statement in April 2024 was direct: “By filing bogus patent listings, pharma companies block competition and inflate the cost of prescription drugs, forcing Americans to pay sky-high prices for medicines they rely on.” [17] Most companies that received challenges did not voluntarily delist their patents. Some did.
9.2 The Generic Manufacturer’s Offensive Move
Generic manufacturers can use the same regulatory dispute process independently of the FTC. Under 21 C.F.R. § 314.53, any person — including an ANDA applicant — can submit a patent delisting request to the FDA asserting that a listed patent does not meet the statutory listing criteria. If the FDA agrees and delists the patent, the automatic 30-month stay triggered by a Paragraph IV certification on that patent falls away.
This creates a two-front strategy: file the ANDA with a Paragraph IV certification to preserve first-filer status, and simultaneously submit a patent delisting request for patents that appear to be improperly listed. If the delisting succeeds, you eliminate a stay that would otherwise delay final approval by up to 30 months. If the delisting fails, you are in the same position as a standard Paragraph IV litigation track.
The FTC’s campaign also surfaced a broader intelligence opportunity. The commission’s public identification of patents it believes are improperly listed — including device patents for autoinjectors listed against GLP-1 receptor agonists — provides generic manufacturers with a curated list of potentially vulnerable Orange Book listings that may not have been obvious targets from a standard patent analysis perspective. Teams monitoring DrugPatentWatch and FTC enforcement activity can use this information to identify products where the patent barrier may be softer than the Orange Book listing count suggests.
10. Competitive Intelligence: Reading the Paragraph IV Filing Landscape
The FDA publishes a biweekly list of ANDA applications containing Paragraph IV certifications. This is public information, but interpreting it quickly requires structured data access and the ability to cross-reference filings against Orange Book patent listings, litigation records, and brand sales data. That is precisely what platforms like DrugPatentWatch are built to do.
10.1 Monitoring for First-Filer Availability
When a brand drug approaches a period of patent vulnerability — compound patent expiration within a five-to-seven year window, formulation patents with questionable validity, method of use patents with narrowly claimed indications — the FDA’s biweekly Paragraph IV listing provides the earliest public signal of generic manufacturer interest. A single Paragraph IV filing tells you that at least one competitor has completed bioequivalence testing, assembled an ANDA, and made a legal judgment that the patents are challengeable.
If same-day filings have already occurred, the window for shared first-filer status on those patents is closed. But the landscape is not static. Brand manufacturers frequently list additional patents as they prosecute continuation applications, and a new Orange Book listing can re-open the Paragraph IV race. Monitoring new Orange Book listings for target drugs — and assessing whether those new patents are valid, infringed, and properly listed — is ongoing intelligence work, not a one-time analysis.
10.2 Reading Litigation Outcomes for Prior Art Intelligence
Every ANDA case that goes to trial produces a public record of the prior art arguments that were made, the claim constructions that were adopted, and the ultimate validity and infringement findings. For a generic manufacturer building a Paragraph IV position on a drug that has already been litigated, that record is invaluable. It tells you which prior art references the court found credible, which invalidity theories failed and why, and what claim constructions the brand will argue in the next round.
A patent that has survived one validity challenge at trial is not unchallengeable — courts in different districts may adopt different claim constructions, and new prior art that was not in front of the first court may produce a different result. But the prior litigation record tells you what arguments you cannot simply repeat and what new theory you need to develop.
DrugPatentWatch aggregates this litigation history alongside current patent and ANDA status data, allowing teams to build a complete picture of a drug’s Paragraph IV history without manually searching multiple court databases and FDA public records. For a business development team evaluating whether to enter a specific generic drug program, that integrated view is the foundation of any serious commercial assessment.
11. The Competitive Generic Therapy (CGT) Designation: A Separate Exclusivity Path
The FDA Reauthorization Act of 2017 created the Competitive Generic Therapy (CGT) designation for drug products that have no approved generic and are in shortage or have inadequate competition. CGT designation carries a 180-day exclusivity period of its own — analogous to first-filer Paragraph IV exclusivity — for the first ANDA applicant to receive approval for a designated CGT product.
