The Hatch-Waxman Act created the modern generic drug industry in 1984. Forty years later, the Paragraph IV certification it established remains the most powerful and most contested legal mechanism in pharmaceutical commerce. Annual filings have grown from 77 in 2005 to over 400, and the financial stakes attached to each challenge routinely exceed nine figures. This is not a regulatory curiosity. It is the central engine of generic drug economics, and every major brand company in the world engineers its patent strategy around it.
What follows is a technically dense, commercially grounded analysis of how Paragraph IV litigation actually works, which court decisions define its current terrain, how the landmark case outcomes translate into portfolio and investment decisions, and where the legal and commercial risks are concentrated heading into 2028 and beyond.
The Hatch-Waxman Architecture: Why Paragraph IV Exists and What It Actually Does
What the Pre-1984 Generic Market Tells You About Drug Pricing Power
Before Hatch-Waxman, generics held 19% of U.S. prescription volume. Today that figure sits at roughly 90%. The legislative intervention was that direct. The pre-1984 framework required any company seeking to market a generic to conduct full clinical trials independently, duplicating the safety and efficacy work already completed by the innovator. The cost made most generics commercially impossible to develop. Beyond economics, the legal environment compounded the problem: even conducting the bioequivalence studies necessary to prepare an FDA submission was classified as patent infringement, exposing generics to damages liability before they had a product to sell.
Hatch-Waxman resolved both problems simultaneously. The Abbreviated New Drug Application (ANDA) pathway eliminated the trial duplication requirement, allowing generics to establish equivalence through bioequivalence testing against the innovator’s reference listed drug (RLD). The 35 U.S.C. § 271(e)(1) safe harbor provision exempted activities “reasonably related to the development and submission of information under a Federal law which regulates the manufacture, use, or sale of drugs” from infringement classification. The effect was to make generic development economically viable and legally defensible for the first time.
How the Orange Book Controls Market Entry for Every Small-Molecule Drug
The FDA’s “Approved Drug Products with Therapeutic Equivalence Evaluations,” universally called the Orange Book, is the operative map of the patent landscape for all small-molecule drugs subject to ANDA approval. Innovator companies submitting an NDA must list all patents claiming the drug substance, the drug product, or an approved method of using the drug. This listing obligation creates a publicly searchable database that simultaneously serves as a defensive tool for brands and an intelligence asset for generic challengers.
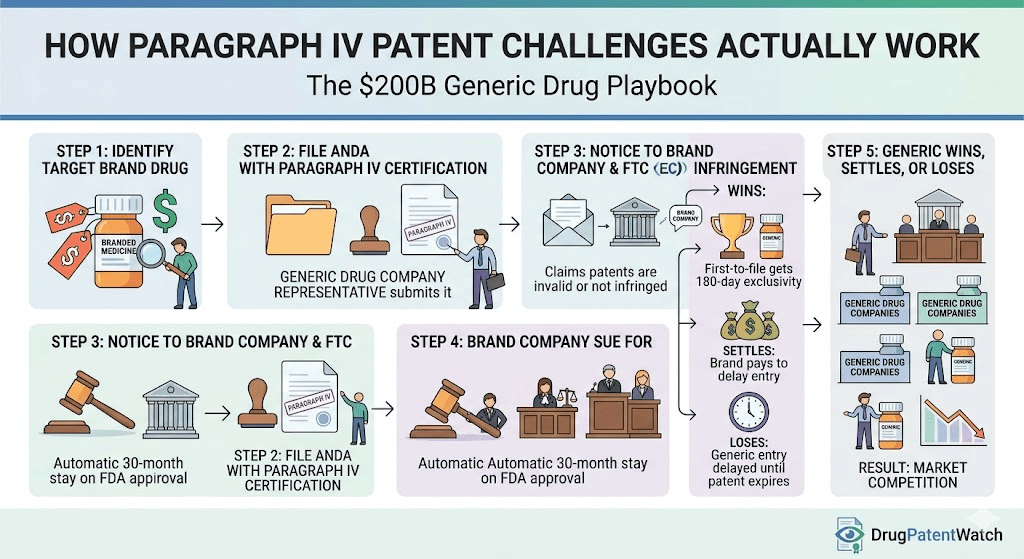
When an ANDA filer addresses the Orange Book, it must certify against every listed patent. Three of the four available certifications are passive: Paragraph I (no patent filed), Paragraph II (patent expired), and Paragraph III (will wait for expiration). Only the Paragraph IV certification is active. Filing it asserts that the listed patent is invalid, unenforceable, or will not be infringed by the proposed generic.
