In pharmaceutical patent litigation, the 180-day generic drug exclusivity is the ultimate prize. It is the “brass ring” 1, a congressionally mandated period of market protection awarded to the first generic applicant that successfully challenges a brand-name drug’s patents. This six-month window, during which the Food and Drug Administration (FDA) is barred from approving subsequent generic applications, can be extraordinarily lucrative. For the first-to-file (FTF) generic, it represents a period of duopoly with the brand manufacturer, a chance to capture significant market share at a price point far above what is possible in a fully commoditized generic market. The financial implications are staggering; this exclusivity can be worth hundreds of millions, sometimes even billions, of dollars in revenue, often accounting for the majority of a generic drug’s total profits.2 In 2020 alone, generics launched with this exclusivity saved the U.S. healthcare system nearly $20 billion.1
But this coveted prize is not a guaranteed trophy. It is a fragile asset, surrounded by a complex minefield of statutory “bear traps.” The very legislation that fortified this incentive, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (MMA), also introduced a series of rigorous forfeiture provisions designed to ensure this powerful tool of competition is not misused to block the very market access it was intended to create.4 For the unprepared or unwary first applicant, this golden opportunity can vanish in an instant, lost to a missed deadline, a strategic miscalculation, or the savvy maneuvering of a competitor.
This report is not a mere recitation of legal statutes. It is a strategic playbook for the intellectual property, business development, and research and development teams on the front lines of the pharmaceutical industry. We will dissect the seven distinct pathways by which a generic company can forfeit its 180-day exclusivity. We will move beyond the letter of the law to explore the FDA’s interpretation, analyze the real-world precedents set by landmark court cases, and distill these complex scenarios into actionable intelligence. For first applicants, this is a guide to defending your most valuable asset. For subsequent filers, this is a manual for identifying and capitalizing on the vulnerabilities of your competitors. In the intricate game of Hatch-Waxman, understanding how to win exclusivity is only half the battle; knowing the seven ways it can be lost is the key to ultimate victory.
The Bedrock of Competition: A Strategic Primer on 180-Day Exclusivity
To master the nuances of forfeiture, one must first grasp the architecture of the system itself. The 180-day exclusivity is not an isolated rule but the central gear in a complex machine designed by Congress to balance two competing, yet essential, public policy goals: fostering brand-name drug innovation and ensuring timely public access to affordable generic medicines.7 This machine, first built in 1984 and significantly overhauled in 2003, operates as an “engineered market,” where success is dictated not by traditional market forces alone, but by a deep, strategic understanding of its intricate legal and regulatory framework.
The Grand Bargain: Hatch-Waxman’s Original Intent
Before 1984, the pharmaceutical market was caught in a paradox. Brand-name drug companies faced a “patent cliff” where their market protection ended abruptly, while generic companies faced a “drug lag,” forced to wait until after a patent expired to even begin the lengthy clinical trials needed for FDA approval. The result was a system that served neither innovation nor access particularly well.
The Drug Price Competition and Patent Term Restoration Act of 1984, universally known as the Hatch-Waxman Act, was a landmark piece of legislation designed to break this deadlock.4 It struck a “grand bargain.” For brand innovators, it offered the restoration of patent life lost to the lengthy FDA approval process, providing a more meaningful period of market protection to recoup massive R&D investments.7 For generic manufacturers, it created the Abbreviated New Drug Application (ANDA) pathway, allowing them to rely on the brand’s original safety and efficacy data, dramatically reducing the time and cost of bringing a generic to market.9
The linchpin of this new system was the Paragraph IV certification. Under Hatch-Waxman, an ANDA filer must make a certification for each patent listed by the brand manufacturer in the FDA’s “Orange Book”.1 While Paragraphs I, II, and III are straightforward statements about a patent’s non-existence, expiration, or the applicant’s intent to wait for expiration, the Paragraph IV certification is an act of commercial warfare.7 By filing a Paragraph IV, the generic applicant asserts that the brand’s patent is either invalid, unenforceable, or will not be infringed by the proposed generic product.7
This was a high-risk, high-reward proposition. Filing a Paragraph IV is considered an act of patent infringement, and it typically triggers an immediate and costly lawsuit from the brand manufacturer, along with an automatic 30-month stay on the ANDA’s approval while the case is litigated.7 To incentivize generics to take on this risk and expense—to act as private attorneys general weeding out weak or invalid patents—Congress created the 180-day exclusivity period.1 The first applicant to file a “substantially complete” ANDA with a Paragraph IV certification would be rewarded with six months of market exclusivity against any other generic competitor who also filed a Paragraph IV challenge.6 This reward was designed to ensure that the trailblazer who bore the cost of litigation would be the first to reap the financial benefits, opening the floodgates for further competition only after their initial reward period.
The MMA’s Course Correction: Plugging the Loopholes
For years, the Hatch-Waxman system worked, spurring a vibrant generic drug industry. However, a significant and anti-competitive loophole emerged. The original statute was clear on how to earn exclusivity but vague on the obligations of the winner. This ambiguity allowed first applicants, sometimes in collusion with brand manufacturers, to “park” their exclusivity.8 A first-filer could win the right to exclusivity but then delay its own market entry, often as part of a “pay-for-delay” settlement with the brand company. Because the 180-day clock didn’t start until the first applicant’s commercial launch, this “parking” strategy could effectively block
all subsequent generic competitors from entering the market indefinitely, perverting the pro-competitive intent of the law.12
Congress responded directly to these abuses with the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (MMA).4 The MMA’s amendments to the Hatch-Waxman Act represented a fundamental philosophical shift. The focus moved from simply
granting exclusivity to actively policing its use. The legislation introduced a series of forfeiture provisions that created a new “use it or lose it” paradigm.9 These provisions, which we will analyze as seven distinct risks, were designed to ensure that the 180-day exclusivity could no longer be warehoused. They imposed a set of time-sensitive obligations on the first applicant, transforming the FDA’s role from a passive gatekeeper to an active enforcer of the law’s pro-competitive spirit.