The CGT pathway does not require a Paragraph IV certification. The brand drug typically has limited or no unexpired Orange Book patents, making the CGT product a Paragraph I, II, or III ANDA rather than a Paragraph IV challenge. The exclusivity is granted based on first-to-approval, not first-to-file with a patent challenge.
The CGT forfeiture rules are similar to the Paragraph IV forfeiture provisions: failure to commercially market within 75 days of final approval triggers forfeiture. FDA approval letters for CGT products explicitly notify the applicant of this forfeiture risk and require notification of the date of first commercial marketing. [18]
For generic manufacturers whose pipeline focuses on drugs with limited patent exposure rather than blockbuster patent challenges, CGT designation can deliver a comparable period of exclusivity with substantially less litigation cost. The strategic question is whether the drugs qualifying for CGT designation generate sufficient revenue during the 180-day window to justify the development investment — and whether the shortage or inadequate competition status that triggered CGT designation reflects a permanent commercial opportunity or a temporary market condition that competitors will quickly correct.
12. The BLOCKING Act and Legislative Risk to First-Filer Economics
Congressional interest in reforming the 180-day exclusivity provisions has been persistent. The BLOCKING Act — formally the Bringing Low-cost Options for Americans Now Act — was introduced to address “parking” of exclusivity by first filers who obtain ANDA approval but delay commercial launch for extended periods, thereby blocking all subsequent ANDA approvals without generating any generic price competition.
The FDA’s FY 2023 Legislative Proposals included a related provision: amending the exclusivity statute so that the FDA could approve subsequent ANDAs unless and until a first applicant begins commercial marketing, at which point approval of subsequent applications would be blocked for 180 days from the actual launch date. [19] Under current law, the 30-month stay on subsequent approvals can run concurrently with or independently of the 180-day exclusivity, creating scenarios where the total period of blocked competition extends well beyond 180 days.
Neither the BLOCKING Act nor the FDA’s legislative proposal has been enacted as of mid-2025. But the reform pressure reflects a real policy concern: first-filer exclusivity was designed to incentivize patent challenges, not to create an extended exclusivity period for manufacturers who file, obtain approval, and then delay launch to maximize the remaining brand market share before generic entry. Generic manufacturers who build business plans around extended approval-to-launch delays are building on legislative risk that Congress and the FDA have repeatedly flagged as a reform target.
The risk management answer is straightforward: launch as promptly as possible after final approval. The 75-day failure-to-market window that triggers forfeiture is a minimum; commercial and legal logic both support launching at or before that deadline. The revenue concentration in the 180-day window means that every week of launch delay is a week of peak pricing given away — and a week closer to forfeiture or legislative reform that cuts the exclusivity short.
13. Evergreening Defense: Recognizing and Challenging Patent Stacking
Patent stacking — the practice of listing multiple patents with overlapping claims on successive formulations, salts, polymorphs, dosing regimens, and delivery mechanisms — is the brand industry’s primary defense against generic entry. Understanding how to evaluate a stacked patent portfolio is essential for any Paragraph IV strategy targeting a drug that has been commercially successful for a decade or more.
13.1 The Lifecycle of a Stacked Patent Portfolio
A brand manufacturer typically lists its compound patent first. As the compound patent approaches expiration, it prosecutes and lists formulation patents claiming the approved dosage form. Then it lists patents on specific salt forms, polymorphic crystal structures, and methods of producing them. Later it lists patents on specific dosing regimens that have been validated in clinical trials. Finally, it lists device patents if the drug is delivered via an inhaler, autoinjector, or prefilled syringe.
Purdue Pharma’s OxyContin strategy is the canonical example. After the original oxycodone compound patent expired, Purdue listed formulation patents covering the abuse-deterrent extended-release matrix, creating a new Orange Book barrier that required generics to be bioequivalent to the reformulated product — a substantially harder target than the original formulation. [20] The abuse-deterrent formulation patents extended effective market exclusivity for years beyond the compound patent’s expiration date.