That assertion constitutes statutory infringement under 35 U.S.C. § 271(e)(2), even though no product has been sold and no commercial harm has occurred. This legal fiction, deliberate and consequential, shifts patent disputes from post-launch enforcement actions to pre-launch proceedings. The brand knows the threat is coming. The generic has structured its commercial timeline around the litigation outcome. Both parties enter the dispute with clear financial stakes before a single prescription is written for the generic.
The 30-Month Stay: What It Actually Delays and What It Doesn’t
After receiving a Paragraph IV notice letter, a brand has 45 days to file an infringement action. Filing within that window triggers an automatic 30-month stay of FDA final approval on the ANDA. The stay gives the parties time to litigate before the generic enters the market.
The 30-month stay is widely misunderstood as the primary determinant of generic entry timing. Empirical data shows otherwise. A cohort study of first-to-file generics approved between 2013 and 2020 found a median gap of 3.2 years between the expiration of the 30-month stay and the actual generic launch date. In nearly all cases studied, the stay expired years before the drug reached pharmacy shelves. The delay driver is not the stay itself but what follows it: ongoing litigation, additional patent families filed after the original ANDA, negotiated settlement terms, and remaining exclusivity periods on patents the generic did not challenge.
For a generic strategist, the expiration of the 30-month stay is a commercial inflection point, not a launch authorization. It marks the moment when the generic can choose to launch “at risk” while litigation continues. An at-risk launch immediately generates revenue and applies maximum commercial pressure on the brand, but it exposes the generic to lost-profits damages if the case ultimately resolves against it.
The 180-Day Exclusivity: How the First-to-File Premium Is Earned and Lost
The 180-day first-to-file exclusivity is the financial rationale for most Paragraph IV challenges. The first company to file a substantially complete ANDA containing a Paragraph IV certification against at least one Orange Book patent earns a period during which the FDA cannot approve any other ANDA for the same drug. The result is a duopoly between brand and first-filer generic, a market structure that allows the generic to price at a modest discount, typically 15-25% below brand, rather than the 80-95% discount that follows full generic market entry.
The financial magnitude of this window is decisive. On a drug generating $2 billion annually, a 180-day exclusivity period with a 20% generic discount and 50% market share capture produces approximately $500 million in generic revenue in six months. Industry analysis consistently finds that first-filer generics earn 60-80% of their total product lifetime revenue during this window. The entire economic model of aggressive Paragraph IV litigation depends on it.
The 2003 Medicare Prescription Drug, Improvement, and Modernization Act introduced forfeiture provisions that complicate the first-filer’s position. Failure to market the drug within a specified period after approval, or entering into an agreement the FTC considers anticompetitive, triggers forfeiture. When forfeited, the exclusivity is extinguished entirely. It does not transfer to the next filer. This matters for portfolio strategy: a first-filer that delays its launch while holding a contested exclusivity position can inadvertently open the door to immediate multi-party competition.
Landmark Paragraph IV Decisions: What Each Case Changed and Why It Matters Now
Why the Prozac Challenge Redefined Generic Drug Strategy for Good
Before the Barr Laboratories v. Eli Lilly case resolved in 2000, attacking a blockbuster patent was a theoretical option, not a standard business model. Eli Lilly’s Prozac (fluoxetine) generated several billion dollars in annual revenue and was protected by what appeared to be a solid patent position. Barr filed a Paragraph IV challenge against that position in 1996, asserting invalidity of Lilly’s key patents. The Federal Circuit vacated the district court decision and invalidated the patent in August 2000.
The commercial result was immediate and disproportionate. When Barr launched generic fluoxetine in August 2001, it captured 65% of the total Prozac market within two months. By the end of Barr’s 180-day exclusivity period, the Prozac brand held 16% of its former prescriptions. In the eleven months following launch, Barr’s generic fluoxetine sales reached $367.5 million, representing roughly a third of the company’s total product revenue. Barr’s quarterly gross margin nearly doubled, from 16.8% to 28.7%. Its share price gained over 35% in the month the favorable appellate ruling was announced.