These new forfeiture rules apply to any drug for which the first Paragraph IV ANDA was submitted after the MMA’s enactment on December 8, 2003.6 If even one Paragraph IV ANDA for a given drug was filed before that date, all subsequent ANDAs for that same drug, regardless of their filing date, are governed by the pre-MMA rules.9 Today, however, virtually all new patent challenges fall under the MMA’s rigorous forfeiture regime. This means that for modern generic strategists, winning exclusivity is no longer the end of the game; it is merely the beginning of a new, high-stakes challenge to maintain it.
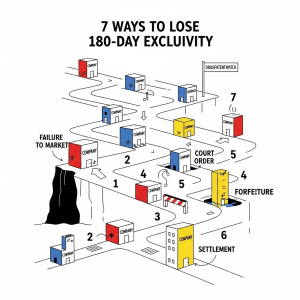
The Forfeiture Gauntlet: A Deep Dive into the 7 Pathways to Losing Exclusivity
The MMA introduced six statutory forfeiture provisions, codified at section 505(j)(5)(D) of the Federal Food, Drug, and Cosmetic (FD&C) Act. However, for strategic purposes, it is essential to analyze them as seven distinct risks. The “Failure to Market” provision, in particular, contains two separate and fundamentally different triggers: one controlled by the first applicant’s own timeline and another that can be activated by a competitor. Understanding each of these pathways in detail is critical for both protecting an earned exclusivity and for identifying opportunities to challenge a competitor’s claim.
The following matrix provides a high-level strategic dashboard of this complex landscape, framing each provision in terms of the primary risk it poses to a first-filer and the opportunity it presents to a subsequent filer.
The 180-Day Exclusivity Forfeiture Matrix
Forfeiture Pathway
Statutory Reference
“Plain English” Trigger
Primary Risk to First-Filer
Primary Opportunity for Subsequent Filer
1. Failure to Market (Self-Imposed)
505(j)(5)(D)(i)(I)(aa)
Fails to market within 75 days of own approval or 30 months of own submission.
Operational delays post-approval can nullify a legal victory.
Limited direct opportunity, but highlights the need for a rival’s operational excellence.
2. Failure to Market (Competitor Trigger)
505(j)(5)(D)(i)(I)(bb)
Fails to market within 75 days of a competitor’s favorable court decision.
Forced into an “at-risk” launch or must cede the market.
Actively control the market timeline by winning own litigation and forcing the FTF’s hand.
3. Withdrawal of Application
505(j)(5)(D)(i)(II)
Affirmatively withdraws ANDA or FDA deems it “constructively withdrawn.”
Loss of exclusivity due to unaddressed application deficiencies or compliance issues.
Monitor competitor’s regulatory filings (e.g., CRLs, 483s) for signs of non-diligence.
4. Amendment of Certification
505(j)(5)(D)(i)(III)
Amends or withdraws P-IV certification for all qualifying patents.
A complete loss in litigation on all challenged patents eliminates exclusivity.
Capitalize on a rival’s total litigation failure.
5. Failure to Obtain Tentative Approval
505(j)(5)(D)(i)(IV)
Fails to get Tentative Approval (TA) within 30 months of ANDA submission.
Most common forfeiture; risk of losing exclusivity due to review delays.
Track the 30-month clock; a competitor’s delay without a clear “excuse” is a major opportunity.
6. Collusive Agreements
505(j)(5)(D)(i)(V)
Enters an agreement found to violate antitrust laws by FTC/AG.
Significant legal and financial risk from “pay-for-delay” type settlements.
Limited direct opportunity due to high bar, but can use antitrust scrutiny to pressure rivals.
7. Expiration of All Patents
505(j)(5)(D)(i)(VI)
All patents that formed the basis for exclusivity have expired.
Pyrrhic victory if the challenged patent expires before a meaningful exclusivity period can run.
A competitor’s short-sighted patent challenge strategy can open the market for all.
Forfeiture 1: Failure to Market – The Self-Imposed Deadline (FDC Act § 505(j)(5)(D)(i)(I)(aa))
This first forfeiture pathway acts as an internal clock, a deadline imposed on the first applicant by its own progress through the regulatory and commercialization process. It ensures that a company cannot receive its final approval and then sit on it, delaying market entry for strategic or operational reasons.
The Letter of the Law
The statute is quite specific. A first applicant forfeits its exclusivity if it “fails to market the drug by the later of” two possible dates. The first of these dates, defined in subpart (aa), is itself a calculation: it is the earlier of:
75 days after the date on which the first applicant’s ANDA is approved and made effective.
30 months after the date of submission of the first applicant’s ANDA.14
Let’s break this down with a practical example. Suppose a company, Genericorp, submits its first-to-file ANDA on January 1, 2024. The 30-month deadline would be July 1, 2026. Now, imagine the FDA grants final approval to Genericorp’s ANDA on March 1, 2026. The 75-day clock would start ticking, setting a marketing deadline of May 15, 2026. Since May 15, 2026, is earlier than the 30-month mark of July 1, 2026, the operative deadline for Genericorp under this subpart is May 15, 2026. If they fail to begin commercial marketing by that date, they risk forfeiture.
