The average cost to develop a new drug reached $2.23 billion in 2024, with an expected ROI of just 5.9%. A first-to-file Paragraph IV ANDA challenger on a $2 billion product, by contrast, can earn $300 million to $700 million in net present value from 180 days of exclusivity alone. For every dollar invested in IP analysis, returns can exceed ten dollars. This article explains how that asymmetry reshaped the entire competitive structure of the pharmaceutical industry.
What Is Paragraph IV Litigation and Why Does It Outcompete Traditional Drug Development?
Paragraph IV of the Hatch-Waxman Act allows a generic manufacturer to file an Abbreviated New Drug Application (ANDA) certifying that a brand drug’s listed patents are either invalid or not infringed. If the brand files a lawsuit within 45 days, the first generic filer earns 180 days of market exclusivity upon FDA approval. The financial returns from a successful first-to-file challenge routinely exceed the economics of developing a new molecular entity, which costs over $2 billion and fails roughly 90% of the time in clinical trials.
The Drug Price Competition and Patent Term Restoration Act of 1984, commonly called the Hatch-Waxman Act, created two distinct economies inside the pharmaceutical industry. The first economy, drug discovery, is the one taught in business schools and celebrated in earnings calls. It runs on clinical trials, regulatory filings, and a decade-plus of capital deployment before a dollar returns. The second economy, Paragraph IV litigation, operates on a different logic entirely: find a flawed patent, file fast, survive 30 months of litigation, and collect a statutory prize worth hundreds of millions of dollars with zero clinical risk.
Describing Paragraph IV as the ‘new R&D’ is not hyperbole. It is a precise description of where sophisticated capital in the generic pharmaceutical space now concentrates. The top five generic manufacturers, Teva, Viatris, Sandoz, Amneal, and Hikma, captured 58% of all 180-day exclusivity periods awarded between 2018 and 2023. Those exclusivity windows represent billions of dollars in above-commodity margins distributed to companies that discovered not a single new compound.
This article maps the mechanics, economics, litigation patterns, and competitive implications of a system that now drives more strategic decision-making in pharmaceuticals than most laboratory programs do. It draws on data from the FDA Orange Book, PTAB trial statistics, Lex Machina case counts, FTC enforcement records, and commercial patent intelligence platforms including DrugPatentWatch.
The Hatch-Waxman Architecture: How the 1984 Act Created a Litigation Economy
The ANDA Pathway: Bypassing Clinical Risk
Before 1984, a generic manufacturer seeking to market a copy of an approved drug had to conduct its own clinical trials demonstrating safety and efficacy. The regulatory burden was the same as for an original new drug application. That requirement made generic development expensive enough that branded companies maintained effective market monopolies well past their patent terms. Congress, observing both the consumer cost of delayed generic entry and the innovator companies’ complaint that patent terms eroded during the FDA review process, threaded a legislative needle with the Hatch-Waxman Act.
The Act created the ANDA pathway. A generic manufacturer can reference the brand’s original clinical data rather than generating new safety and efficacy trials, provided it demonstrates that its product is pharmaceutically equivalent (same active ingredient, dosage form, route of administration, and strength) and bioequivalent (similar rate and extent of absorption) to the reference listed drug. The FDA review is scientific and regulatory, not clinical. Development costs for an ANDA run in the low millions of dollars compared to the billions required for a new molecular entity.
The tradeoff was patent protection. Congress required brand manufacturers to list their relevant patents in the FDA’s Orange Book: Official Compendium of Approved Drug Products with Therapeutic Equivalence Evaluations. When a generic filer submits an ANDA, it must certify one of four positions with respect to each listed patent. A Paragraph I certification says the patent has not been filed; Paragraph II says the patent has expired; Paragraph III says the generic will not launch until the patent expires. Each of those positions avoids litigation. Paragraph IV is different.
What Paragraph IV Certification Means Legally
A Paragraph IV certification is a legal allegation. The generic filer asserts that each listed patent it cites under Paragraph IV is either invalid, unenforceable, or will not be infringed by the proposed generic product. Filing this certification constitutes an act of technical infringement under 35 U.S.C. Section 271(e)(2), the so-called artificial act of infringement provision. This statutory fiction allows courts to adjudicate patent disputes before any product enters the market.
The procedural trigger runs as follows. Within 20 days of FDA accepting the ANDA for filing, the generic manufacturer must notify the patent owner and NDA holder of its Paragraph IV certification, including a detailed statement of the factual and legal basis for its invalidity or non-infringement positions. The patent owner then has 45 days to file an infringement lawsuit. If it does, a 30-month automatic stay of FDA approval kicks in, preventing the agency from granting final approval to the ANDA during that period, unless a court rules on the patents first or the 30 months expires.
The brand’s incentive to sue is essentially automatic. Filing within 45 days costs the brand nothing except legal fees and activates a 30-month moratorium on generic entry. Not filing forfeits any chance of securing that delay. The system produces near-certain litigation on any commercially significant product, which is exactly what it was designed to do.
The 180-Day Exclusivity Incentive: Why Paragraph IV Is a Competition for the Prize
Congress needed a mechanism to incentivize generics to accept the litigation risk of filing Paragraph IV certifications rather than simply waiting for patents to expire. The solution was the 180-day exclusivity period. The first ANDA applicant, or group of applicants filing on the same day, to submit a Paragraph IV certification against a given patent earns a 180-day period of marketing exclusivity upon final FDA approval. During that window, no other ANDA for the same drug can receive final approval, giving the first filer a statutory duopoly (itself and the brand) before multi-generic commoditization begins.
The economics of that window are the engine of the entire Paragraph IV system. On a drug with $2 billion in annual sales, a generic priced at a 20 to 25 percent discount to the brand can generate $300 million to $500 million in revenue during its exclusive period, with gross margins that, absent a competing authorized generic, can reach 50 to 60 percent. Net present value estimates for first-to-file exclusivity on large primary-care products routinely run between $300 million and $700 million.
‘A generic manufacturer that identifies a credible invalidity argument on a brand product’s primary patent 18 months before competing generic filers, uses that intelligence to execute a first-to-file ANDA, and prevails in Paragraph IV litigation earns returns that can exceed $10 for every $1 invested in IP analysis.’DrugPatentWatch, ‘How Prescription Drug Prices Are Set,’ March 2026
That ratio — ten dollars returned per dollar of IP intelligence invested — frames why Paragraph IV now functions as a capital allocation category in its own right, separate from and often preferred over traditional generic formulation development or, for that matter, new drug discovery.
The Litigation Economics: Paragraph IV ROI vs. NME Development Costs
New Drug Development Costs in 2024: $2.23 Billion Per Asset
The Deloitte annual survey of pharmaceutical R&D returns, published in early 2025, put the average cost for a major pharmaceutical company to develop a drug in 2024 at $2.23 billion, up from $2.12 billion the previous year. That figure includes the costs of failed compounds allocated across the successful ones. The same cohort of 20 companies with the highest R&D budgets spent $7.7 billion on clinical trials for candidates that were ultimately terminated in 2024 alone.
Expected ROI on pharmaceutical R&D was 5.9% in 2024, an improvement from 4.1% in 2023, but still thin by the standards of most capital allocation decisions. For context, the S&P 500’s average annual return over the past decade exceeded 12%. The pharmaceutical R&D model delivers a return below inflation-adjusted cost of capital in most years.
The clinical failure rate explains the economics. Roughly 90% of drug candidates that enter Phase I clinical trials fail before reaching approval. The 10% that succeed must carry the burden of all the failures. That burden, spread across a portfolio, produces the $2.23 billion average cost per approved product. Small companies without diversified portfolios face failure costs that are not diluted by a pipeline of successes; they face binary outcomes on bets that kill them if they lose.
Paragraph IV Cost Structure: ANDA Preparation, Litigation Expenses, and IP Intelligence
The cost profile of a Paragraph IV strategy looks nothing like NME development. ANDA preparation costs for a solid oral dosage form typically run between $2 million and $8 million, depending on formulation complexity, bioequivalence study requirements, and manufacturing readiness. For complex generics or products with challenging bioequivalence requirements, that number may reach $15 million to $25 million. But it does not carry clinical risk. If the bioequivalence study fails, the manufacturer revises its formulation and retests. There is no Phase II or Phase III equivalent to fund for years before discovering the asset doesn’t work.
Litigation costs in a Hatch-Waxman case run higher than in most commercial patent disputes because of the volume of patents, the technical complexity of invalidity arguments, and the compressed 30-month timeline. A contested Paragraph IV trial involving four to six Orange Book patents, with expert witnesses, claim construction briefing, and a full bench trial, typically costs the generic defendant between $8 million and $20 million in legal fees and expert costs. The brand spends comparable amounts. Both sides spend more if the case involves biologics or complex formulations requiring extensive expert testimony.
The total outlay for a competitive first-to-file Paragraph IV campaign on a major product, from IP scouting through ANDA preparation through trial, typically runs $15 million to $40 million. Against a potential NPV prize of $300 million to $700 million, the risk-adjusted expected value calculation strongly favors the challenge, particularly when the patent analysis suggests meaningful invalidity arguments.
The $236 Billion LOE Opportunity Window: Where Paragraph IV Capital Is Flowing
The strategic importance of Paragraph IV litigation is inseparable from what the industry calls the patent cliff, the cluster of branded drug patent expirations that creates the opportunity landscape for generic entry.
An estimated $236 billion opportunity for generic and biosimilar manufacturers will open between 2025 and 2030 as major drugs lose exclusivity. More than $300 billion in prescription drug revenues will lose patent exclusivity in this window, covering nearly 200 drugs including approximately 70 blockbusters generating over $1 billion each in annual sales. The previous patent cliff, in 2016, eroded about $100 billion in brand-name sales; the current one is three times that size.
Among the most exposed companies are Merck and Pfizer, whose portfolios include several blockbuster small-molecule drugs reaching loss of exclusivity, including Merck’s diabetes franchise. By 2026, eight of the 13 largest pharmaceutical firms, representing 55% of global market value, could see 30% or more of their revenue jeopardized, with losses ranging from $6 billion to $38 billion per company.