These figures, circulated throughout the generic industry, had an effect equivalent to a proof-of-concept publication. The number of Paragraph IV filings began a sustained climb that continued for the next decade. The model was validated not as a legal theory but as a repeatable, high-return business strategy.
What Investors Are Watching: Pre-LOE Revenue Vulnerability by Market Category
The Prozac case established that therapeutic dominance, physician loyalty, and brand equity offer no protection once a core composition-of-matter patent falls. The larger the revenue base, the larger the incentive for a challenger. Academic research confirms the pattern: drugs in the top decile of market value face a Paragraph IV challenge approximately 90% of the time. Drugs in the bottom deciles are challenged only about 24% of the time.
For investors holding equity in brand pharmaceutical companies, this creates a systematic risk that appears roughly 12-24 months before the patent expiration date actually arrives, when first-filer generics are typically receiving ANDA approvals and settlement timelines are resolving. Drugs approaching loss of exclusivity (LOE) with weak or narrow patent coverage are the most acute near-term exposure.
Revenue at Risk Through 2030: Drugs Facing Primary Patent Expiry
Drug
Company
Primary Patent Expiry
2024 Revenue (Est.)
Lead Biosimilar/Generic Filer
Keytruda (pembrolizumab)
Merck
2028 (biologics exclusivity)
$25B+
Multiple biosimilar developers
Eliquis (apixaban)
Bristol Myers Squibb/Pfizer
2026-2028 (settlement-dated)
$12B
Multiple ANDA filers settled
Ozempic/Wegovy (semaglutide)
Novo Nordisk
2031-2033+
$21B+
Contested; GLP-1 thicket active
Entresto (sacubitril/valsartan)
Novartis
2025-2028
$7B
Multiple challengers
Dupixent (dupilumab)
Sanofi/Regeneron
Biologics exclusivity 2027+
$14B+
Pre-exclusivity
Jardiance (empagliflozin)
Boehringer Ingelheim/Lilly
2025-2028
$7B
ANDA filed
These LOE timelines are active risk variables, not static figures. Settlement negotiations, IPR outcomes, and FDA decisions on new formulation patents all shift the effective entry date.
How FTC v. Actavis Killed Pay-for-Delay and What Replaced It
The Supreme Court’s 2013 decision in FTC v. Actavis is the most consequential judicial intervention in the Paragraph IV settlement market since the Hatch-Waxman Act itself. The case arose from a settlement in the AndroGel (testosterone gel) litigation. Solvay Pharmaceuticals had paid several generic challengers, including Actavis, substantial sums to drop their Paragraph IV challenges and delay entry until 2015, well before the patent’s natural expiry.
The FTC argued that payments from brand to generic in exchange for delayed entry were per se anticompetitive. The Supreme Court rejected per se treatment but also rejected the “scope of the patent” test that had protected most reverse payment settlements in circuit courts. Instead, the Court held that these agreements must be evaluated under the antitrust rule of reason.
The core logic was economic. A patent gives its holder the right to try to exclude, not a guaranteed right to exclude. That right is contingent on validity. A large, unjustified payment from a brand to a generic challenger signals that the brand has doubts about its own patent’s validity and is purchasing market protection it may not be legally entitled to. The Court described this as potential “sharing of monopoly profits” at consumer expense.
Actavis did not ban reverse payments. It mandated that any large, otherwise-unexplained transfer of value from brand to generic be scrutinized for anticompetitive purpose. The FTC subsequently reported a substantial decline in settlements structured around direct cash payments. The risk calculus for both parties changed permanently.
Why No-Authorized-Generic Agreements Are Now Subject to Antitrust Review
FTC v. Actavis left one critical question open: does it apply to non-cash forms of consideration? The Third Circuit answered in King Drug Co. of Florence, Inc. v. SmithKline Beecham Corp. (2015), a case involving GSK’s lamotrigine (Lamictal) settlement with Teva.
The settlement included no direct cash payment. Instead, GSK agreed not to launch an authorized generic during Teva’s 180-day exclusivity period. An authorized generic is the brand company’s own generic version, which it can launch to compete with the first-filer generic during the exclusivity window. One FTC study found that the presence of an authorized generic reduces first-filer revenues during the exclusivity period by an average of 50%.