FDA’s Interpretation
The FDA’s interpretation of this provision focuses on two key terms: “submission” and “market.” The “date of submission” is generally considered the date the FDA receives and accepts a “substantially complete” application for review.16 The term “commercial marketing” is also critical. While not explicitly defined in the statute, the FDA generally considers it to be the introduction or delivery for introduction into interstate commerce of the drug product. This requires more than token shipments; it involves a genuine effort to make the product available to the public.
This provision effectively transforms the ANDA approval letter from a finish line into a starting pistol. The moment that approval is granted, a 75-day countdown begins. This structure is a direct response to the pre-MMA era of “parking,” making it impossible for a company to gain approval and then indefinitely delay launch.
In the Trenches: Strategic Implications & Mitigation
The primary risk posed by this provision is operational. A company can execute a brilliant legal and regulatory strategy, secure first-to-file status, win or settle its patent litigation, and obtain final FDA approval, only to lose its multi-million dollar exclusivity because of a last-minute failure in the supply chain, a manufacturing issue, or a poorly planned commercial launch.
For a first applicant, mitigation of this risk requires a paradigm shift. Legal, regulatory, and commercial teams cannot operate in sequential silos. The commercial launch plan must be developed in parallel with the regulatory approval strategy. Key mitigation steps include:
Manufacturing Readiness: Ensuring that the designated manufacturing facility is fully compliant with current Good Manufacturing Practices (cGMP), has passed recent FDA inspections, and is prepared to produce launch quantities at scale.
Supply Chain Integrity: Securing the supply of active pharmaceutical ingredients (API) and other raw materials well in advance of anticipated approval.
Commercial Launch Planning: Having packaging, distribution networks, and marketing materials prepared and ready to deploy the moment final approval is received.
For a subsequent filer, this provision offers a more limited, indirect opportunity. While you cannot trigger this specific deadline, you can and should monitor the operational readiness of the first applicant. News of manufacturing delays, FDA Form 483 inspection observations, or API shortages at a first-filer’s facility are all critical pieces of competitive intelligence. They signal a heightened risk that the first filer may stumble after approval, potentially forfeiting exclusivity and opening the market sooner than anticipated.
Forfeiture 2: Failure to Market – The Competitor’s Trigger (FDC Act § 505(j)(5)(D)(i)(I)(bb))
If the first forfeiture pathway is an internal clock, this second pathway is an external one that can be wound up and set ticking by a competitor. This provision is one of the most powerful tools the MMA gave to subsequent ANDA filers, transforming them from passive observers into active agents who can directly influence the market entry timeline.
The Letter of the Law
This provision establishes the second “bookend” date for the “failure to market” calculation. Exclusivity is forfeited if the first applicant fails to market by the later of the date calculated under subpart (aa) (the self-imposed deadline) and the date calculated under this subpart (bb).
The subpart (bb) date is triggered by a legal victory. It is defined as 75 days after a date on which, for each patent that qualified the first applicant for exclusivity, at least one of the following events has occurred:
A court enters a final, non-appealable decision that the patent is invalid or not infringed. This can happen in a case brought against the first applicant or in a case brought by or against any other applicant who has received tentative approval.
A court signs a settlement order or consent decree that includes a final judgment that the patent is invalid or not infringed.
The patent information is withdrawn by the brand company.14
The crucial element here is the phrase “or any other applicant.” This means a second- or third-to-file generic company, by pursuing its own patent litigation to a successful conclusion, can start the 75-day marketing clock on the first applicant.14
FDA’s Interpretation
The FDA’s interpretation has significantly amplified the power of this provision. A key question was one of timing: did the subsequent applicant need to have tentative approval before it secured the favorable court decision? In a pivotal July 2018 letter concerning generic buprenorphine and naloxone sublingual film, the FDA answered with a resounding “no”.17
The agency clarified that the subsequent applicant’s tentative approval can be obtained at any time—before, during, or after the court case—as long as it is in hand by the time the FDA makes the forfeiture determination. This interpretation dramatically expands the circumstances under which a subsequent filer can trigger forfeiture. A subsequent filer can litigate its case and, upon receiving a favorable final court decision, can then work to resolve any remaining issues with its ANDA to secure tentative approval, retroactively activating the 75-day clock on the first filer.17
In the Trenches: Case Study – The Teva/Cozaar Precedent
The strategic complexity of this provision is vividly illustrated by the litigation surrounding Teva’s exclusivity for generic Cozaar and Hyzaar.16 In that case, the brand sponsor, Merck, attempted to use a component of this trigger—the withdrawal of patent information—to unilaterally strip Teva of its exclusivity. After Teva filed its Paragraph IV, Merck chose not to sue and instead simply requested that the FDA delist the challenged patent from the Orange Book. Merck’s strategy was to argue that this delisting triggered a forfeiture event for Teva, which had not yet marketed its product.16
The D.C. Circuit Court of Appeals rejected this maneuver. The court ruled that a forfeiture based on patent withdrawal or delisting can only occur if the withdrawal is the result of an adverse court ruling or an “unfavorable settlement” from the brand’s perspective.16 A brand company cannot, through its own unilateral action, “vitiate a generic’s exclusivity”.16 This decision was critical because it prevented brand sponsors from gaming the system, but it also affirmed the underlying principle: a legal event that resolves the patent dispute can indeed trigger forfeiture. Later, when the same patent expired because Merck failed to pay maintenance fees, the FDA, following the court’s logic, ruled that this unilateral inaction also did not cause a forfeiture.16
Strategic Implications & Mitigation
This provision creates a high-stakes game of chicken. For a first applicant, it introduces the “Catch-22” risk: winning a patent case too early can be just as dangerous as losing.18 If a first filer wins a decisive victory on one patent but is still blocked from final approval by another, later-expiring patent (against which it filed a Paragraph III certification), a subsequent filer could get its own favorable ruling on the first patent, trigger the 75-day clock, and force the first filer to forfeit because it cannot legally market its product yet. The mitigation strategy for the first filer is complex, involving careful litigation timing and potentially seeking stays in litigation to avoid a premature court decision.18
For a subsequent filer, this is a powerful offensive weapon. The strategy is clear: litigate your own patent case aggressively and independently. A victory doesn’t just clear your own path; it puts immense pressure on the first applicant. It forces them to either launch their product “at-risk” (if appeals of the court decision are still pending) or forfeit their exclusivity, clearing the way for all other subsequent filers who are ready for approval. This provision ensures that the litigation strategy of the second and third filers can be just as impactful as that of the first.