For generic manufacturers with active Paragraph IV programs, this wave represents not just volume but target quality. The products facing LOE include primary-care blockbusters with established patient populations, predictable payer formulary dynamics, and stable demand. These are exactly the products where 180-day exclusivity has the highest NPV, because the market is large, demand is inelastic, and brand loyalty among prescribers decays quickly once pharmacy substitution becomes available.
How the 30-Month Stay Works — and How Brands Weaponize It
The Mechanics of Patent-Triggered Litigation Delay
When a brand manufacturer receives a Paragraph IV notice and sues within 45 days, FDA cannot grant final approval to the ANDA for 30 months from the date of the notice letter. This automatic stay operates as a regulatory injunction. The brand does not need to post a bond, demonstrate likelihood of success on the merits, or satisfy any of the traditional preliminary injunction factors. The stay applies automatically upon timely filing of the suit.
The value of the 30-month stay to a brand company is straightforward to calculate. For a drug generating $3 billion in annual sales, 30 months of protected revenue equals $7.5 billion in gross sales before accounting for the price increases brands routinely implement in the months preceding patent expiry. That $7.5 billion substantially exceeds the cost of a contested Hatch-Waxman trial, even a long and expensive one. The litigation is a financial instrument as much as a legal proceeding.
Orange Book Patent Stacking: How Brands Multiply the Delay
A brand manufacturer can list multiple patents in the Orange Book. Each listed patent that the generic certifies under Paragraph IV triggers a separate 30-month stay, though under Hatch-Waxman amendments, only the first timely suit triggers the stay. The strategic play is not to multiply stays but to list enough patents with sufficient scope diversity that the generic faces a technically demanding multi-patent invalidity campaign, increasing litigation cost and complexity.
The FDA requires that Orange Book patents ‘claim the drug substance (active ingredient), drug product (formulation or composition), or a method of using the drug.’ The statute is specific. Patents on manufacturing equipment, packaging, stability testing methods, or ancillary components do not qualify. In practice, brand manufacturers have pushed the boundaries of eligible listings for decades, listing patents whose connection to the approved drug product is tenuous.
The regulatory environment for Orange Book listings shifted dramatically between 2023 and 2025. The FTC, under Chair Lina Khan, disputed over 500 patent listings held by major pharmaceutical companies including Teva, GSK, and Boehringer Ingelheim. The agency’s core argument was that patents on drug-device combination components, such as inhaler caps, dose counters, straps, and similar mechanical components, do not claim the ‘drug product’ as defined by 21 U.S.C. Section 355(b)(1) and cannot legitimately trigger the 30-month stay.
In December 2024, the Federal Circuit affirmed a lower court decision ordering Teva to delist five Orange Book patents. The ruling gave teeth to the FTC’s position and prompted Teva to agree to remove more than 200 patent listings shortly thereafter, along with a $35 million settlement. For generic manufacturers challenging inhaler and auto-injector products, this regulatory shift removes significant 30-month stay exposure and potentially accelerates market entry.
What Happens When the 30-Month Stay Expires Before Trial
When the 30-month clock runs out before the court decides the case, FDA can grant tentative or final approval to the ANDA, and the generic can launch ‘at risk.’ At-risk launch means the generic enters the market while the patent case is still pending. If the brand wins the litigation after an at-risk launch, it can collect damages, including potentially enhanced damages if the court finds willful infringement. The commercial math on at-risk launch is analyzed carefully by every sophisticated generic company: the revenue from early market entry must justify the damages exposure if the brand ultimately prevails.
At-risk launches occur more often than the public record suggests, because brand manufacturers do not always pursue damages aggressively when the patent case was already close. The commercial reality is that by the time a post-launch damages case works through the system, the patent may be near expiry, the damages calculation is complex, and settlement becomes the rational outcome for both parties.
The 180-Day Exclusivity Prize: Value Calculation, Authorized Generics, and Forfeiture Risk
How to Calculate the NPV of 180-Day Exclusivity
The NPV of first-to-file 180-day exclusivity depends on four variables: the branded drug’s annual revenue, the generic’s pricing relative to brand, whether an authorized generic is launched during the exclusivity period, and the probability-weighted litigation outcome. A simplified model for a $1.5 billion annual revenue product:
Variable
Assumption
Result
Branded annual revenue
$1.5 billion
Base
Generic price discount
22% below brand
~$1.17B generic WAC equivalent run-rate
Generic market share during exclusivity
70% of prescriptions
~$408M in 6 months
Gross margin (no authorized generic)
55%
~$224M gross profit
With authorized generic (price competition)
Generic market share 45%, price 40% below brand
~$108M gross profit
Litigation costs (ANDA + trial)
$20M
Deducted from both scenarios
Risk-adjusted NPV (70% win probability)
No AG scenario
~$142M
Risk-adjusted NPV (70% win probability)
With AG competition
~$62M
The authorized generic variable is where settlement negotiations center. A brand company can offer a ‘no-AG agreement’ as a form of valuable, non-cash consideration to a generic challenger. In exchange for the generic agreeing to a later market entry date, the brand promises not to launch an authorized generic during the eventual 180-day exclusivity period, thereby preserving the full value of the prize for the generic. A generic manufacturer with no authorized generic competition can price at 20 to 30 percent below brand rather than the 40 to 60 percent discount that authorized generic competition forces.
The Authorized Generic Countermeasure: Brand’s Most Effective Weapon Against the 180-Day Exclusivity
The authorized generic (AG) is a brand-name drug sold in generic packaging, typically through a distribution partner, at generic prices. It is not a separate ANDA; the brand simply uses its own approved NDA to market a version of its drug at a price competitive with the first-to-file generic. No regulatory approval is needed beyond what the brand already holds.
An AG launched during the 180-day exclusivity window does not break the exclusivity; no other ANDA applicant can get final approval during that period. But the AG does break the first filer’s commercial monopoly. Instead of a duopoly between the brand (at brand price) and the generic (at 20 percent discount), the market has three participants: the brand at brand price, the AG at a competitive generic price, and the first-to-file generic at a similar competitive price. Pharmacy price competition between two low-cost options compresses the generic’s margins significantly.
This dynamic means that the real value of a no-AG commitment, which the FTC tracks and scrutinizes carefully, is the difference between the duopoly scenario and the three-way competition scenario, often $100 million to $300 million in additional gross profit for the first-to-file generic. When brands offer no-AG commitments as part of settlement agreements, the FTC treats them as a form of reverse payment, even if no cash changes hands.
The 180-Day Forfeiture Traps: Use It or Lose It
Congress amended the forfeiture rules for 180-day exclusivity under the Medicare Modernization Act of 2003 to prevent first-to-file generics from warehousing their exclusivity rights, blocking all other generics without actually launching. There are six forfeiture triggers, and the most commercially consequential is failure to launch. About 35% of first applicants lose their exclusivity because they never launch. Sometimes it’s because they can’t scale production. Sometimes it’s because they strike a deal with the brand-name company to delay entry.
The failure-to-obtain-tentative-approval (FTOTA) trigger is the most common trap for well-intentioned generic companies that simply lose control of their ANDA review timeline. If an ANDA applicant does not receive tentative approval within 30 months of filing, its exclusivity is forfeited unless the delay was caused by FDA review requirements or patent/exclusivity litigation. Generic companies managing large Paragraph IV portfolios track each ANDA’s tentative approval timeline obsessively, because forfeiture eliminates the entire commercial thesis of a first-to-file challenge.
Paragraph IV Settlement Mechanics: The Real Outcome of Most Cases
Why 76% of Paragraph IV Challenges Succeed — and What That Number Actually Measures
The 76% generic success rate in Paragraph IV challenges includes wins at trial, favorable settlements granting early entry dates, and settlements with no-AG commitments. It does not mean 76% of cases go to trial and the generic wins. Most cases settle, and settlements often grant the generic entry before patent expiry — that is the ‘success’ being measured.
One comprehensive study found that the overall success rate for generics in Paragraph IV challenges was a remarkable 76%. However, this number comes with a critical caveat: the study defined ‘success’ to include not only court victories but also favorable settlements.
The framing matters enormously. A 2024 analysis of terminated Hatch-Waxman cases found that innovator companies prevailed on the merits 20% of the time, whereas generic companies won a mere 2% of the time. These statistics are profoundly misleading because they ignore the single most important factor in Paragraph IV litigation: settlement. The vast majority of ANDA disputes never reach a final verdict. They are resolved through settlement agreements.
A settlement is a success from the generic manufacturer’s perspective if it grants an entry date before the challenged patents expire. On a product where the compound patent runs to 2031 and the settlement grants entry in 2028, the generic has effectively invalidated three years of exclusivity through legal strategy alone. The economic value of that advance entry, discounted to present value, can be hundreds of millions of dollars for a blockbuster drug.
How Paragraph IV Settlements Are Structured: Entry Date, Volume Caps, and No-AG Terms
A typical Paragraph IV settlement involves four elements. First, a licensed entry date, the date before patent expiry on which the generic can launch. Second, a covenant not to sue, by which the brand agrees not to assert the settled patents against the generic’s ANDA product. Third, often a no-AG commitment, structured as the brand’s promise not to sell an authorized generic during the first-to-file’s exclusivity window. Fourth, in some cases, a supply or licensing arrangement under which the brand provides API, formulations data, or co-promotion rights to the generic.
Volume-limited settlements represent a separate strategic category. Innovators like Celgene (BMS) learned to avoid the patent cliff through volume-limited settlements. For Revlimid (lenalidomide), they negotiated agreements that cap generic market share until 2026. This structure preserves brand revenue while offering generics a guaranteed, if limited, revenue stream. It avoids the ‘flash crash’ of brand revenue, allowing for a ‘controlled descent.’