The Third Circuit held that a no-authorized-generic agreement is economically equivalent to a cash payment. It transfers substantial market value to the generic challenger by protecting its duopoly profits during the exclusivity period. That transfer can serve the same anticompetitive purpose as direct cash: inducing the generic to abandon its patent challenge and agree to delayed entry.
The implication for every subsequent settlement negotiation is direct. Any concession a brand makes to a generic challenger must be justified on pro-competitive grounds or bear the risk of antitrust scrutiny. License agreements, supply deals, side contracts, and no-AG commitments are all subject to economic substance analysis. The form of the transfer is irrelevant. What matters is whether the value transferred, whatever its structure, is explained by legitimate commercial considerations or serves primarily to keep the generic off the market.
Key Patent Expiry Dates and Settlement Entry Windows: The Commercial Map
Settlement agreements in Paragraph IV cases typically specify an authorized entry date, the date on which the generic is permitted to launch regardless of remaining patent life. These negotiated dates represent the commercial resolution of the underlying patent dispute, and they function as de facto LOE dates for revenue modeling purposes.
For Eliquis (apixaban), the major Paragraph IV settlements with generic challengers including Mylan, Teva, and others specify entry dates between 2026 and 2028, depending on the patent claims at issue and the strength of each challenge. Bristol Myers Squibb and Pfizer hold co-promotion rights, and both face proportional revenue erosion as those settlement windows open.
For Xarelto (rivaroxaban), which faced earlier LOE pressure, the settlement structure allowed Bayer and Johnson & Johnson to manage entry timing, though generic competition is now active and the market structure reflects post-LOE erosion.
The critical intelligence exercise for investors and IP teams is mapping actual settlement-authorized entry dates, not nominal patent expiry dates. These two figures often diverge significantly, sometimes by years, and the settlement-authorized date is the operative commercial variable.
Skinny Label Strategy: How GSK v. Teva Changed the Risk Profile of Generic Entry
What the Skinny Label Pathway Actually Allows and Why Brands Use Method-of-Use Patents to Block It
When a drug is approved for multiple indications and some of those indications are protected by method-of-use patents while others are not, a generic manufacturer can file an ANDA that carves out the patented indications from its label, a process called skinny labeling or “section viii” labeling. The generic’s label covers only the unpatented uses. The brand’s method-of-use patent remains nominally intact, but the generic competes for the substantial portion of prescriptions written for non-patented indications.
Brands have responded to this pathway by submitting use codes to the Orange Book that describe their method-of-use patents as broadly as possible. An inflated use code implies patent coverage extends across all approved indications, including unpatented ones, which the FDA uses as a basis for blocking section viii submissions.
The Supreme Court’s 2012 decision in Caraco Pharmaceutical Laboratories, Ltd. v. Novo Nordisk A/S addressed this directly. Novo Nordisk’s repaglinide (Prandin) was approved for three uses, but only one, combination therapy with metformin, was covered by a patent. Novo submitted an overbroad use code to the FDA that implied all three uses were patented. The Court unanimously held that the ANDA counterclaim provision at 21 U.S.C. § 355(j)(5)(C)(ii)(I) allows generics to seek a court order compelling the brand to correct inaccurate patent information, including inflated use codes.
Caraco is essential precedent for any generic pursuing a skinny label strategy against a drug with multiple approved indications. It prevents brands from using administrative overreach in the Orange Book to substitute for substantive patent protection they do not actually hold.
How GSK v. Teva Turned Skinny Labels Into Enterprise Compliance Issues
The Federal Circuit’s 2021 ruling in GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc. fundamentally altered the induced infringement analysis for skinny label products. Teva had carved carvedilol’s congestive heart failure indication (covered by GSK’s method-of-use patent) from its generic Coreg label. The skinny label was procedurally correct. Teva’s problem was its marketing.
GSK presented evidence that Teva had issued press releases describing its product as an “AB-rated equivalent” to Coreg and had used promotional language that did not limit the equivalence claim to the approved non-patented indications. A jury awarded $235 million in damages. The Federal Circuit, applying a “totality of the circumstances” standard, reinstated the verdict after an intermediate reversal. The Supreme Court declined to hear Teva’s appeal, leaving the Federal Circuit’s analysis as controlling law.