Forfeiture 3: Withdrawal of Application – The Unforced Error (FDC Act § 505(j)(5)(D)(i)(II))
This forfeiture pathway addresses situations where the first applicant either gives up its quest for approval or fails to maintain its application in an approvable state. It acts as a backstop to ensure that an applicant who is no longer seriously pursuing a product cannot indefinitely block competitors.
The Letter of the Law
The statute outlines two distinct conditions for forfeiture under this provision:
The first applicant affirmatively withdraws its ANDA.
The Secretary of Health and Human Services (through the FDA) considers the application to have been withdrawn because it does not meet the requirements for approval under section 505(j)(4) of the Act.19
The first condition is straightforward—it’s a voluntary surrender. The second condition, known as “constructive withdrawal,” is where the strategic complexity lies. It grants the FDA discretionary power to declare an application abandoned if the applicant is not diligently pursuing its approval.
FDA’s Interpretation
The concept of “constructive withdrawal” is the FDA’s ultimate tool to prevent the indefinite warehousing of exclusivity by a non-performant applicant. This is not about minor deficiencies or the normal back-and-forth of the review process. Constructive withdrawal comes into play when an applicant shows a persistent failure to address major deficiencies outlined by the FDA in communications like a Complete Response Letter (CRL). If an applicant repeatedly fails to correct significant issues, such as those related to manufacturing compliance, bioequivalence, or labeling, the FDA can conclude that the applicant is not “actively pursuing approval” and deem the application withdrawn for the purposes of forfeiture.21
In the Trenches: Case Study – The Ranbaxy Precedent
While not a formal forfeiture decision under this specific clause, the saga of Ranbaxy Laboratories in the early 2010s is the canonical example of the problem this provision is meant to solve. Ranbaxy was the first applicant for several blockbuster drugs, including generic versions of valsartan (Diovan), esomeprazole (Nexium), and valganciclovir (Valcyte).21 However, the company became embroiled in a massive scandal involving data integrity and systemic violations of cGMP at multiple manufacturing facilities.
This led to a consent decree with the Department of Justice and placed Ranbaxy on the FDA’s Application Integrity Policy List, which effectively froze the agency’s ability to approve its pending ANDAs.21 Because Ranbaxy held the 180-day exclusivity for these key products, its internal compliance failures created a market logjam. Competitors with approvable ANDAs were blocked, not by patents, but by the “parked” exclusivity of a company that could not get its own products approved. The situation became so dire that the FDA eventually had to find alternative grounds to force forfeiture of Ranbaxy’s exclusivities to allow other generics to enter the market. The Ranbaxy case serves as a powerful illustration of why the “constructive withdrawal” authority is so critical; it provides the FDA with a mechanism to break such a logjam and prevent one company’s failures from holding the entire generic market for a drug hostage.
Strategic Implications & Mitigation
For the first applicant, the lesson is clear: maintaining a high-quality, approvable application is not just a regulatory requirement; it is a core component of defending your exclusivity. This means promptly and thoroughly responding to all FDA communications and maintaining impeccable manufacturing and quality control standards. Any sign of non-diligence is a potential crack in the armor that a competitor can exploit.
For subsequent filers, this provision turns a competitor’s regulatory and compliance status into a key source of competitive intelligence. Diligently monitoring a rival’s public disclosures, using the Freedom of Information Act (FOIA) to request communications between the FDA and the first applicant, and tracking FDA inspection reports (such as Form 483s and Warning Letters) can provide early indicators of an applicant’s risk of constructive withdrawal. If a first filer is struggling with significant, unresolved cGMP issues, they are not just a company with production problems; they are a company with a vulnerable multi-million dollar asset. A well-timed citizen petition to the FDA, highlighting the first applicant’s lack of diligence and the harm to public health from delayed generic entry, can be a powerful tool to encourage the agency to exercise its constructive withdrawal authority.
Forfeiture 4: Amendment of Certification – The Strategic Retreat (FDC Act § 505(j)(5)(D)(i)(III))
This forfeiture provision is tied directly to the outcome of patent litigation. It addresses the scenario where a first applicant, after challenging a brand’s patents, effectively concedes defeat by changing its certification.
The Letter of the Law
Forfeiture under this clause is triggered when the first applicant “amends or withdraws the certification for all of the patents with respect to which that applicant submitted a certification qualifying the applicant for the 180-day exclusivity period”.9
The key phrase here is “all of the patents.” To understand this, one must recall that eligibility for exclusivity is established by filing a Paragraph IV certification against at least one Orange Book-listed patent.22 A generic company might challenge multiple patents for the same drug. This provision states that forfeiture only occurs if the company abandons its challenge to every single one of those patents that formed the basis of its first-applicant status.
For example, if a generic company files an ANDA with Paragraph IV certifications against Patent A and Patent B, it is the first applicant. If it subsequently loses its court case regarding Patent A and amends its certification for that patent to a Paragraph III (stating it will wait for the patent to expire), it does not forfeit its exclusivity, provided it continues to maintain its Paragraph IV challenge to Patent B. It would only forfeit if it also amended its certification for Patent B, thereby abandoning its challenge entirely.