Volume caps attract FTC scrutiny because they limit the price-depressive effect of generic competition without providing the consumer savings the Hatch-Waxman Act intended. The FTC has characterized sufficiently restrictive quantity caps as functional equivalents of full market exclusivity. Whether a volume cap rises to the level of an illegal reverse payment under the FTC v. Actavis ‘rule of reason’ standard depends on whether the value it transfers to the generic exceeds what could be justified by avoided litigation costs.
FTC v. Actavis and the Post-2013 Antitrust Environment for Settlement Agreements
The Supreme Court’s 2013 decision in FTC v. Actavis established the governing antitrust framework for pharmaceutical patent settlements. The Court rejected both extreme positions: it declined to make reverse payments per se illegal, as the FTC advocated, and equally declined to immunize them from antitrust scrutiny as long as the entry date fell within the patent term, as the pharmaceutical industry argued. The rule-of-reason standard it adopted asks whether the payment to the generic exceeds the value of what the generic provided to the brand in the settlement.
In practice, post-Actavis settlements evolved to minimize obvious cash payments in favor of non-cash value transfers: no-AG commitments, supply agreements, licensing fees framed as fair market value, and business partnerships. The FTC has continued to pursue cases where non-cash consideration is large enough to function as an effective payment for delay. In June 2024, a federal court in New Jersey allowed antitrust claims to proceed against a brand manufacturer over disputed Orange Book listings, and multiple members of Congress have signaled continued interest in the issue.
Litigation Geography: Why Delaware and New Jersey Dominate Hatch-Waxman Cases
District of Delaware and D.N.J.: The Two Courts That Decide Generic Competition
The District of Delaware and the District of New Jersey collectively handle the overwhelming majority of Hatch-Waxman ANDA cases. Delaware draws cases because most pharmaceutical brand companies incorporate there. New Jersey draws them because it is home to major pharmaceutical company operational headquarters, creating general personal jurisdiction.
The world of Hatch-Waxman litigation is geographically concentrated to an extraordinary degree. The U.S. District Courts for the District of Delaware and the District of New Jersey are the undisputed epicenters, consistently handling the overwhelming majority of all ANDA cases filed nationwide.
This concentration has practical consequences. Both courts have accumulated deep institutional knowledge of pharmaceutical patent issues. Judge Richard Andrews in Delaware and the specialized patent judges in New Jersey have presided over scores of Hatch-Waxman trials and developed consistent approaches to claim construction, objective indicia of non-obviousness, and written description requirements. Litigants who practice regularly in these courts develop informational advantages over less experienced opponents.
The Federal Circuit has exclusive appellate jurisdiction over all patent cases from these courts. The Federal Circuit issued seven Hatch-Waxman decisions in 2024, compared to five in both 2022 and 2023, with several reshaping prosecution strategy, portfolio management, and PTAB practice in ways that will govern behavior for years. Federal Circuit decisions are the law of the land for pharmaceutical patent questions, and understanding recent Federal Circuit doctrine is a prerequisite for any serious Paragraph IV strategy.
The 2024 Litigation Volume Surge: 312 ANDA Cases Filed
In 2024, 312 complaints were filed initiating Hatch-Waxman litigation, compared to 259 in 2023. The overwhelming majority of ANDA complaints were filed in the District of Delaware and the District of New Jersey.
ANDA case filings reached 312 in 2024, a 20% increase from the 259 filed in 2023 and the third consecutive annual increase, according to Lex Machina data. The increasing caseload, combined with the compressed timelines imposed by the 30-month stay, creates significant docket management pressure in both primary venues.
After peaking in 2014 and 2015, case filings generally trended downward. This decline, which saw a 36% drop between 2017 and 2021, did not signal a retreat from patent challenges. Instead, it reflected a maturing and consolidating generic industry becoming more selective, focusing on more complex and profitable drug products rather than pursuing every possible target. The recent data showing 312 complaints in 2024 indicates that the strategic importance of these challenges remains high.
The 2024 volume increase likely reflects two factors. The first is the approaching $236 billion LOE wave, which is pulling forward ANDA filings by companies positioning for first-to-file status on high-value targets. The second is consolidation in the branded pharmaceutical industry through acquisitions, which frequently triggers patent portfolio reviews that create new Paragraph IV filing opportunities as patents are reassigned, narrowed, or abandoned.
PTAB vs. District Court: Choosing Your Forum in a Two-Front Patent War
Inter Partes Review vs. Hatch-Waxman Litigation: A Strategic Comparison
Factor
IPR at PTAB
District Court (Hatch-Waxman)
Burden of proof for invalidity
Preponderance of evidence
Clear and convincing evidence
Typical cost
$500K–$2M
$8M–$20M+
Timeline to decision
~18 months post-institution
30+ months (30-month stay period)
Claims invalidation rate
70–78% of challenged claims (2024)
~24% complete invalidation of Orange Book patents
Scope of challenge
Validity only (prior art grounds)
Validity, infringement, unenforceability
180-day exclusivity preservation
Does not generate exclusivity rights
First-to-file earns 180-day exclusivity
Court stay of ANDA case
Courts rarely stay Hatch-Waxman for IPR
N/A
Estoppel risk
IPR estoppel prevents re-arguing same grounds in district court
No estoppel generated for PTAB purposes
Why Generic Manufacturers Use Both Forums Simultaneously
The modern challenger’s strategy should almost always include a parallel proceeding at the Patent Trial and Appeal Board. The PTAB uses a lower burden of proof (‘preponderance of the evidence’) than district courts (‘clear and convincing evidence’) and has a significantly higher rate of invalidating patent claims, often in the 60 to 70 percent range. An IPR is faster and cheaper than district court litigation and places immense pressure on the brand company, often forcing them to the settlement table on more favorable terms for the generic.
The pressure mechanics work as follows. When a generic company files an IPR within 12 months of receiving the Paragraph IV notice, the PTAB proceeding runs on a parallel track to the district court litigation. An IPR institution decision comes within six months of filing; a final written decision comes 12 months after institution. If the PTAB institutes on strong prior art grounds and the case looks likely to result in claim invalidation, the brand faces the prospect of losing its patents entirely on an administrative record before the district court trial even starts. Settlement becomes more attractive for the brand at that point.
Across all technologies, the rate at which PTAB final written decisions find all challenged claims unpatentable has been steadily increasing, rising from 55% in 2019 to 70% in 2024. On a per-claim basis, in 2024, nearly 78% of all claims that went to a final decision were found invalid.
However, the IPR’s power has an important limitation for Paragraph IV purposes. An IPR does not generate 180-day exclusivity. Only the first ANDA filer with a Paragraph IV certification earns that right. A company that invalidates a patent through IPR but was not the first ANDA filer gets no exclusivity benefit; it simply opens the market to all waiting ANDA applicants simultaneously. This means IPR is most powerful as a settlement lever when combined with a first-to-file ANDA position, not as a standalone strategy.
Estoppel Risk: The Strategic Cost of Filing IPR Petitions
Filing an IPR petition carries a statutory estoppel consequence. Under 35 U.S.C. Section 315(e), if an IPR proceeds to a final written decision, the petitioner cannot assert in subsequent district court proceedings any ground that it raised or reasonably could have raised in the IPR. This estoppel applies to prior art grounds and can foreclose a generic company from using its best invalidity arguments at trial if the PTAB proceeding concludes first.
Managing this estoppel risk requires careful prior art partitioning at the outset of the litigation campaign. Sophisticated generic companies file IPR petitions on prior art grounds they consider strong but not their strongest, reserving the most potent invalidity arguments for district court where the estoppel does not apply and where the full record of the brand’s prosecution history can be used for prosecution history estoppel arguments.
The Fintiv Problem: When District Court Timelines Block IPR Institution
The PTAB’s 2020 Apple Inc. v. Fintiv decision established a framework under which the PTAB can deny IPR institution on discretionary grounds when a district court trial is scheduled soon. The six Fintiv factors include the proximity of the trial date, investment by the court and parties in the parallel litigation, and overlap between the IPR grounds and district court invalidity contentions.
For pharmaceutical IPRs, Fintiv is less problematic than in other technologies because Paragraph IV litigation timelines are set by the 30-month stay clock rather than by normal civil docket scheduling. A Paragraph IV case where trial is three years out does not present the ‘nearby trial date’ concern that Fintiv was designed to address. A Paragraph IV case filed on a biologic patent may be years away from trial, which weakens the Fintiv argument that a nearby district court trial date warrants deferring to the court forum.
Orange Book Patent Thickets: How Brands Engineer IP Portfolios to Deter Challenges
The Four-Layer Orange Book Portfolio Strategy
Brand pharmaceutical companies no longer rely on a single composition-of-matter patent to protect a product. The standard defensive IP architecture now involves multiple layers of patents covering different aspects of the same drug. Build Orange Book portfolios in four layers: compound, polymorph/salt, formulation, and method-of-use. Each layer adds incremental complexity and exclusivity duration.
The compound patent is the foundational protection. It covers the novel chemical entity, its stereoisomers and salts, and related structural analogs. Compound patents typically expire 20 years from filing, often 10 to 12 years from approval after accounting for clinical development time. Patent Term Extension under 35 U.S.C. Section 156 restores up to five years of patent term lost to FDA regulatory review, potentially extending compound patent life to 14 years post-approval. Pediatric exclusivity adds six months of market protection on top of patent expiry.
Polymorph and salt patents cover specific crystalline forms or salt versions of the active ingredient. These are frequently challenged as obvious variants of the compound, but they require significant formulation science to invalidate. A generic that needs to use a specific polymorph of an API because its manufacturing process produces that form, but that polymorph is patented, faces a non-infringement problem rather than an invalidity problem — harder to solve.
Formulation patents cover the specific drug product: the inactive ingredients, the tablet structure, the drug delivery system. Controlled-release formulations attract layered patent protection covering the matrix technology, the coating polymers, the release kinetics, and sometimes the manufacturing process. A generic must achieve bioequivalence with the brand, but it does not need to use the same formulation. Designing around formulation patents while maintaining bioequivalent release profiles is core generic formulation science.