The decision’s practical implications extend well beyond Teva’s specific facts. It means that even a technically correct skinny label is not a complete defense against induced infringement if the generic’s broader commercial conduct, press releases, formulary communications, sales representative scripts, pharmacy benefit manager presentations, or investor materials, can be construed as encouraging prescribers to use the product for the patented indication.
From 2015 to 2019, roughly half of all new generic drugs for products with multiple indications entered the market using a skinny label. After the final Federal Circuit ruling in the GSK case in 2021, the rate of skinny label approvals dropped from 56% of susceptible new generics to 20% by 2023. That statistical shift represents a genuine chilling effect, one that delays competition and maintains elevated prices for brand drugs that would otherwise face earlier generic pressure.
What Happens After Loss of Exclusivity When Skinny Labels Are Involved
When a brand drug faces skinny label competition for its unpatented indications, the commercial impact follows a distinct pattern from full LOE. The generic captures volume in unpatented indication segments while the brand retains pricing power in the patented segment. Prescriber-level data typically shows rapid generic penetration in primary care settings, where the unpatented indication often dominates prescription volume, while the brand maintains share in specialist settings where the patented indication is more commonly targeted.
This creates a bifurcated market structure that brands can exploit. By heavily marketing the patented indication through specialist channels, the brand defends its highest-value segment while the generic absorbs commodity-level competition in primary care. The net revenue impact depends on the relative prescription volumes across indications, which varies significantly by drug class.
For investors, the key metric is the indication-level revenue split at LOE entry. A brand where 70% of prescriptions are for unpatented indications faces near-total revenue erosion from day one of skinny label entry. A brand where the patented indication represents 70% of volume maintains substantial pricing power for the duration of that method-of-use patent.
How Patent Thickets Work and Why They Complicate Generic Entry Timelines
AbbVie’s Humira Patent Strategy: 250 Applications and Two Decades of Exclusivity
AbbVie’s adalimumab (Humira) is the most documented example of a patent thicket used to extend exclusivity beyond the natural life of the core composition-of-matter patent. The drug was first approved in 2002. AbbVie accumulated over 250 patent applications covering formulations, dosage regimens, manufacturing processes, methods of use, and device features of the prefilled syringe delivery system. The result was that biosimilar competition in the United States did not begin until 2023, more than two decades after launch.
The commercial value of that two-decade exclusivity is measured in the hundreds of billions. Humira generated over $20 billion annually at peak. The difference between a 2015 biosimilar entry date and the actual 2023 date represents roughly $160 billion in additional branded revenue over the intervening period.
The Humira situation is extreme in scale but not unusual in structure. Data compiled from FDA Orange Book submissions shows that 66% of all patent applications filed for top-selling drugs are submitted after FDA approval of the drug. These are secondary patents, not innovation patents. They are systematically filed as lifecycle management tools designed to extend market protection into periods where the original inventive contribution would otherwise be public domain.
Which Drugs Face the Largest Revenue Cliffs from Patent Thicket Erosion
The challenge in thicket analysis is distinguishing between patents that provide commercially meaningful protection and patents that are primarily litigation impediments. A 300-patent thicket does not necessarily represent 300 distinct barriers. Many secondary patents overlap or share vulnerabilities. An invalidation of one key formulation patent can effectively clear multiple claims simultaneously.
For generic challengers, the standard due diligence process now requires mapping the entire secondary patent portfolio, not just the primary composition-of-matter patents, before committing to ANDA preparation and litigation. This requires evaluating: the prosecution history of each patent to identify double-patenting or continuation-in-part vulnerabilities; the commercial relevance of each claimed modification (some thicket patents cover formulations or dosage strengths that are commercially irrelevant to the generic’s planned product); and the feasibility of engineering around method-of-use claims through skinny labeling.
For GLP-1 drugs, specifically semaglutide (Novo Nordisk’s Ozempic and Wegovy) and tirzepatide (Lilly’s Mounjaro and Zepbound), the patent landscape extends into the early 2030s and is actively growing. Novo holds active patents on the GLP-1 molecular structure, the subcutaneous injection device, the oral formulation, and specific dosing regimens. Challenging this portfolio requires a multi-patent, multi-forum strategy that most generic filers are only beginning to execute.