FDA’s Interpretation
This is one of the more mechanically straightforward forfeiture provisions, and the FDA’s interpretation aligns closely with the plain language of the statute. The agency’s guidance confirms that as long as a first applicant lawfully maintains at least one Paragraph IV certification that qualified it for exclusivity, forfeiture under this clause does not occur.9 The forfeiture is an all-or-nothing event tied to a complete capitulation in the patent challenge. Because of its clarity, this has been one of the least frequently invoked forfeiture provisions; as of early 2010, there were no reported FDA decisions based on this clause.23
Strategic Implications & Mitigation
The strategic implications of this provision underscore the critical importance of the initial patent selection and challenge strategy. A company’s decision on which patents to challenge is not just a litigation tactic; it is the foundational act upon which the entire exclusivity asset is built.
The primary mitigation strategy against this forfeiture risk is the diversification of patent challenges. Relying on a single Paragraph IV certification to secure exclusivity is an incredibly high-risk, all-or-nothing proposition. If that single challenge fails, the path to forfeiture under this provision is swift and certain.
A more robust strategy involves challenging multiple patents listed for the reference drug, even if the arguments against some patents are stronger than others. This creates a firewall. A loss on one patent does not lead to a total loss of exclusivity. This approach provides insulation and strategic flexibility, allowing the company to continue pursuing its exclusivity based on the remaining challenges. This forces the brand company to defend its intellectual property on multiple fronts and significantly reduces the generic’s risk of a catastrophic, single-point failure that would trigger this forfeiture provision. The initial due diligence conducted by the generic’s legal and scientific teams is therefore a crucial, and largely irreversible, step in building a defensible exclusivity position.
Forfeiture 5: Failure to Obtain Tentative Approval – The 30-Month Gauntlet (FDC Act § 505(j)(5)(D)(i)(IV))
This is, by a significant margin, the most common and most complex pathway to forfeiture. It establishes a hard deadline for the first applicant to demonstrate to the FDA that its application is scientifically sound and approvable, but it pairs this deadline with a highly nuanced and frequently litigated exception.
The Letter of the Law
The core of the provision is a simple clock: a first applicant forfeits its exclusivity if it “fails to obtain tentative approval of the application within 30 months after the date on which the application is filed”.11 Tentative Approval (TA) is a formal notification from the FDA that an ANDA has met all the scientific and regulatory requirements for approval but cannot be granted
final approval because of unexpired patents or other exclusivities blocking the market.24
However, the statute provides a critical escape hatch: “…unless the failure is caused by a change in or a review of the requirements for approval of the application imposed after the date on which the application is filed”.23 This “exception clause” is the heart of the strategic battleground for this provision. Additionally, the FDA Amendments Act of 2007 clarified that if an ANDA’s approval is delayed by the agency’s review of a citizen petition, the 30-month clock is extended by the amount of time the petition was under review.23
FDA’s Interpretation & Case Law
The 30-month rule has been the basis for the majority of forfeiture decisions since the MMA was enacted.23 However, the FDA has also recognized a wide range of circumstances that qualify for the “change in requirements” exception, effectively saving a first applicant’s exclusivity. This has created a parallel track of regulatory disputes where the focus shifts from the science of the drug to the minutiae of the FDA’s review process.
A review of FDA decisions reveals several categories of “excuses” the agency has accepted 25:
Changes in Bioequivalence (BE) Standards: This is a common reason. If the FDA issues new BE guidance or changes its expectations for BE studies after an ANDA has been filed, it will typically excuse a resulting delay. In the case of generic Lialda, the FDA found that Zydus’s failure to get TA within 30 months was caused by a change in BE methodology that was communicated to the company while its application was pending, thus preserving its exclusivity.26 Similarly, in an early case involving Acarbose, the FDA ruled that its own change in requirements for which tablet strength to use in BE studies was a valid excuse.23
Changes to the Reference Listed Drug (RLD): If the brand company changes the RLD’s labeling or formulation after the ANDA is filed, and this change requires the generic applicant to conduct new work, the FDA will generally consider this a valid reason for delay.25
FDA-Initiated Reviews: The agency has excused delays caused by its own internal review of approval standards, such as a review of requirements related to the acceptable size of generic tablets compared to the brand product.25
Citizen Petitions: As now codified, delays caused by the FDA’s need to address a citizen petition that raises scientific or legal questions about a generic’s approvability will toll the 30-month clock.25
This body of precedent shows that while the 30-month deadline is real, the exception clause provides significant room for interpretation. The key is demonstrating a clear causal link between an action or change initiated by the FDA (or a third party, via a citizen petition) and the applicant’s inability to secure TA on time.
Strategic Implications & Mitigation
For a first applicant, navigating this gauntlet requires meticulous record-keeping and proactive communication with the FDA. The goal is to build a comprehensive administrative record that can support an “exception” argument if needed. Every piece of correspondence from the FDA, every change in a USP monograph, every new draft guidance, and every shift in the RLD’s status must be documented and analyzed for its potential to impact the 30-month clock. If a delay seems likely, the applicant should not wait for the deadline to pass but should proactively communicate with the agency to establish the reason for the delay.
For a subsequent filer, this provision represents a prime hunting ground. The 30-month date for any first applicant is a fixed point that can be easily tracked using public information or competitive intelligence services. As a first filer approaches this deadline without a TA, the subsequent filer should intensify its monitoring. If the 30-month mark passes, and there is no obvious, publicly known “excuse” (like a new BE guidance or a pending citizen petition), the first applicant becomes highly vulnerable. The subsequent filer can then prepare to argue to the FDA that forfeiture has occurred, clearing the path for its own approval once its application is ready.