Method-of-use patents are the most strategically complex layer. They cover specific indications, patient populations, dosing regimens, and combination therapies. A generic manufacturer can carve a specific indication out of its label (a ‘skinny label’) to avoid infringing a method-of-use patent on one indication while still marketing the product for other approved uses. Skinny label strategies are now under pressure following litigation over induced infringement liability for off-label use when the omitted indication drives a significant portion of prescribing.
AbbVie’s Humira Blueprint: The Patent Thicket Playbook
The biosimilar patent thicket strategy pioneered by AbbVie on Humira (adalimumab) illustrates the extremity of what is achievable through layered IP. AbbVie maintained more than 130 patents on Humira at its peak, covering the antibody composition, manufacturing processes, formulation, drug delivery devices, dosing regimens, and multiple indication-specific methods of treatment. Biosimilar applicants who settled with AbbVie agreed to launch dates of January 2023 in the U.S. market, nearly 20 years after adalimumab’s original FDA approval in 2002. The aggregate value of that exclusivity extension ran into the tens of billions of dollars in retained revenue.
The Humira thicket is instructive because it illustrates both the power and the limits of layered patent protection. AbbVie successfully delayed all U.S. biosimilar entry until 2023, but by the time biosimilars launched, pricing pressure was severe. By mid-2024, multiple adalimumab biosimilars had received formulary placement at prices 80 to 85 percent below AbbVie’s list price, and Humira’s U.S. revenue had fallen sharply. The 20-year exclusivity extension was commercially valuable, but it did not prevent eventual pricing collapse when competition arrived.
Post-Amgen v. Sanofi: How the Written Description Doctrine Is Reshaping Antibody Patents
The Supreme Court’s 2023 decision in Amgen Inc. v. Sanofi fundamentally altered the landscape for broad antibody patents. The Court unanimously held that a patent claiming an entire genus of antibodies defined by their function, rather than their structure, must enable the full scope of the claimed genus. Amgen’s patents on antibodies that bind to PCSK9 to reduce LDL cholesterol were invalidated because they claimed all antibodies that achieved the functional result without adequately enabling a person skilled in the art to make and use the full scope of that claim.
Amgen v. Sanofi directly affects Paragraph IV strategy in the biologic space. Brand companies whose antibody patents claim broad functional categories now face heightened enablement challenges. Generic and biosimilar challengers can argue that a patent claiming ‘any antibody that binds X and blocks function Y’ is invalid under the post-Amgen enablement standard unless the specification discloses representative structural examples across the claimed scope. For brand companies prosecuting follow-on antibody patents, narrow structural claiming is now legally safer than the broad functional claiming that characterized the early biologic patent era.
Evergreening: The Brand Strategy to Outlast Paragraph IV Challenges
Reformulation, New Dosage Forms, and the Lifecycle Management Playbook
Lifecycle management is the brand industry’s answer to Paragraph IV exposure. When a compound patent is challenged successfully, the brand’s response is to have already established a next-generation product: a new formulation, a new delivery system, a new indication, or a combination product. The goal is to migrate patients to the protected successor product before generic competition arrives on the original formulation.
Extended-release formulations are the most common lifecycle management tools. AstraZeneca’s conversion of omeprazole (Prilosec) patients to esomeprazole (Nexium), the single-isomer active metabolite, before Prilosec’s generic entry was the template for an entire generation of pharmaceutical lifecycle management strategy. By the time generic omeprazole arrived, AstraZeneca had successfully repositioned large portions of the patient population on Nexium, which had years of remaining patent protection.
The FTC and generic industry characterize this as ‘product hopping’ when the brand uses payor and prescriber channel pressure to switch patients rather than allowing organic generic substitution. The antitrust question is whether the brand’s conduct constitutes an illegal attempt to maintain monopoly through product switching, not through the merits of the new product. Courts have split on this question. The Second Circuit’s 2015 decision in New York v. Actavis (donepezil/Aricept) found that a product hop can violate antitrust law if the brand withdraws the original product to eliminate pharmacist substitution rights, denying generic manufacturers the benefit of state automatic substitution laws.
Skinny Labels and the GSK v. Teva Fallout
A generic manufacturer that files a Paragraph IV challenge faces method-of-use patents covering specific approved indications. The standard solution is the ‘skinny label’ or ‘carve-out’: the generic omits the patented indication from its proposed label and seeks approval only for the non-patented uses. If the FDA accepts the carve-out, the generic’s label does not include the patented method, and the generic can argue it does not directly infringe.
Induced infringement is the complication. Even with a carved-out label, a brand can argue that the generic induces infringement because it knows physicians will prescribe the generic off-label for the patented indication, and the generic is facilitating that use through its general marketing presence. The Federal Circuit’s 2021 decision in GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc. held that Teva’s skinny-label carvedilol tablets induced infringement of GSK’s method-of-use patent covering the heart failure indication, even though Teva had carved that indication from its label.
GSK v. Teva caused significant strategic recalculation throughout the generic industry. Companies with skinny-label ANDAs that cover high-prescribing patented indications now face potential induced infringement liability they thought their carve-outs avoided. Several companies withdrew skinny-label ANDAs and refiled under Paragraph III certification to avoid at-risk induced infringement exposure. The Federal Circuit subsequently granted en banc rehearing and issued a modified opinion, but the case remains the most-discussed Hatch-Waxman decision of the early 2020s.
The Investor Lens: How to Analyze Paragraph IV Pipeline Risk for Branded Pharma
Reading the Orange Book to Forecast Generic Entry Dates
The Orange Book lists every patent covering an FDA-approved drug and its expected expiration date. For each listed patent, the FDA shows whether any ANDA applicants have filed Paragraph IV certifications. The presence of Paragraph IV certifications tells you which drugs are actively challenged. Platforms like DrugPatentWatch aggregate this data and layer on ANDA filing counts, litigation status, expected approval dates, and settlement terms to give analysts a complete competitive entry picture.
The Orange Book is the starting point for any generic entry timeline analysis, but it is not sufficient alone. Several exclusivity categories operate outside the patent framework and can delay generic entry even after patent expiry. Five-year new chemical entity (NCE) exclusivity prevents ANDA filing during the first four years and prevents approval for the first five years after brand approval. Three-year new clinical investigation exclusivity protects specific new conditions of use supported by new clinical trials. Orphan drug exclusivity provides seven years of protection for rare disease treatments.
DrugPatentWatch collects and integrates Orange Book patent data, exclusivity expiration dates, ANDA filing counts by product, Paragraph IV certification status, PTAB petition filings against Orange Book patents, and settlement terms disclosed under the Medicare Modernization Act’s mandatory filing requirements. For an institutional investor or sell-side analyst modeling a brand company’s revenue trajectory, tracking this data systematically produces materially different LOE forecasts than relying on FDA public records alone.
What Paragraph IV Certification Counts Signal for Investors in Branded Drugs
When a drug receives its first Paragraph IV certifications, the clock starts on generic entry uncertainty. The number of certifications matters for competitive reasons: if ten generic companies file simultaneously, none earns first-to-file exclusivity (they all share it), the litigation involves more defendants, and settlement is more complex. For the brand, multi-defendant litigation means higher legal costs but also more potential settlement partners whose interests may diverge from each other.
For investors in brand companies, the relevant questions are: how many Paragraph IV filers are there, what is the patent strength of the challenged patents, when does the 30-month stay expire, is there pending IPR activity at PTAB, and what entry date do settlement probabilities point toward? A brand drug with a strong compound patent, no prior art problems, and a settlement history of granting entry at patent expiry faces a very different LOE profile than one whose compound patent is weak and whose formulation patents are the primary barrier.
For payers analyzing a drug that may be generating a 20:1 return on R&D investment, the key question is not whether competition will eventually arrive, but when. A drug whose compound patent expires next year but whose method-of-use patents run for another 12 years is in a fundamentally different commercial position than a drug facing a clean patent cliff. That distinction does not appear in any single public source; it requires synthesis of patent data, exclusivity dates, litigation status, and biosimilar development intelligence.
How Paragraph IV Activity Affects Branded Drug Valuations: A Case Study Framework
Case Study: Merck’s Januvia/Janumet Paragraph IV Timeline
Merck’s sitagliptin franchise generated approximately $2.25 billion in 2023 revenue. Merck’s core compound patent expired in 2023, but a related salt/polymorph patent ran into 2027. Facing Paragraph IV challenges, Merck settled in 2020 so that generics could launch in mid-2026.
The Januvia settlement is a typical late-franchise strategic capitulation. With the compound patent already expired and the remaining IP of questionable validity, Merck’s best outcome was an orderly licensed entry rather than contested at-risk launches that would have compressed its pricing power immediately. The 2026 entry date allowed Merck to execute price increases on Januvia through 2025, capturing additional revenue before the controlled generic entry. Merck disclosed this LOE timeline to investors years in advance, allowing orderly capital reallocation planning.
Case Study: Eliquis (Apixaban) — The $13 Billion Question
Eliquis, which generated $13.3 billion in combined sales for Bristol-Myers Squibb and Pfizer, is among the first drugs subject to Medicare price negotiations and will see its list price drop to $231 per month in 2026. U.S. patent protection for the drug is set to expire in 2028.
Eliquis faces a dual revenue compression that makes it one of the most scrutinized LOE cases in the industry: IRA price negotiation reducing its Medicare price starting in 2026, followed by patent expiry and generic entry in 2028. The compound patent expiry timing and the strength of Eliquis’ remaining patent portfolio, which includes formulation and method-of-use claims, will determine how aggressively generics can challenge ahead of 2028. Any successful Paragraph IV challenge settling for early 2027 entry would compress Eliquis’ remaining exclusivity revenue by hundreds of millions of dollars for BMS and Pfizer.