Why Manufacturing Complexity Functions as a Non-Patent Barrier
Patent thickets are the legal dimension of brand defense strategy. Manufacturing complexity is the commercial dimension. For large-molecule biologics, including monoclonal antibodies, GLP-1 receptor agonists, and fusion proteins, replicating the reference biologic requires sophisticated cell culture infrastructure, proprietary purification processes, and demonstration of structural similarity across a range of analytical tests that the FDA’s 351(k) biosimilar pathway requires.
The manufacturing barrier is separate from patent protection and does not expire. A biosimilar developer that successfully clears the patent thicket through litigation or settlement still faces years of process development and clinical bridging studies before reaching the market. The combination of patent thicket and manufacturing complexity creates a compound barrier that explains why biosimilar penetration rates remain lower than small-molecule generic penetration rates even after exclusivity ends.
For investors assessing the post-LOE revenue model for biologic drugs, the realistic market share erosion trajectory is substantially different from the small-molecule generic experience. Humira lost approximately 35% of its U.S. volume in the first year of biosimilar competition, compared to the 65-80% volume losses that small-molecule brands typically experience in the first quarter after generic entry.
IPR Strategy: Opening a Second Front Against Brand Patents
How Inter Partes Review Changes the Leverage Dynamic in Paragraph IV Litigation
The America Invents Act of 2011 created Inter Partes Review, a trial proceeding before the Patent Trial and Appeal Board that allows challengers to contest patent validity outside the federal district court system. For generic manufacturers, IPR opened a second litigation venue with two structurally favorable differences from district court.
First, the burden of proof for invalidity is lower. District court requires clear and convincing evidence to invalidate a patent. PTAB requires only a preponderance of the evidence. Second, PTAB historically applies a broader claim construction standard. Broader construction makes it easier to find prior art that anticipates or renders obvious the patent’s claims.
These procedural advantages translated into high invalidation rates in the PTAB’s early years. Institution denial rates have increased and invalidation rates have moderated somewhat, but the PTAB remains a significantly more favorable forum for patent challengers than the federal district courts. Early data showed invalidation of challenged claims in over 70% of instituted proceedings.
For a generic company in active Paragraph IV litigation, the IPR creates direct settlement leverage. A brand facing both a district court trial and a PTAB proceeding is managing two timelines simultaneously. A PTAB decision invalidating key claims arrives faster and on more favorable terms than a district court verdict. The threat of a swift IPR victory accelerates settlement discussions and typically shifts the negotiated entry date earlier.
Most Important Ongoing Litigation: Estoppel Risk and the IPR Decision Framework
The IPR’s leverage comes with a structural risk that must be factored into any filing decision. The estoppel provisions of the America Invents Act bar an IPR petitioner, after the PTAB institutes the proceeding and reaches a final written decision, from raising in subsequent district court proceedings any invalidity argument it raised or reasonably could have raised in the IPR. A lost IPR can permanently eliminate the generic’s best invalidity arguments in the parallel litigation.
PTAB discretionary denial adds further complexity. Under the Fintiv factors, the PTAB may decline to institute an IPR petition if the parallel district court case is at an advanced stage, on the theory that duplicative proceedings waste agency resources. This creates a timing constraint: IPR petitions should be filed early, before the district court schedule advances to a point that triggers denial.
The decision to file an IPR therefore requires a multi-variable analysis: the strength of the invalidity arguments, the availability and quality of prior art, the district court timeline, the specific judge’s history with parallel proceeding stays, and the company’s risk tolerance for the estoppel consequence of a PTAB loss. For patent thickets where multiple secondary patents require invalidation, coordinating IPR filings across the portfolio while managing estoppel risk across all challenged claims is a specialized strategic discipline.
Invalidity Doctrines: How Prosecution History Becomes a Generic’s Weapon
How Obviousness-Type Double Patenting Invalidated a Key Celebrex Patent
The Federal Circuit’s 2008 ruling in Pfizer, Inc. v. Teva Pharmaceuticals USA, Inc. concerning celecoxib (Celebrex) illustrates how a brand’s procedural choices during patent prosecution can create a fatal invalidity argument years later.