This provision sits at the intersection of patent law, drug regulation, and antitrust enforcement. It was created as a direct legislative response to the “pay-for-delay” settlements that plagued the pre-MMA landscape, where brand and generic companies would agree to delay generic entry in exchange for a payment.
The Letter of the Law
The statute provides for forfeiture if the first applicant “enters into an agreement with another applicant…, the holder of the application for the listed drug, or an owner of the patent… [and] the Federal Trade Commission or the Attorney General files a complaint, and there is a final decision from which no appeal can be taken that the agreement has violated the antitrust laws”.9
The procedural hurdles here are extremely high. Forfeiture requires:
An agreement between the generic and the brand (or another generic).
A formal complaint filed by the FTC or the Department of Justice (DOJ).
A final, unappealable judgment from a court or administrative body that the agreement was illegal.
FDA’s Interpretation and the High Bar
The FDA’s role in this process is passive. The agency has made it clear that it will not conduct its own antitrust analysis of settlement agreements.28 It will only act upon receiving notice of a final, unappealable decision from the proper authorities (FTC or DOJ). This high bar is the primary reason why this provision, as of the dates in the available research, had never been the basis for an actual forfeiture determination by the FDA.13
The timeline is the main obstacle. A complex antitrust lawsuit can take many years to move through the courts and the appeals process. By the time a “final, unappealable decision” is rendered, the 180-day exclusivity period in question has often already been triggered, run its course, and expired, making the forfeiture issue moot.13
In the Trenches: The Post-Actavis World
While this specific forfeiture clause has not been a direct enforcement tool, its landscape was fundamentally altered by the 2013 Supreme Court decision in FTC v. Actavis, Inc..27 Prior to
Actavis, many courts had ruled that a patent settlement was immune from antitrust scrutiny as long as it did not restrict competition beyond the scope of the patent itself. Actavis rejected this view, holding that large, unexplained “reverse payments” from a brand to a generic could be subject to “rule of reason” antitrust analysis to determine if they were an unreasonable restraint on trade.27
The Actavis decision did not automatically make pay-for-delay settlements illegal, but it opened the door for the FTC to challenge them much more aggressively. This has had a profound chilling effect on the most blatant forms of pay-for-delay. While it hasn’t led to forfeitures under this specific clause, it has dramatically increased the risk associated with such agreements. The FTC’s $1.2 billion settlement with Cephalon over its drug Provigil, while not a forfeiture case, demonstrates the massive financial penalties that can result from these arrangements.31
Strategic Implications
The true power of this forfeiture provision lies not in its direct application by the FDA, but in its existence as a deterrent that shapes settlement behavior across the entire industry. The risk of a massive FTC lawsuit and treble damages in private antitrust litigation acts as a Sword of Damocles hanging over any patent settlement negotiation.
This has forced companies to become more creative and subtle in how they structure settlements. Blatant cash payments have become less common, replaced by more complex arrangements that are harder to value and prosecute. A common form of non-cash consideration is a “no-AG commitment,” where the brand company promises not to launch its own “authorized generic” (AG) during the first filer’s 180-day exclusivity period.31 This promise has significant value to the generic—an FTC study found that AG competition can reduce a first-filer’s exclusivity revenues by 40% to 52%—but it is not a direct cash payment, making the antitrust analysis more complex.31
For legal and business development teams, this provision means that every patent settlement must be scrutinized through an antitrust lens. The question is no longer just “can we end the litigation?” but “does the structure of this deal create an unacceptable risk of an FTC investigation or a forfeiture of our most valuable asset down the line?”
Forfeiture 7: Expiration of All Patents – The Ticking Clock (FDC Act § 505(j)(5)(D)(i)(VI))
This final forfeiture provision ties the life of the 180-day exclusivity directly to the life of the intellectual property that gave rise to it. It ensures that the special market protection afforded by exclusivity cannot outlive the underlying patents that were challenged.
The Letter of the Law
The statute is concise and clear: forfeiture occurs if “all of the patents as to which the applicant submitted a certification qualifying it for the 180-day exclusivity period have expired”.9
As with the “Amendment of Certification” provision, the operative word is “all.” If a first applicant qualified for exclusivity by challenging Patent A (expiring in 2028) and Patent B (expiring in 2030), the expiration of Patent A in 2028 does not trigger forfeiture. The exclusivity remains viable based on the continuing challenge to Patent B. Forfeiture would only occur upon the expiration of Patent B in 2030, assuming it had not been invalidated by a court in the interim.
FDA’s Interpretation
The FDA’s interpretation of this provision has been tested by complex, real-world scenarios that highlight the critical importance of timing. The key question is: what happens when the patent landscape shifts during the ANDA submission and review process?
In the Trenches: Case Study – The Restasis Conundrum
The case of generic Restasis (cyclosporine ophthalmic emulsion) presented the FDA with a matter of first impression for this provision.16 The brand sponsor, Allergan, had one patent listed in the Orange Book, which was set to expire on May 17, 2014. A generic company, InnoPharma, submitted the first ANDA with a Paragraph IV certification against this single patent.
However, the patent expired before the FDA had even formally accepted any ANDA for review. Shortly before the original patent’s expiration, Allergan listed new patents for Restasis. This created a conundrum. Did InnoPharma’s exclusivity, based on the now-expired patent, simply vanish? And if so, did that mean all 180-day exclusivity for the product was forfeited forever, even for subsequent companies that were the first to challenge the newly listed patents?