Biosimilar Patent Disputes: The BPCIA Patent Dance vs. Paragraph IV Mechanics
How the Biologics Price Competition and Innovation Act Differs from Hatch-Waxman
The BPCIA’s ‘patent dance’ is a structured information exchange in which the biosimilar applicant shares its manufacturing process with the reference product sponsor, and the two parties negotiate which patents to litigate. Unlike Hatch-Waxman, the BPCIA does not create automatic stays, does not have a precise equivalent to the 180-day exclusivity, and allows broader information exchange before litigation crystallizes. The FDA’s 12-year reference product exclusivity period is the functional analog to a compound patent under Hatch-Waxman.
The Biologics Price Competition and Innovation Act of 2009 (BPCIA), enacted as part of the Affordable Care Act, created the regulatory pathway for biosimilar applications. Rather than listing patents in an Orange Book analog, the BPCIA requires the biosimilar applicant and reference product sponsor to exchange information about the proposed product and applicable patents in a defined pre-litigation sequence called the patent dance. The parties then negotiate a list of patents for immediate litigation and reserve others for later rounds.
The BPCIA’s 12-year reference product exclusivity period is a regulatory exclusivity, not a patent. It prevents FDA from relying on the reference product’s safety and efficacy data for 12 years from the reference product’s approval, regardless of patent status. For Humira (approved 2002), this exclusivity ran until 2014, well before the patent thicket created by AbbVie’s prosecuted portfolio. For Keytruda (approved September 2014), the reference product exclusivity period means no biosimilar can rely on the FDA’s pembrolizumab safety data until September 2026.
AbbVie Humira, Keytruda, and the Next Wave of Biologic LOE Events
Merck’s pembrolizumab (Keytruda) is the most commercially consequential biologic facing LOE in the 2025 to 2030 window. With $29.5 billion in 2023 revenue, Keytruda is effectively the financial engine of the entire company. Merck has built a patent estate around pembrolizumab that includes composition-of-matter patents on the antibody structure, method-of-use patents covering its approved indications (pembrolizumab has over 40 FDA-approved indications), and formulation patents on its IV formulation and concentration.
The biosimilar applicants targeting Keytruda will not file Paragraph IV certifications; they will navigate the BPCIA patent dance. But the commercial and financial analysis is structurally identical: which patents are strong, when does the strongest IP expire, what is the probability-weighted entry date, and what is the NPV of early entry through successful patent challenge? Samsung Bioepis, Formycon, and other biosimilar developers have already filed 351(k) applications for pembrolizumab biosimilars, initiating the BPCIA process.
The FTC’s Expanding Role: Orange Book Challenges, Pay-for-Delay Enforcement, and What Changes in 2025–2026
The FTC’s 2023–2024 Orange Book Delisting Campaign
In September 2023, the FTC issued a policy statement warning pharmaceutical companies that improper Orange Book listings could constitute an antitrust violation. The FTC sent letters directly to brand manufacturers flagging specific patents it viewed as potentially improper listings.
The delisting campaign targeted primarily patents on drug-delivery device components for inhalers and auto-injectors: patents covering inhaler caps, dose counters, lanyards, and similar components that are not part of the active drug product or its formulation. The FTC’s theory was that listing such patents in the Orange Book, and then suing generic filers who certified against them under Paragraph IV, triggered improper 30-month stays that delayed generic competition beyond what valid patent protection justified.
In December 2024, the Federal Circuit affirmed the lower court decision ordering Teva to delist five Orange Book patents, and Teva subsequently agreed to remove more than 200 patent listings and pay $35 million in settlement. The decision is the most concrete enforcement outcome of the FTC’s Orange Book campaign and creates clear precedent: device-component patents that do not claim the drug substance, drug product as formulated, or an FDA-approved method of use are not properly listable in the Orange Book.
Will the IRA’s Drug Price Negotiation Accelerate or Reduce Paragraph IV Activity?
The Inflation Reduction Act of 2022 introduced Medicare drug price negotiation for a targeted set of high-expenditure drugs without generic or biosimilar competition. The first ten drugs subject to negotiation had their negotiated prices take effect January 2026. The IRA also imposes inflation rebates when drug prices rise faster than inflation and caps Medicare Part D out-of-pocket costs.
The IRA creates a complex interaction with Paragraph IV strategy. For drugs subject to negotiation, generic entry no longer confers the same premium avoidance benefit for federal payers, because CMS has already negotiated the brand price down. Generic entry still eliminates the drug’s negotiated brand price for non-negotiated purchasers and commercial plans, but the delta between brand and generic pricing in the negotiated Medicare market is compressed. Some analysts argue this weakens the incentive for brand manufacturers to fight generic entry aggressively on IRA-selected drugs, because the drug’s commercial value has already been capped by negotiation.
The counter-argument is that the IRA actually increases the incentive for brand manufacturers to secure every possible day of exclusivity before generic entry, because each month of exclusivity before negotiation-depressed prices take full effect represents maximum-price revenue. Brands subject to the IRA’s 11-year no-competition eligibility threshold (9 years for biologics) have an additional structural incentive to use patent portfolio depth to push generic entry past that threshold, since drugs facing generic competition before the threshold becomes eligible are not selected for negotiation.
The Data Intelligence Layer: How Paragraph IV Professionals Use Patent Analytics
DrugPatentWatch and Commercial Patent Intelligence Platforms
Paragraph IV litigation strategy starts not in the courtroom but in the data. The key intelligence questions at the outset of any Paragraph IV target assessment are: What patents are listed in the Orange Book for this product? When do they expire? Have any Paragraph IV certifications already been filed? If so, by how many competitors? What is the patent prosecution history suggesting about the scope of the claims? Are there pending or completed PTAB proceedings against any of the listed patents? What are the FDA approval timelines for competing ANDAs already on file?
DrugPatentWatch aggregates Orange Book data, ANDA filing information, PTAB proceedings, Paragraph IV notice letter data from mandatory MMA filings, and FDA exclusivity records into a unified commercial intelligence database. For a generic manufacturer’s business development team assessing whether to invest in an ANDA targeting a specific product, these aggregated data points feed directly into the NPV model: expected approval date, first-to-file probability, litigation cost estimate, and probability of settlement versus trial verdict.
The competitive advantage in generic drug development is not chemistry. It is IP intelligence combined with litigation risk assessment. The first generic manufacturer to file a successful Paragraph IV ANDA on a $2 billion product earns 180-day market exclusivity worth potentially $300 million to $700 million in net present value. The second filer earns a fraction of that; the tenth earns commodity-level margins from day one.
How to Use Patent Filing Dates, Prosecution History, and Prior Art to Score Invalidity Risk
The invalidity risk analysis for a specific Orange Book patent begins with the prosecution history: the record of communications between the patent applicant and the USPTO examiner during the examination process. Prosecution history reveals what claims were originally filed, what rejections the examiner made, and what arguments and amendments the applicant made in response. Claim scope is interpreted in light of prosecution history through the doctrine of prosecution history estoppel; arguments made to overcome rejections limit the scope of the granted claims.
For obviousness analysis, the key prior art search identifies publications, patents, and public disclosures that existed before the patent’s priority date and that, individually or in combination, might have made the claimed invention obvious to a person skilled in the art at the time. The validity of secondary factors, ‘objective indicia’ of non-obviousness, including commercial success, long-felt need, and teaching away, became substantially harder to establish after the Federal Circuit’s post-KSR International Co. v. Teleflex Inc. jurisprudence, which expanded the range of prior art combinations courts consider in obviousness analysis.
The Role of USPTO Patent Examination Quality in Generating Paragraph IV Targets
Not all Orange Book patents are equal. Some represent genuine innovations with strong prosecution histories and clean prior art landscapes. Others were prosecuted narrowly, cover peripheral aspects of the drug product, and rest on prior art art that a motivated challenger can readily identify. The quality of USPTO examination varies, and patents that issued without adequate prior art searching or with overbroad claims relative to the disclosed examples are more likely to face successful invalidity challenges.
Post-grant proceedings at the PTAB, including IPR, post-grant review (PGR), and the now-expired inter partes reexamination, have historically been used to attack Orange Book patents that survived initial examination but whose claims prove weaker when subjected to adversarial prior art scrutiny. A comprehensive 2018 study found that the rate of complete invalidation for Orange Book patents at the PTAB was 23%, almost identical to the complete invalidation rate of 24% for the same types of patents in district court litigation over the same period. That parity, across two forums with very different procedural structures, suggests that patent quality, not forum selection, is the primary predictor of invalidity outcomes in pharmaceutical cases.
The Generic Industry Consolidation Effect: Fewer Players, Bigger Bets
How M&A Reshaped the Paragraph IV Competitive Landscape
The generic pharmaceutical industry has consolidated sharply since the early 2000s. Teva acquired Barr Pharmaceuticals, PLIVA, Ivax, and Allergan Generics to become the world’s largest generic manufacturer. Viatris was formed from the merger of Mylan and Upjohn (Pfizer’s generic and off-patent branded business). Sandoz, the generics division of Novartis, has operated as a standalone company since its 2023 spinoff. Sun Pharma acquired Ranbaxy. Amneal merged with Impax. This consolidation has had direct consequences for Paragraph IV strategy.
Larger generic companies can spread Paragraph IV litigation costs across a broader revenue base, accept lower probability bets on difficult patents, and sustain multi-year litigation programs on complex cases. They also face more regulatory scrutiny on settlement agreements, because the FTC monitors large generic companies’ settlement terms more closely than small players’. The consolidation has reduced the number of first-to-file competitors on many products; instead of 15 independent generic companies filing simultaneously, a market might now have seven, and three of those might be part of the same consolidated group whose ANDAs are coordinated.
The consolidation effect is also visible in target selection. The post-2017 decline in ANDA case filings reflects a maturing and consolidating generic industry becoming more selective, often focusing on more complex and profitable drug products rather than pursuing every possible target. Smaller generic companies that cannot sustain multi-year litigation programs on major products increasingly focus on Paragraph III certifications (wait for patent expiry) or on complex generics where the ANDA exclusivity operates independently of 180-day Paragraph IV exclusivity.