The FTC’s 2023-2024 Orange Book challenge campaign specifically targeted this kind of patent stacking in the device patent context. Device patents for autoinjectors — the delivery mechanism for GLP-1 receptor agonists like semaglutide — do not meet the Hatch-Waxman listing criteria for drug product patents if they claim only the device and not the drug itself. By challenging these listings, the FTC was attempting to strip away a layer of the patent stack that it argued had no legitimate basis under the statute.
13.2 The Generic Manufacturer’s Response to Stacking
Facing a stacked patent portfolio, a generic manufacturer has two options that can be combined: challenge each listed patent under Paragraph IV and build invalidity and non-infringement arguments for each, or design the generic product to avoid the formulation, polymorph, and device patent claims while achieving bioequivalence to the RLD.
The design-around approach is more powerful than it looks. If the brand’s extended-release formulation patent claims a specific polymer matrix, and the generic can achieve equivalent release kinetics using a different polymer system that does not fall within the claim language, the generic avoids the patent entirely — no litigation needed on that patent, no 30-month stay on that certification. The challenge is that design-around approaches require early-stage formulation development decisions that constrain subsequent CMC work. Teams that integrate IP strategy into formulation development from the beginning of the ANDA program have a material advantage over teams that treat patent analysis as a post-formulation exercise.
For polymorph patents, the analysis centers on whether the generic’s API — typically sourced from an API manufacturer, often in India or China — uses a crystalline form that falls within the brand’s polymorph patent claims. API sourcing decisions have direct IP consequences that many generic manufacturers do not fully appreciate until they are already in litigation.
14. Building the Team: Legal, Regulatory, and Commercial Integration
Paragraph IV strategy fails most often not because the legal theory is wrong, but because the legal, regulatory, and commercial tracks are not integrated. The attorneys building the invalidity case do not talk to the regulatory team managing the FDA review cycle. The regulatory team does not talk to the commercial team planning launch readiness. The commercial team does not understand the forfeiture triggers that determine when launch must occur.
14.1 The Integrated Timeline
A functional Paragraph IV program runs on a single integrated timeline that tracks: bioequivalence study completion dates, ANDA submission target, FDA review cycle milestones (Day 74 letter, Complete Response Letter, tentative approval, final approval), litigation milestones (claim construction, fact discovery close, expert reports, dispositive motions, trial date), forfeiture trigger dates (75-day marketing deadline from tentative approval under certain scenarios, 30-month submission anniversary), and commercial launch readiness milestones (inventory build, distribution agreements, trade activation).
Each of these tracks has dependencies that cross functional lines. The FDA’s response to a Complete Response Letter on a chemistry question affects the timeline to tentative approval, which affects the forfeiture trigger analysis, which affects the commercial team’s inventory build planning. Treating these as independent workstreams creates gaps that competitors exploit.
14.2 Outside Counsel Selection
ANDA patent litigation concentrates in the District of Delaware and the District of New Jersey, with some volume in the Northern District of Illinois and the Eastern District of Texas. The bench in Delaware — where most major pharmaceutical companies are incorporated and where brand manufacturers routinely choose to sue — has decades of experience with Hatch-Waxman litigation. Judges like Colm Connolly, Maryellen Noreika, and their colleagues have developed specific procedural preferences for ANDA cases that make local experience genuinely valuable, not just a billing line item.
The lead outside counsel team needs demonstrated experience trying ANDA cases to judgment, not just settling them. A team that has only settled its cases has not actually tested its invalidity arguments under cross-examination, and the deference that brand manufacturers give to generic counsel in settlement negotiations is partially a function of whether the generic’s legal team has a credible trial record.