The FDA solicited public comment on the issue, with some subsequent filers arguing that InnoPharma had indeed become a first applicant and had immediately forfeited its exclusivity upon the patent’s expiration. They argued that in the post-MMA world, “once that exclusivity is forfeited, it is gone forever,” and no other patents can give rise to a new period of exclusivity for the same product.16 This case demonstrates how this seemingly simple provision can lead to incredibly complex outcomes when patent portfolios evolve.
Strategic Implications & Mitigation
This forfeiture provision directly links a company’s patent analysis to its commercial strategy. It punishes myopic thinking. A generic company’s goal is not merely to find a patent that is easy to invalidate; it is to find a patent that can be invalidated and has enough remaining patent life to support a commercially meaningful 180-day exclusivity period.
The strategic implication is that target drug selection must be a holistic process. The legal team may identify a patent that is highly vulnerable to an invalidity challenge, but if that patent is set to expire in 18 months, the commercial value of a successful challenge is minimal. The 30-month stay on approval alone could consume the entire remaining patent life. A victory in court would be pyrrhic, as the exclusivity would be forfeited upon the patent’s expiration, potentially before the generic could even launch.
The ideal target for a Paragraph IV challenge is a patent that is both legally weak and has several years of life remaining. This ensures that if the litigation is successful, the resulting 180-day exclusivity period will occur at a time when it provides a genuine and profitable head start against other competitors. This requires a deep integration of legal, regulatory, and commercial analysis long before an ANDA is ever filed.
The Strategic Overlay: Turning Forfeiture Risk into Competitive Intelligence
Understanding the seven forfeiture provisions is foundational, but for the most sophisticated players in the generic pharmaceutical space, this knowledge is not merely a defensive shield. It is an offensive weapon. The forfeiture framework, with its web of deadlines and triggers, creates a predictable, data-driven timeline of a competitor’s potential failure. For a well-prepared subsequent filer, a first applicant’s forfeiture risk is a market opportunity waiting to be exploited.
Building a Proactive Monitoring System
A passive, “wait-and-see” approach is a recipe for being left behind. A successful strategy requires building a proactive, systematic competitive intelligence program focused on monitoring the forfeiture vulnerabilities of every first applicant in your target markets. This system should track several key data streams:
The 30-Month Tentative Approval Clock: This is the most important and predictable data point. For every first-to-file competitor, the date of their ANDA submission should be logged, and a 30-month countdown should be initiated. This is their primary forfeiture deadline.
Litigation Status: Closely monitor the progress of all patent litigation involving the first applicant and other subsequent filers. A final court decision in any of these cases could trigger the 75-day “failure to market” clock.
FDA Regulatory Status: Keep a close watch on the first applicant’s regulatory health. This includes monitoring for the issuance of Complete Response Letters, FDA Form 483 inspection observations at their manufacturing facilities, and any Warning Letters. These are all strong indicators of potential delays that could jeopardize their ability to meet the 30-month TA deadline or achieve operational readiness for launch.
Patent Portfolio Changes: Track any changes to the Orange Book listing for the reference drug. The expiration of a key patent, or the delisting of one following a court decision, can directly impact a first filer’s exclusivity status.
Leveraging Competitive Intelligence Platforms: A DrugPatentWatch Deep Dive
Manually tracking this data across dozens of products and competitors is a monumental task. This is where specialized competitive intelligence platforms become indispensable. A service like DrugPatentWatch aggregates this critical information, transforming it from a scattered collection of public records into a structured, actionable dataset.
Here is how a strategic team can operationalize its monitoring system using such a platform:
Track the 30-Month Clock: The FDA’s Paragraph IV Certification List, easily accessible and searchable within DrugPatentWatch, provides the “Date of First PIV Submission” for each drug product.10 This is the starting gun for the 30-month TA countdown. By setting up alerts for key products, an analyst can be automatically notified as a competitor approaches the 24-month, 28-month, and 30-month marks, prompting a deeper dive into their regulatory status.
Assess the Competitive Landscape: The list also details the “Number of Potential First Applicant ANDAs Submitted”.10 This is a goldmine of intelligence. If there is only one first applicant, their forfeiture opens the market to everyone. If there are multiple first applicants who filed on the same day, they share the exclusivity.11 In this scenario, the forfeiture of one applicant merely consolidates the power of the remaining partners. Understanding this dynamic is crucial for forecasting market entry.
Integrate Data for Predictive Analysis: The true power comes from integrating these data points. Imagine this scenario: Using DrugPatentWatch, your team identifies a first applicant for a blockbuster drug that is now 28 months post-submission with no public announcement of a Tentative Approval. A separate search of FDA’s inspection database reveals that their primary manufacturing facility received a Form 483 with significant observations six months ago. This is no longer just a collection of facts; it is a powerful predictive signal. It indicates a high probability of forfeiture under the 30-month rule and serves as a trigger to accelerate your own launch preparations, ensuring you are ready to enter the market the moment the FDA declares the first applicant’s exclusivity extinguished.
By systematically leveraging these tools, a subsequent filer can move from being a price-taker on the timeline to an actor that anticipates and prepares for market-opening events long before they happen. This transforms regulatory data from a compliance burden into a potent source of competitive advantage.
Conclusion: From Defense to Offense in the Exclusivity Endgame
The 180-day exclusivity remains the most powerful incentive driving patent challenges and early generic entry in the pharmaceutical industry. However, the landscape architected by the Medicare Modernization Act of 2003 is one of constant peril for the first applicant. The seven forfeiture provisions have transformed this coveted prize from a static trophy into a dynamic, high-stakes asset that must be continuously defended against a battery of internal deadlines and external threats.
This report has detailed the intricate mechanics of each forfeiture pathway, from the operational pressures of the “failure to market” clocks to the procedural gauntlet of the 30-month tentative approval rule. We have seen how FDA interpretation and landmark court cases have shaped the strategic realities of these provisions, creating a complex environment where a simple misstep can have multi-million dollar consequences.