The Top 5 Generic Manufacturers’ Paragraph IV Portfolio Strategy
Company
2024 Hatch-Waxman Position
Key Paragraph IV Tactics
Notable Targets (Recent)
Teva Pharmaceuticals
Largest global generic manufacturer; significant U.S. first-to-file portfolio
Large U.S. generics presence; active Paragraph IV filer across primary care
First-to-file on primary care blockbusters; authorized generic deals
Multiple GLP-1 adjacent products
Sandoz (Novartis spinoff)
Strong European and U.S. presence; active biosimilar patent dance filer
BPCIA patent dance on biologics; ANDA first-to-file on small molecules
Post-2023 spin independence strategy
Amneal Pharmaceuticals
Aggressive first-to-file culture; active complex generics program
IP analysis-driven target selection; willing to litigate to trial
Complex oral solids and specialty generics
Hikma Pharmaceuticals
U.S. injectables and oral solids; growing Paragraph IV program
Injectable ANDAs with Paragraph IV; 505(b)(2) hybrid applications
Oncology injectables
What Happens After Generic Entry: The LOE Revenue Cliff and Brand Pricing Strategy
How Fast Does Revenue Erode After Paragraph IV Generic Launch?
For small-molecule oral drugs with multiple generic competitors, branded revenue typically falls 70 to 90 percent within the first six months of generic entry. During the 180-day exclusivity period with only one or two generic competitors, the brand retains 30 to 50 percent of volume at premium pricing. After exclusivity ends and multi-generic competition begins, commoditization is rapid.
During previous cliffs, small-molecule drugs typically lost up to 90 percent of their revenues to generic competition after LOE, a trend reflected in analysis using GPI pulse 2.0. The speed of erosion depends on therapeutic area, prescriber behavior, payor formulary strategy, and whether the brand launches an authorized generic or a ‘soft brand’ successor product.
Payor formularies accelerate generic adoption. Most commercial and government payers move branded drugs to non-preferred tiers immediately upon generic availability, increasing patient cost-sharing substantially for those who choose the brand. Pharmacy benefit managers negotiate steep rebates from generic manufacturers in exchange for preferred placement. The net result is that generic market share in major therapeutic classes reaches 80 to 90 percent within 12 months of multi-competitor entry.
Pre-LOE Pricing Strategy: The Price Increase Before the Cliff
Surge pricing is usually adopted 12 to 18 months before LOE to maximize earnings prior to patent expiry. Most brands implement systematic price increases multiple times a year, as opposed to one large increase. One major pharmaceutical company gradually increased the whole acquisition cost (WAC) of its nerve pain medication starting as early as three years before LOE in anticipation of the looming revenue cliff. Another raised the price of its multiple myeloma drug by more than 50 percent between 2016 and 2022 to maximize profits before the product’s LOE in 2022.
This pricing behavior is strategically rational within the current system but has become politically toxic. Congressional hearings on drug pricing frequently feature specific examples of brands raising prices in the final years before patent expiry. The IRA’s inflation rebate provision, which requires manufacturers to pay rebates to Medicare when their drug prices rise faster than inflation, creates a partial check on pre-LOE price increases for drugs covered by Medicare Part D and Part B.
Paragraph IV Strategy for Specialty and Orphan Drugs: Different Economics, Same Logic
When 180-Day Exclusivity Is Worth More on Specialty Products
The financial case for Paragraph IV challenges is not limited to primary care blockbusters. Specialty pharmaceuticals, including oncology, neurology, immunology, and rare disease treatments, can generate Paragraph IV economics that equal or exceed primary care products, despite smaller patient populations, because of higher unit prices and less price-elastic payor behavior.
An oncology drug with $800 million in annual U.S. sales and a specialty drug price of $15,000 to $40,000 per patient per year presents a different generic entry economics than a primary care drug at the same revenue level. Generic oncology competitors price at smaller discounts to the brand (30 to 40 percent rather than 70 to 80 percent) because the market is specialist-driven and formulary management by oncology pharmacy is less aggressive than in primary care. During 180-day exclusivity, the first generic captures a meaningful share at a relatively high price, generating strong margins.
Orphan Drug Exclusivity as a Patent Challenge Complication
Orphan Drug Designation and the seven-year marketing exclusivity it provides under the Orphan Drug Act present a distinct challenge for generic manufacturers. Orphan exclusivity is regulatory, not patent-based, and it runs from the date of NDA approval. An ANDA for an orphan drug product cannot receive final approval during the seven-year exclusivity period, regardless of the patent landscape. This exclusivity cannot be challenged through Paragraph IV; it operates independently of Orange Book patents.
For a drug that received orphan designation, the generic entry timeline is therefore the later of: patent expiry (accounting for any Patent Term Extension and pediatric exclusivity), plus any Hatch-Waxman regulatory exclusivities, or seven years from orphan drug approval. A Paragraph IV challenge that invalidates the compound patent still cannot produce generic entry before the orphan exclusivity expires. Generic manufacturers targeting orphan products must model this regulatory barrier separately from the patent analysis.
Global Paragraph IV Equivalents: How Other Markets Handle Generic Patent Challenges
European Patent Challenges: SPCs, National Courts, and the Unified Patent Court
The European generic entry system operates through national courts rather than a centralized administrative pathway. Supplementary Protection Certificates (SPCs) extend patent protection in European Union member states for up to five years beyond the basic patent expiry, analogous to U.S. Patent Term Extension but calculated differently and subject to national variation in implementation. Generic manufacturers challenge SPCs and underlying patents through national court proceedings, which differ substantially in procedure, timeline, and outcome across member states.
The Unified Patent Court, operational since June 2023, created a new pan-European litigation forum that can issue decisions with effect across all participating UPC member states. A single UPC decision invalidating a patent eliminates that patent’s protection in every participating country simultaneously. The launch and growing influence of the Unified Patent Court will introduce new strategic dimensions. A single UPC decision can invalidate a patent across numerous European countries, raising the stakes of global patent disputes and requiring more coordinated international strategies.
Canadian Prohibition Proceedings and the PMNOC Regulations
Canada’s Patented Medicines (Notice of Compliance) Regulations create a Hatch-Waxman analog in which generic applicants file a Notice of Allegation against patents listed in the Patent Register for a reference drug, and brand patentees can initiate prohibition proceedings in the Federal Court. The prohibition period is 24 months rather than 30. Canadian Paragraph IV-equivalent litigation runs in parallel with U.S. ANDA proceedings for multinational generic companies; the strategic considerations of claim construction and invalidity analysis are similar, though Canadian patent law has meaningful doctrinal differences from U.S. law.
The Future of Paragraph IV: AI Patent Analysis, Biosimilar Complexity, and IRA Reshaping
How AI Is Changing Pharmaceutical Patent Validity Analysis
Machine learning tools are beginning to change the front-end intelligence work of Paragraph IV strategy: patent landscaping, prior art searching, invalidity argument generation, and claim chart mapping. Large language models trained on patent databases can generate obviousness arguments and prior art combinations faster than human associates, reducing the time between ANDA filing decision and preliminary invalidity theory development. This acceleration matters in a competitive first-to-file race where weeks of analytical advantage can determine whether a generic company files before competitors.
The impact on patent prosecution is symmetric. Brand companies use AI-assisted claim drafting to generate more defensible claim language, informed by patterns of claims that survive versus fail invalidity challenges. The arms race dynamic means that AI is simultaneously strengthening the offense and the defense. The first-order effect, however, may favor challengers, because the marginal cost of prior art discovery and invalidity hypothesis generation falls, lowering the barriers to Paragraph IV filing on complex multi-patent products.
AI is beginning to reshape both the drug discovery process and the legal standards that govern its patentability. The teams that track these changes systematically, integrating scientific, legal, regulatory, and commercial intelligence into a unified strategy, will continue to outperform those that treat pharmaceutical IP as a pure legal problem.
The Next Five Years: Which Products Face the Highest Paragraph IV Pressure Through 2030
2025–2026
Januvia/Janumet (sitagliptin, Merck) generic entry under 2020 settlements. Xeljanz (tofacitinib, Pfizer) first generic tofacitinib approved in August 2025. Ozempic (semaglutide, Novo Nordisk) core patent expires early 2026, though follow-on patents extend monopoly through 2031 on current Ozempic formulation.
2026–2027
Eliquis (apixaban, BMS/Pfizer) compound patent expiry 2026–2028 window depending on challenge success. Multiple Paragraph IV ANDAs actively being assessed. Combined brand revenue at risk: $13+ billion. Trulicity (dulaglutide, Lilly) approaching exclusivity threshold in 2027; biosimilar candidate in development.
2028–2030
Keytruda (pembrolizumab, Merck) BPCIA reference product exclusivity expires September 2026 but compound and method-of-use patents extend well beyond; biosimilar filers need to clear the patent dance and litigation. Multiple Paragraph IV certifications expected on remaining small-molecule blockbusters including Entresto (sacubitril/valsartan) and Xarelto (rivaroxaban).
GLP-1 Agonists: The Most Watched Paragraph IV Battleground of the Decade
The GLP-1 receptor agonist class, anchored by Ozempic (semaglutide) and Mounjaro (tirzepatide), represents the most commercially consequential set of patent disputes heading into the late 2020s. Ozempic’s compound patent for semaglutide faces expiry in 2026, but Novo Nordisk has built a formulation and delivery patent portfolio that includes the current pen device, the specific injectable formulations, and dosing regimens. Generic and biosimilar challengers will need to navigate that portfolio.
Tirzepatide (Mounjaro, Zepbound) is a newer molecule with a later compound patent expiry, but its commercial success, over $11.5 billion in 2024 revenue for Mounjaro alone, already makes it a target for forward-looking generic ANDA planning. The first Paragraph IV certifications against tirzepatide’s Orange Book patents will generate significant commercial intelligence about the strength of Lilly’s IP position in this class.