14.3 Expert Witnesses
Pharmaceutical patent trials are expert-intensive. The key expert roles are a person of ordinary skill in the art (POSITA) on formulation chemistry, a pharmacologist or pharmacokineticist on bioequivalence and method of use issues, a patent agent expert on prosecution history and claim interpretation, and a damages expert. For invalidity cases, the formulation chemistry expert’s ability to explain prior art to a non-specialist judge and jury determines the outcome as much as the prior art itself does.
Expert witness identification and retention should happen early — ideally before the notice letter is finalized, so that the invalidity arguments in the letter reflect the expert’s actual views on the science. Experts who are surprised by arguments they are asked to defend at trial rarely make credible witnesses.
15. The Data Layer: How Intelligence Tools Shape Strategy
The information advantage in Paragraph IV strategy has become a competitive differentiator in its own right. A generic manufacturer that tracks Orange Book updates daily, monitors ANDA filing activity weekly, and has litigation outcome data integrated into patent analysis is working with a materially different decision-making foundation than a team relying on periodic manual searches.
DrugPatentWatch provides exactly this kind of integrated intelligence infrastructure for the generic industry. Its database combines Orange Book patent listings, ANDA application history (including Paragraph IV filing dates, same-day filer identification, and tentative and final approval dates), litigation records, and patent expiration timelines into a single searchable platform. For a business development team evaluating a pipeline of 20 potential generic targets, the ability to quickly identify first-filer availability, active litigation status, and upcoming patent expiration dates — without manually cross-referencing multiple government databases — directly compresses the timeline from target identification to program initiation decision.
The platform’s value is particularly acute in the same-day filer scenario. Because the FDA’s biweekly publication of Paragraph IV filings lags the actual submission dates, a team relying solely on the public disclosure may not learn that a competitor filed on the same day until weeks after the fact. DrugPatentWatch’s real-time patent monitoring can surface signals — changes in brand patent prosecution activity, new continuation applications, NDA supplement filings that may trigger new Orange Book listings — that allow teams to anticipate patent events before they become public, reducing the risk of a surprise same-day filing situation.
For litigation strategy, the platform’s integration of prior Paragraph IV litigation outcomes by drug and by patent provides the prior art intelligence discussed above: which arguments have been tried, what prior art was before the court, and what outcomes suggest about the vulnerability or durability of the surviving patent claims.
16. Putting the Playbook Together: A Program Architecture
The Paragraph IV program from target selection to commercial launch follows a predictable architecture, though the timelines vary significantly by drug complexity.
16.1 Phase 1: Target Assessment (Months 1-3)
Target selection incorporates brand sales data, remaining patent life, Orange Book patent count, existing ANDA filing history (is first-filer status still available?), and preliminary IP strength assessment. DrugPatentWatch and IQVIA sales data anchor the commercial analysis. Preliminary patent review by IP counsel anchors the legal risk assessment. The output is a go/no-go decision on initiating a full formulation development program.
16.2 Phase 2: Formulation Development and Bioequivalence (Months 3-30)
Formulation development proceeds with IP strategy as a co-input — not as an afterthought. The target product profile specifies not only the required bioequivalence performance but also the IP constraints: formulation components and configurations to avoid, API polymorph specifications, and manufacturing process parameters that have Paragraph IV implications. Bioequivalence study design is reviewed for consistency with the ANDA regulatory requirements and for any formulation features that might create labeling issues relevant to Section viii analysis.
16.3 Phase 3: Prior Art Search and Legal Theory Development (Months 6-18)
The comprehensive prior art search runs in parallel with formulation development, not sequentially after it. The invalidity arguments must be developed early enough to inform both the ANDA formulation strategy (design-around opportunities) and the notice letter content. The FTO analysis is updated as the formulation finalizes, ensuring that the actual product to be manufactured matches the non-infringement positions being argued.
16.4 Phase 4: ANDA Assembly and Submission (Months 24-36)
ANDA assembly is a project management exercise that requires coordination between regulatory, CMC, and legal teams. The target submission date is determined by the competitive filing timeline — when is first-filer status most at risk? The notice letter is drafted, reviewed by litigation counsel, and finalized for delivery within 20 days of the FDA’s acknowledgment of the ANDA.