But the most critical takeaway is that a purely defensive, compliance-oriented mindset is no longer sufficient. The intricate rules of the forfeiture game create vulnerabilities, and where there are vulnerabilities, there are opportunities. The most successful generic companies of the next decade will be those that master these rules not only to protect their own first-to-file assets but also to go on the offensive. They will build robust competitive intelligence systems to monitor their rivals’ progress, using data from platforms like DrugPatentWatch to create predictive models of forfeiture risk. They will weaponize the “competitor’s trigger” by aggressively litigating their own cases to force a first filer’s hand. They will scrutinize a competitor’s regulatory filings and manufacturing compliance, ready to argue for a “constructive withdrawal” when a rival falters.
In this new era, victory belongs to the prepared. It requires a holistic strategy that seamlessly integrates legal, regulatory, and commercial intelligence. It demands a shift in thinking—from simply winning the race to file, to mastering the entire, complex endgame of forfeiture that follows.
Key Takeaways
“Use It or Lose It” is the New Paradigm: The 2003 MMA fundamentally changed the 180-day exclusivity game from a “win and hold” strategy to a “use it or lose it” imperative, enforced by seven distinct forfeiture provisions.
The 30-Month Clock is the Most Common Trap: The “Failure to Obtain Tentative Approval” within 30 months of submission is the most frequent cause of forfeiture. However, its “change in requirements” exception clause is a critical and highly contested strategic battleground.
Subsequent Filers Are Active Players: Subsequent ANDA filers are not passive observers. The “Competitor’s Trigger” for failure-to-market allows a subsequent filer who wins their own patent case to start a 75-day marketing clock on the first applicant, giving them the power to force the timeline.
Operational Readiness Equals Legal Success: Forfeiture can occur 75 days after final approval if a company is not ready for commercial launch. This elevates the importance of manufacturing readiness and supply chain integrity to the same level as legal and regulatory strategy.
Antitrust Risk Shapes Settlement Strategy: While the “Collusive Agreement” forfeiture is rarely invoked directly, the underlying antitrust risk, amplified by the Supreme Court’s Actavis decision, acts as a powerful deterrent that shapes the structure of all patent settlements.
Forfeiture Risk is Actionable Intelligence: Each forfeiture provision is tied to a predictable date or event. By systematically tracking these triggers using competitive intelligence platforms like DrugPatentWatch, companies can transform public regulatory data into predictive models of competitor vulnerability and market entry opportunities.
Frequently Asked Questions (FAQ)
1. If there are multiple “first applicants” who filed on the same day, how does the forfeiture of one applicant affect the others?
Forfeiture is determined on an applicant-by-applicant basis. Based on the FDA’s decision regarding Nateglinide, if one of several first applicants forfeits its eligibility, the remaining first applicants retain their right to share the 180-day exclusivity.11 The 180-day clock will still be triggered by the first commercial marketing by
any of the remaining eligible first applicants. This creates a complex strategic environment where the actions (or failures) of one partner can significantly impact the timing and value of the exclusivity for all others.
2. Can a first applicant “cure” a forfeiture event after it has occurred? For example, if they miss the 30-month TA deadline but get TA at 32 months, can they regain eligibility?
Generally, no. A forfeiture event, once it has occurred, is considered permanent for that ANDA. The statute does not provide a mechanism for “curing” a missed deadline or reversing a forfeiture. The only viable strategy is to argue that the forfeiture event never legally occurred in the first place. For the 30-month rule, this would involve successfully convincing the FDA that the delay was “caused by a change in or a review of the requirements for approval,” thereby invoking the statutory exception and arguing the 30-month clock was never breached.25
3. How does the launch of an “Authorized Generic” (AG) by the brand company impact the 180-day exclusivity and forfeiture analysis?
An AG launch has no legal impact on the 180-day exclusivity or the forfeiture provisions. The 180-day exclusivity only blocks the FDA from approving other ANDAs with Paragraph IV certifications; it does not prevent the brand from marketing its own generic version under its existing New Drug Application (NDA).31 However, the
strategic and financial impact is immense. An FTC study found that AG competition reduces a first-filer’s revenues during the exclusivity period by an average of 40% to 52%.33 This drastically reduces the value of the exclusivity prize and can heavily influence a generic’s decisions regarding settlement or at-risk launches.
4. What is the FDA’s current stance on making forfeiture determinations? Do they rule proactively?
The FDA’s established practice is to defer making a forfeiture determination until it becomes practically necessary. The agency typically does not rule on forfeiture at the moment a potential trigger occurs. Instead, it addresses the issue only when a subsequent ANDA is otherwise ready for final approval and is blocked solely by a potential first applicant’s 180-day exclusivity.10 This practice of “punting” on the decision creates a period of strategic uncertainty and reinforces the need for all parties to continuously monitor the status of the first applicant.
5. If a first filer successfully uses the “change in requirements” exception to avoid the 30-month TA forfeiture, is there any new deadline for them to obtain tentative approval?
This is a point of significant legal nuance. The statute itself does not impose a new, secondary deadline once the initial 30-month clock is waived via the exception.14 However, this does not give the applicant unlimited time. The applicant is still subject to the general requirement to diligently pursue its application. An excessive or unexplained delay, even after the 30-month clock has been waived, could still expose the applicant to forfeiture under the “constructive withdrawal” provision if the FDA concludes they are no longer actively pursuing approval.21 Therefore, while the hard deadline is removed, the regulatory pressure to advance the application remains.


















")