What the IRA’s 13-Year Biologic Negotiation Eligibility Threshold Means for BPCIA Strategy
The IRA makes biologics eligible for Medicare price negotiation after 13 years on the market (versus 9 years for small molecules). Biologic reference product sponsors whose drugs have been on the market for over 13 years without biosimilar competition face price negotiation. This creates a perverse incentive: if a biologic’s reference product sponsor can trigger biosimilar competition before the 13-year mark, the drug becomes ineligible for IRA negotiation. Some analysts have argued this creates an incentive for reference product sponsors to license biosimilar entry strategically, avoiding the negotiation mechanism while controlling the biosimilar entry date.
Whether this dynamic materially changes BPCIA settlement behavior is an open empirical question for the 2025 to 2028 period. What is clear is that the IRA adds another layer of economic analysis to biologic patent strategy that did not exist before 2022.
Key Takeaways
Key Takeaways
Paragraph IV litigation generates returns that exceed $10 per $1 invested in IP analysis for first-to-file challengers on blockbuster products, making it one of the highest-ROI capital deployment strategies in the pharmaceutical industry.
The cost to develop a new molecular entity averaged $2.23 billion in 2024 with a 5.9% expected ROI. A contested Paragraph IV campaign costs $15 million to $40 million with NPV upside of $300 million to $700 million for first-to-file exclusivity on major products.
ANDA case filings rose 20% to 312 in 2024, a third consecutive annual increase, driven by the $200–$300 billion LOE wave opening through 2030.
Settlement, not trial, resolves most Paragraph IV cases. The 76% generic ‘success rate’ includes settlements granting early entry dates well before patent expiry. Only 2% of cases produce a final court verdict favoring the generic.
The PTAB invalidates 70% of all challenged claims at final written decision, with 78% of specific claims found invalid on a per-claim basis in 2024. This makes parallel IPR a near-mandatory component of any serious Paragraph IV campaign.
Orange Book patent stacking — four layers of compound, polymorph, formulation, and method-of-use patents — is the brand industry’s primary defense. The FTC’s 2023–2025 delisting campaign is eroding the device-component segment of these stacks.
The Teva/FTC Orange Book settlement in December 2024, resulting in over 200 delisted patents and a $35 million payment, marks the most significant enforcement action on improper Orange Book listings in the Act’s history.
The 180-day exclusivity forfeiture mechanism means first-to-file winners must launch or lose their prize. Approximately 35% of first applicants forfeit their exclusivity, often because they cannot meet the use-it-or-lose-it launch requirement.
DrugPatentWatch and comparable patent intelligence platforms allow generic manufacturers to assess first-to-file probability, patent strength, and settlement probability before committing ANDA preparation resources, materially improving the precision of capital allocation decisions.
AI patent analysis tools are beginning to reduce the cost and time of invalidity argument generation, potentially lowering barriers to Paragraph IV filing on complex multi-patent products and accelerating the arms race dynamic between brand patent prosecutors and generic challengers.
Frequently Asked Questions
What is the Paragraph IV certification and when should a generic company file one?
A Paragraph IV certification is a generic manufacturer’s legal assertion, submitted as part of its ANDA, that one or more Orange Book-listed patents for the reference drug are invalid, unenforceable, or will not be infringed by the generic product. A company should consider filing when patent analysis suggests a credible invalidity or non-infringement argument, the first-to-file exclusivity prize has positive expected value after accounting for litigation cost and settlement probability, and no other ANDA applicant has already secured first-to-file status. The key is filing before competitors, because the first-to-file advantage is binary: once another company files on the same day or earlier, the shared exclusivity splits the economic prize.
How long does Paragraph IV ANDA litigation typically take?
From notice letter to trial or settlement, Paragraph IV litigation typically runs 24 to 36 months. The 30-month stay clock starts from the date of the Paragraph IV notice letter. District court scheduling varies by venue; the District of Delaware has historically moved cases to trial within the 30-month window. Cases that settle early, as many do, can resolve within 12 to 18 months of the initial filing. Cases involving multiple patents, large numbers of defendants, or complex technical issues may extend beyond 30 months, allowing at-risk launch considerations to enter the calculation.
What is the difference between Orange Book patent expiry and actual generic entry date?
Orange Book patent expiry is when the patent’s legal term ends. Actual generic entry depends on: whether any Paragraph IV certifications have been filed and what their litigation status is; whether any 30-month stays are active; whether FDA has granted tentative or final ANDA approval; and whether any settlement agreements have established a contractual entry date. The effective market exclusivity can extend well beyond the Orange Book expiry date if remaining formulation or method-of-use patents are not yet expired and no Paragraph IV certifications cover them. DrugPatentWatch tracks both the patent expiry and the expected entry date based on ANDA filings and litigation status, which often differ substantially.
What is the authorized generic and how does it affect Paragraph IV economics?
An authorized generic is a version of a brand drug sold in generic packaging by the brand manufacturer or its authorized distributor, without any new regulatory approval beyond the existing NDA. It can be launched at any time, including during the first-to-file generic’s 180-day exclusivity period. An authorized generic reduces the first-to-file generic’s revenue by competing for price-sensitive pharmacy substitutions, typically forcing the generic’s price down by an additional 15 to 25 percent relative to a duopoly scenario. A no-AG commitment in a settlement agreement preserves the first-to-file generic’s duopoly position and is treated by the FTC as quantifiable economic value that can constitute a reverse payment if large enough.
Can a Paragraph IV ANDA be filed before all clinical studies are complete?
Yes. An ANDA can be filed when the generic manufacturer’s bioequivalence studies are complete and the formulation is ready for review. The ANDA does not require clinical safety or efficacy trials beyond bioequivalence. This means a generic company can prepare, conduct bioequivalence testing, and file an ANDA in parallel with litigation planning, without waiting for any clinical endpoint. The timeline from ANDA filing to first-to-file position is driven by IP preparation speed, not clinical development timelines, which is a core reason Paragraph IV economics differ so fundamentally from NME development economics.
What is the ‘patent dance’ under the BPCIA and how does it differ from Paragraph IV?
The BPCIA patent dance is a structured pre-litigation information exchange between a biosimilar applicant and the reference product sponsor. The biosimilar applicant shares its manufacturing process and product information; the reference product sponsor identifies applicable patents; the parties negotiate a list of patents for immediate litigation and reserve others. Unlike Paragraph IV under Hatch-Waxman, the BPCIA does not create an automatic 30-month stay, does not have an equivalent to the 180-day exclusivity period for the first biosimilar filer, and uses a different notice structure (180-day commercial marketing notice before first commercial sale). The 12-year FDA reference product exclusivity is the primary regulatory barrier, not Orange Book patent stacking.
How does IPR at the PTAB interact with a pending Paragraph IV ANDA case?
Filing an IPR petition against an Orange Book patent while a related Hatch-Waxman case is pending in district court creates a parallel proceeding. If the PTAB institutes the IPR, the proceeding produces a final written decision within 12 months of institution. A favorable IPR outcome (patent claims invalidated) creates substantial settlement leverage. Courts in Hatch-Waxman cases rarely grant stays of the district court proceedings pending IPR completion, meaning both timelines run simultaneously. The strategic risk is IPR estoppel: if the IPR reaches a final written decision, the petitioner is estopped in district court from arguing any invalidity ground it raised or could have raised in the IPR. Sophisticated litigants partition their invalidity arguments between the two forums to avoid foreclosing their strongest district court theories.
What financial metrics do investors use to assess Paragraph IV pipeline risk for branded drug companies?
Investors model LOE impact using: annual revenues of the drug at risk, expected entry date based on settlement probability and patent expiry, revenue erosion speed by therapeutic category, branded revenue retained post-generic entry through lifecycle management or authorized generic, and IRA price negotiation impact for Medicare-covered products. For a drug with $2 billion in annual revenue, a three-year acceleration in generic entry through a successful Paragraph IV challenge reduces NPV by $3 to $6 billion depending on discount rate and market share assumptions. Tracking Paragraph IV certification counts and litigation activity through platforms like DrugPatentWatch provides earlier warning of entry risk than management guidance alone.
Why do generic companies prefer first-to-file status over second or third filer?
The 180-day exclusivity is awarded only to the first ANDA applicant to file a Paragraph IV certification against a given patent. The second filer earns no exclusivity. The commercial gap between first and second filer is large: the first filer earns 180 days of duopoly profits before multi-generic commoditization; the second filer enters a competitive market from day one. For products where 180-day exclusivity NPV exceeds $100 million, the race to first-to-file status can be measured in days or weeks. Generic companies maintain intelligence programs specifically to identify imminent Paragraph IV filing opportunities before competitors, using patent expiry calendars, prosecution monitoring, and competitive ANDA tracking.
What happens to Paragraph IV litigation incentives if the FDA approves more complex generics through non-ANDA pathways?
The 505(b)(2) application pathway, used for drugs that rely partly on existing published literature or on the FDA’s prior findings of safety or effectiveness for an approved drug, is an increasingly important alternative to the ANDA for complex generics and reformulations. 505(b)(2) applications can include Paragraph IV certifications and can generate 180-day exclusivity under certain conditions. For products where bioequivalence cannot be demonstrated through standard ANDA methods because of complex pharmacokinetics, novel delivery systems, or locally acting drugs, the 505(b)(2) route provides a Paragraph IV challenge option that the ANDA pathway does not. The financial economics are similar, but the regulatory preparation cost is higher because 505(b)(2) applications often require additional clinical bridging studies.