16.5 Phase 5: Litigation and Regulatory Review (Months 36-72+)
The 30-month stay period runs concurrently with the FDA’s substantive review of the ANDA. FDA deficiency letters must be answered promptly to maintain tentative approval timelines. Litigation proceeds on the district court schedule, with IPR petitions filed within one year of the brand’s infringement complaint if IPR is part of the strategy. Settlement analysis is conducted continuously as the case develops, with full Actavis antitrust analysis for any settlement term involving payment or commercial benefit flowing from brand to generic.
16.6 Phase 6: Launch Preparation and Execution (Final 6-9 months before expected approval)
Commercial launch preparation is integrated into the program from Phase 1, but the final six to nine months require intensive execution. Inventory build, distribution network activation, pricing and contracting with pharmacy benefit managers and wholesalers, and trade marketing must all be ready at the moment final approval is granted. The 75-day failure-to-market forfeiture window is the clock — missing it forfeits the exclusivity entirely.
17. Common Mistakes That Cost Generic Manufacturers the Prize
Four recurring mistakes account for most lost exclusivity positions in Paragraph IV programs:
Filing without a completed bioequivalence package. Teams who file a preliminary ANDA to claim first-filer status and then plan to supplement with bioequivalence data risk a “not substantially complete” determination from the FDA. The filing date resets to the date of the completed application, potentially ceding first-filer status to a competitor who filed a complete package even slightly later.
Inadequate notice letters. Courts have held that a notice letter failing to provide a “detailed statement” of the factual and legal basis for invalidity or non-infringement is deficient, which can result in dismissal of the brand’s infringement suit — and a loss of the 30-month stay. While dismissal sounds like a generic win, it actually eliminates the litigation that was providing cover during the stay period, leaving the generic without a procedural tool to delay competitors.
Ignoring the forfeiture clock after tentative approval. Teams who obtain tentative approval and then shift focus to litigation, treating the regulatory track as complete, miss the failure-to-market forfeiture triggers that start running based on the tentative approval date under certain competitor-triggered circumstances. Regulatory and legal monitoring must continue through tentative approval and into the commercial launch window.
Treating the notice letter as a compliance exercise rather than a litigation document. The arguments in the notice letter anchor the litigation. A notice letter that makes broad, conclusory invalidity assertions without specific prior art citations gives the brand no actionable infringement target and forces subsequent litigation to operate from a weaker starting position. The notice letter is the opening brief of the Paragraph IV case — invest in it accordingly.
Key Takeaways
First-filer status for 180-day exclusivity is determined by the ANDA submission date for a substantially complete application with a Paragraph IV certification. Same-day filers share the exclusivity. The race is real, and it begins years before the ANDA is filed — when the formulation development program starts.
Brand manufacturers sue approximately 75% of the time within the 45-day window after receiving the Paragraph IV notice letter. The notice letter must be a litigation-quality document, not a compliance form.
Six forfeiture triggers under the MMA can strip exclusivity from a first filer: failure to market within 75 days of final approval (under specified conditions), withdrawal of application, amendment of certification, failure to obtain tentative approval, entry into an anticompetitive agreement, and expiration of all challenged patents. Know the timelines and manage them actively.
FTC v. Actavis (2013) subjects reverse payment settlements to antitrust scrutiny under a rule-of-reason standard. Large, unexplained payments — including no-AG commitments — from brand to generic in settlement of Paragraph IV litigation carry antitrust risk. Settlements structured around agreed entry dates without payment are substantially safer.
GSK v. Teva (2021) established that a Section viii skinny label does not automatically protect a generic manufacturer from induced infringement liability if the manufacturer’s commercial conduct directs physicians to use the product for a carved-out patented indication. Skinny label strategies require rigorous commercial discipline post-launch.