References
LexMachina / National Law Review. (2025, January 15). 2024 Hatch-Waxman Litigation Year in Review. National Law Review. https://natlawreview.com/article/2024-hatch-waxman-year-review
DrugPatentWatch. (2026, February 11). Uncovering the success patterns in modern Paragraph IV litigation. https://www.drugpatentwatch.com/blog/uncovering-the-success-patterns-in-modern-paragraph-iv-litigation/
DrugPatentWatch. (2026, February 23). Top Paragraph IV litigation trends and what they mean for pharma. https://www.drugpatentwatch.com/blog/top-paragraph-iv-litigation-trends-and-what-they-mean-for-pharma/
DrugPatentWatch. (2026, March 22). ANDA litigation: The complete playbook for pharmaceutical patent litigators, IP teams, and institutional investors. https://www.drugpatentwatch.com/blog/anda-litigation-strategies-and-tactics-for-pharmaceutical-patent-litigators/
DrugPatentWatch. (2026, April 17). Paragraph IV certifications: The complete IP strategy. https://www.drugpatentwatch.com/blog/the-paragraph-iv-playbook-turning-patent-challenges-into-market-dominance/
DrugPatentWatch. (2026, April). Multi-district litigation in Paragraph IV cases: The complete litigation & IP strategy playbook. https://www.drugpatentwatch.com/blog/multi-district-litigation-in-para-iv-cases-strategic-considerations/
DrugPatentWatch. (2026, April). Drug patent challenges: The complete strategic playbook for IP teams and portfolio managers. https://www.drugpatentwatch.com/blog/when-science-meets-law-the-art-and-strategy-of-challenging-drug-patents/
Thomas, J. R. (2009). Paragraph IV of the Hatch-Waxman Act and pharmaceutical patent challenges. Congressional Research Service.
DrugPatentWatch. (2026, March 30). How prescription drug prices are determined. https://www.drugpatentwatch.com/blog/how-are-prescription-drug-prices-determined/
DrugPatentWatch. (2026, April 8). Cancer drug ROI: Why pharma makes $14.50 for every $1 spent on R&D. https://www.drugpatentwatch.com/blog/comparison-of-sales-income-and-research-and-development-costs-for-fda-approved-cancer-drugs-sold-by-originator-drug-companies/
Fiercebiotech. (2025, March 25). Drug development cost pharma $2.2B per asset in 2024. https://www.fiercebiotech.com/biotech/drug-development-cost-pharma-22b-asset-2024-plus-how-glp-1s-impact-roi-deloitte
Drug Discovery News. (2026, February 24). Blockbuster drugs face a massive patent cliff in 2026. https://www.drugdiscoverynews.com/blockbuster-drugs-face-a-massive-patent-cliff-in-2026-17019
IntuitionLabs. (2026). Drug patents expiring in 2026: A comprehensive guide. https://intuitionlabs.ai/articles/drug-patent-expirations-2026
Credevo. (2026, March 5). Trending molecules and generic drug opportunities: Blockbuster drugs with upcoming patent expirations (2026–2030). https://credevo.com/articles/2026/03/05/trending-molecules-and-generic-drug-opportunities-blockbuster-drugs-with-upcoming-patent-expirations-2026-2030/
DeepCeutix. (2026, February 2). $300 billion in pharma revenue loses patent protection by 2030. https://deepceutix.com/insights/patent-cliff-reformulation
DrugPatentWatch. (2023, August 5). Pay-for-delay pharma settlements: The complete antitrust & IP strategy guide. https://www.drugpatentwatch.com/blog/the-evolution-of-reverse-payment-cases-courts-and-agencies-approaches-in-pharma-litigation/
DrugPatentWatch. (2026, February 6). The ‘use it or lose it’ rule: Decoding 180-day generic exclusivity forfeiture. https://www.drugpatentwatch.com/blog/the-use-it-or-lose-it-rule-decoding-180-day-generic-exclusivity-forfeiture/
Federal Trade Commission. (n.d.). Pay for delay. https://www.ftc.gov/terms/pay-delay
FDA / BioPharm International. (2026). FDA clarifies how it handles 180-day exclusivity. https://www.biopharminternational.com/view/fda-clarifies-how-it-handles-180-day-exclusivity
DrugPatentWatch. (2026, February 11). The golden six months: Mastering 180-day exclusivity to win the generic drug race. https://www.drugpatentwatch.com/blog/the-golden-six-months-mastering-180-day-exclusivity-to-win-the-generic-drug-race/
FDA-Approved-Rx. (2026, February 8). FDA’s 180-day exclusivity: How first generic applicants gain market advantage. https://fda-approved-rx.net/fda-s-180-day-exclusivity-how-first-generic-applicants-gain-market-advantage
DrugPatentWatch. (2026, February 17). Decoding settlement agreements: How to analyze pay-for-delay and other drug patent deals. https://www.drugpatentwatch.com/blog/decoding-settlement-agreements-how-to-analyze-pay-for-delay-and-other-drug-patent-deals/
DrugPatentWatch. (2025, May 21). Parallel proceedings: Coordinating IPR and Para IV challenges. https://www.drugpatentwatch.com/blog/parallel-proceedings-coordinating-ipr-and-para-iv-challenges/
DrugPatentWatch. (2026, February 11). Uncovering the success patterns in modern Paragraph IV litigation — IPR section. https://www.drugpatentwatch.com/blog/uncovering-the-success-patterns-in-modern-paragraph-iv-litigation/
DrugPatentWatch. (2025, November 19). How safe is your drug patent from PTAB challenges? A strategic guide for pharma leaders. https://www.drugpatentwatch.com/blog/how-safe-is-your-drug-patent-from-ptab-challenges-a-strategic-guide-for-pharma-leaders/
IPWatchdog / Rothwell Figg. (2024, May 22). Winning strategies at the PTAB, Part I: Bring your game. https://ipwatchdog.com/2024/05/22/winning-strategies-ptab-part-bring-game/id=176805/
PTAB Law Blog / Rothwell Figg. (2025, January 6). Trial statistics trends at the PTAB: 2024 edition. https://www.ptablaw.com/2025/01/06/trial-statistics-trends-at-the-ptab-2024-edition/
DrugPatentWatch. (2026, March 22). The Patent Trial and Appeal Board: The definitive analyst’s guide to IPR strategy. https://www.drugpatentwatch.com/blog/understanding-the-patent-trial-and-appeal-board-ptab-a-comprehensive-overview/
DrugPatentWatch. (2025, November 20). Landmark Paragraph IV patent challenge decisions: A strategic playbook for generic manufacturers. https://www.drugpatentwatch.com/blog/landmark-paragraph-iv-patent-challenge-decisions-a-strategic-playbook-for-generic-manufacturers/
DrugPatentWatch. (2026, March 22). Drug patent expiration: The complete strategic guide to loss of exclusivity, lifecycle management, and the $400 billion cliff. https://www.drugpatentwatch.com/blog/the-impact-of-drug-patent-expiration-financial-implications-lifecycle-strategies-and-market-transformations/
EY. (2026, March 19). Navigating pharma loss of exclusivity. https://www.ey.com/en_us/insights/life-sciences/navigating-pharma-loss-of-exclusivity
Labiotech. (2026, March 23). The next pharma patent cliff: How 2026–2032 will reshape revenue. https://www.labiotech.eu/best-biotech/pharma-patent-cliff/
Global Pricing Innovations. (2025, November 4). Patent cliff in pharma: Navigating disruption and creating opportunity. https://globalpricing.com/patent-cliff-in-pharma-navigating-disruption-and-creating-opportunity/
DrugPatentWatch. (2026, March 30). Maximizing ROI on drug development by monitoring competitive patent portfolios. https://www.drugpatentwatch.com/blog/maximizing-roi-on-drug-development-by-monitoring-competitive-patent-portfolios/
Michigan Journal of Economics. (2026, January 11). The pharmaceutical pricing dilemma: Reconciling R&D and manufacturing costs. https://sites.lsa.umich.edu/mje/2026/01/11/the-pharmaceutical-pricing-dilemma-reconciling-rd-and-manufacturing-costs/
ScienceDirect. (2023). Settled: Patent characteristics and litigation outcomes in the pharmaceutical industry. https://www.sciencedirect.com/science/article/abs/pii/S0144818823000479
DrugPatentWatch. (2026, April). Drug patent challenges: When science meets law. https://www.drugpatentwatch.com/blog/when-science-meets-law-the-art-and-strategy-of-challenging-drug-patents/
Supreme Court of the United States. (2013). Federal Trade Commission v. Actavis, Inc., 570 U.S. 136.
Supreme Court of the United States. (2023). Amgen Inc. v. Sanofi, 598 U.S. 594.
Federal Circuit. (2021). GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc., 7 F.4th 1320 (Fed. Cir. 2021).
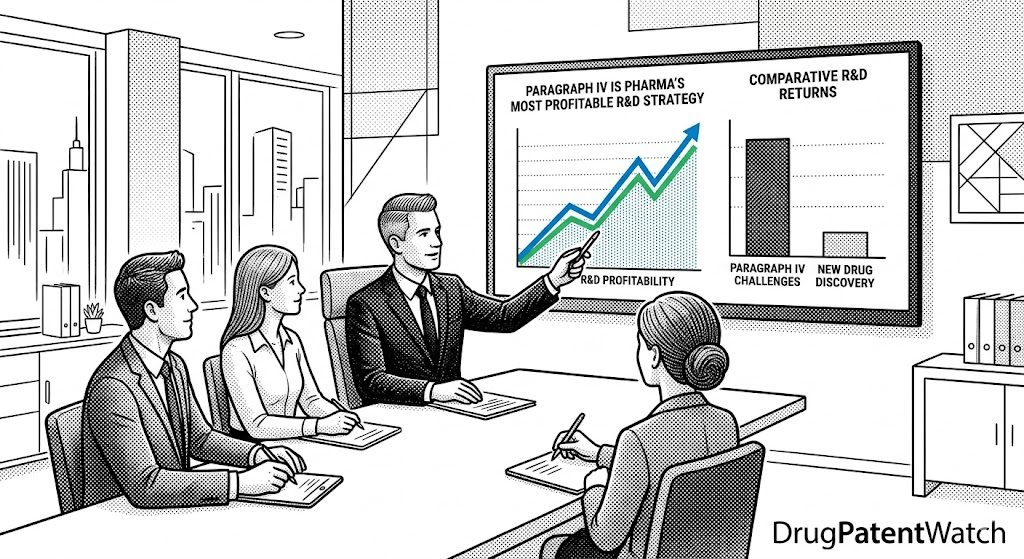
















")