The FTC’s 2023-2024 Orange Book challenge campaign identified over 400 patents as potentially improperly listed. Generic manufacturers can use the same regulatory dispute process to seek delisting of invalid Orange Book listings, potentially eliminating 30-month stays on their ANDAs.
Platforms like DrugPatentWatch provide integrated access to Orange Book patent data, ANDA filing history, litigation records, and patent expiration timelines — intelligence infrastructure that compresses target assessment timelines and surfaces competitive signals earlier than manual public database searches.
Commercial launch readiness must be integrated into the program from the beginning, not treated as a final-phase execution task. The 75-day failure-to-market window is the hard deadline; every week of delay after final approval is peak revenue abandoned.
Frequently Asked Questions
Q1: Can a generic manufacturer file a Paragraph IV certification on only some of the Orange Book-listed patents for a drug, rather than all of them?
Yes. The statute requires “a” Paragraph IV certification — singular — not certifications against every listed patent. A generic manufacturer can file Paragraph IV certifications for patents it believes are invalid or non-infringed, while filing Paragraph III certifications (agreeing to wait for patent expiration) for patents it does not wish to challenge. Critically, the first-filer status for 180-day exclusivity is established by the Paragraph IV certification that is filed first, even if that certification covers only a subset of the Orange Book patents. The first applicant does not need to file a Paragraph IV certification for the latest-expiring patent to qualify as a first applicant. However, Paragraph III certifications on remaining patents do constrain the effective launch date for the product — the generic cannot launch while any Paragraph III-certified patent remains in force unless the brand grants a license or the patent expires.
Q2: What happens to the 180-day exclusivity if the first filer loses the patent litigation at trial?
A first filer who loses in district court — where the court finds the patent valid and infringed — does not automatically lose its first-filer exclusivity status. The exclusivity remains as long as the first filer has not triggered a forfeiture condition. The litigation outcome, however, affects the commercial path. If the patent is found valid and infringed, the first filer cannot launch until the patent expires (absent a settlement granting a license), which may be years away. During that period, the failure-to-market forfeiture trigger can come into play if a subsequent ANDA applicant reaches tentative approval and the first filer fails to market within the applicable 75-day window calculated under the statute. The first filer’s strategic position post-adverse judgment shifts almost entirely to settlement discussions — seeking a license with an agreed entry date — or an appeal.
Q3: How does a generic manufacturer protect itself against authorized generic competition during the 180-day exclusivity window?
There is no statutory mechanism that prevents a brand manufacturer from launching an authorized generic during the 180-day exclusivity window. The 180-day exclusivity blocks the FDA from granting effective approval to other ANDA applicants — it does not block the brand from using its existing NDA approval to sell a generic version of its own drug. The only way to eliminate the authorized generic threat is through settlement terms that include a no-AG commitment, but those commitments carry post-Actavis antitrust risk and must be carefully structured to avoid constituting a “large and unexplained” reverse payment. The commercial modeling for any Paragraph IV program must include authorized generic competition scenarios, as authorized generics have historically captured a substantial portion of the generic market in the months following generic launch.
Q4: What is the strategic difference between filing an IPR petition at the PTAB and pursuing invalidity through ANDA litigation in district court?
IPR and district court litigation use different invalidity standards, apply different claim construction frameworks (though post-SAS Institute, PTAB applies the same Phillips claim construction standard as district courts), and operate on different timelines. IPR applies a preponderance of the evidence standard for invalidity, compared to the clear and convincing evidence standard in district court — which makes IPR somewhat more favorable for invalidity challengers on the merits. IPR is also generally faster than district court litigation, with a PTAB final written decision typically issued within 12 to 18 months of petition institution. However, IPR is limited to challenges based on prior art (patents and printed publications) under §§ 102 and 103; invalidity grounds like lack of written description, enablement, or prior use under § 273 are not available at the PTAB. District court litigation is broader but more expensive and slower. The combination of parallel IPR and district court proceedings creates multiple pressure points on the brand, but coordination between PTAB counsel and district court counsel is essential to manage estoppel consequences and strategic sequencing.
Q5: How does the same-day filing rule work when a generic manufacturer discovers a competitor filed on the same day?
Under the MMA, all ANDA applicants who submit substantially complete applications with Paragraph IV certifications on the same calendar day share the 180-day exclusivity. The FDA determines same-day filing status based on the date of receipt of a substantially complete application — not the date the application was submitted electronically or mailed. When multiple applicants qualify as same-day first filers, each of them individually holds first-filer status for exclusivity purposes, and the FDA cannot grant effective approval to any other ANDA applicant until one of the shared first filers triggers the 180-day clock by commencing commercial marketing. Crucially, a same-day first filer’s exclusivity is forfeited individually if it fails to meet its own forfeiture conditions — one co-first-filer’s forfeiture does not automatically cause the other co-first-filers to forfeit. This means that a generic manufacturer who discovers a same-day competitor must still manage its own forfeiture risk independently, and the commercial launch race between co-first-filers determines which company has the exclusive market position during the first months of the exclusivity window before the other co-first-filers launch.
References
Coughlin, D. F., & Dede, R. A. (2006). Hatch-Waxman game-playing from a generic manufacturer perspective. Biotechnology Law Report, 25(5), 525–536.
Association for Accessible Medicines. (2021). The U.S. generic & biosimilar medicines savings report. AAM. https://accessiblemeds.org
U.S. Food and Drug Administration. (2003). Guidance for industry: 180-day generic drug exclusivity under the Hatch-Waxman Amendments to the Federal Food, Drug, and Cosmetic Act. FDA Center for Drug Evaluation and Research. https://www.fda.gov
Medicare Prescription Drug, Improvement, and Modernization Act of 2003, Pub. L. No. 108-173, 117 Stat. 2066 (2003).
21 C.F.R. § 314.95 (2024). Notice of certification of invalidity or noninfringement of patent.
Drug Price Competition and Patent Term Restoration Act of 1984, Pub. L. No. 98-417, 98 Stat. 1585 (1984) (Hatch-Waxman Amendments).
Actavis Laboratories FL, Inc. v. United States, No. 2024-1029 (Fed. Cir. Mar. 2025) (expert testimony cited in decision).
Federal Trade Commission. (2013). FTC v. Endo Pharmaceuticals: Complaint. FTC. https://www.ftc.gov/terms/pay-delay
21 U.S.C. § 505(j)(5)(D)(i)(I) (2024). Forfeiture of exclusivity — failure to market.
U.S. Food and Drug Administration. (2018, July 13). Letter clarifying 180-day exclusivity forfeiture provisions. FDA CDER. Summarized in Cozen O’Connor, FDA’s Clarification of 180-Day Exclusivity Rules (Aug. 23, 2018). https://www.cozen.com
FTC v. Actavis, Inc., 570 U.S. 136 (2013).
Antitrust Law Blog. (2013, June). FTC v. Actavis: What does it mean for reverse-payment settlements? https://www.antitrustlawblog.com
GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc., No. 2018-1976, 7 F.4th 1320 (Fed. Cir. Aug. 5, 2021).
Jones Day. (2021, September). Court reinstates verdict of induced infringement. https://www.jonesday.com
Federal Trade Commission. (2024, April). FTC challenges over 300 Orange Book patent listings. FTC Press Release. https://www.ftc.gov
U.S. Food and Drug Administration. (2024). ANDA approval letter, ANDA 217671 (Halcinonide Topical Solution). FDA CDER. https://www.accessdata.fda.gov
Hyman, J. (2022, March 31). The good, the bad and the ugly: New FDA legislative proposal on 180-day exclusivity. The FDA Law Blog. https://www.thefdalawblog.com
DrugPatentWatch. (2026, March 18). The complete expert guide to FDA Orange Book and Purple Book patent research. https://www.drugpatentwatch.com/blog/drug-patent-research-expert-tips-for-using-the-fda-orange-and-purple-books/





















")