Obviousness-type double patenting (ODP) is a judicially created doctrine that prevents a patent holder from extending exclusivity by filing a second patent on an obvious modification of an invention already claimed in a first patent. The safe harbor provision at 35 U.S.C. § 121 protects against ODP rejections when the USPTO forces an applicant to split a single application into multiple divisional applications through a restriction requirement.
Teva’s analysis of Pfizer’s prosecution history identified a critical procedural distinction. The challenged patent, the ‘068, was a continuation-in-part (CIP) of the parent application, not a divisional. A CIP, by definition, adds new subject matter not present in the parent. The Federal Circuit drew a categorical line: the § 121 safe harbor applies only to divisional applications. Because Pfizer had filed a CIP rather than a divisional, it had forfeited safe harbor protection. Without the safe harbor, the ‘068 patent was found to be an obvious variant of claims already in an earlier-expiring Pfizer patent and was declared invalid for ODP.
The lesson for generic strategy is specific and actionable. Every target patent’s prosecution history should be examined for CIP filings. A CIP filing that resulted in a patent covering claims that are obvious variations of the parent’s disclosure may be vulnerable to ODP outside the § 121 safe harbor, regardless of its nominal expiry date. This is a pure prosecution-history analysis. It does not require scientific disagreement with the claimed invention. It only requires a careful reading of how the patent application was filed and how it relates to earlier patents in the family.
Why the ANDA Specification Is the Controlling Infringement Document
Sunovion Pharmaceuticals, Inc. v. Teva Pharmaceuticals USA, Inc. (Federal Circuit, 2013) provides the clearest statement of a principle that should govern ANDA preparation from day one. The case involved eszopiclone (Lunesta), where the accused generic’s ANDA product specification described a product that fell within the scope of Sunovion’s patent claims. Dr. Reddy’s Laboratories attempted to resolve the infringement problem not by amending its ANDA, but by submitting a declaration to the district court that it would only market a product with specifications outside the patent scope.
The Federal Circuit rejected this approach categorically. The infringement analysis in a Paragraph IV case addresses the product for which FDA approval is sought. The controlling document is the ANDA. A company cannot “talk its way out of infringement” through courtroom declarations while its FDA submission seeks approval for a product that, as described, meets the patent’s claim limitations.
The practical directive is unambiguous: non-infringement must be engineered into the product and reflected in the ANDA specification before filing. The ANDA is simultaneously a regulatory submission and the primary evidentiary document in the litigation that will follow. Every specification range, every manufacturing tolerance, every analytical test threshold must be reviewed against the claims of all Orange Book patents before the ANDA is filed. Product development, regulatory affairs, and patent counsel must work from a common infringement analysis, not in separate tracks.
How Biosimilar Competition Differs from Small-Molecule Generic Entry
What Makes GLP-1 and Biologic Patent Challenges Different from Traditional Paragraph IV Filings
Biologics do not use the Hatch-Waxman/ANDA pathway. They are governed by the Biologics Price Competition and Innovation Act (BPCIA), which created a biosimilar approval pathway under 351(k) of the Public Health Service Act. The BPCIA has its own “patent dance” procedure, a sequential exchange of patent and product information between the biosimilar applicant and the reference product sponsor that determines which patents will be litigated and when.
The Amgen v. Sandoz case (Federal Circuit 2015, Supreme Court 2017) was the foundational precedent establishing that the patent dance is optional for the biosimilar applicant under some circumstances, though opting out carries specific consequences for the information-sharing process and the litigation timeline. The Supreme Court clarified that Sandoz’s filgrastim biosimilar could provide notice of commercial marketing without completing the full patent dance, while affirming that the 180-day notice period before commercial marketing provides important timing rights to the reference product sponsor.
The commercial dynamics of biosimilar competition differ from small-molecule generic competition in several ways that matter for investment modeling. Biosimilar interchangeability designation, which the FDA grants when a biosimilar has been demonstrated to produce the same clinical result as the reference product in any given patient, allows pharmacy-level substitution without prescriber intervention. Without interchangeability, payer substitution requires prescriber engagement. As of mid-2025, the FDA has granted interchangeability designations for multiple adalimumab biosimilars, which is driving faster formulary substitution for Humira than would otherwise occur.
How Biosimilar Launch Timing Works in the Post-Humira Era
The Humira biosimilar launches, which began in 2023, established the current practical model for biologic LOE transitions. AbbVie had negotiated settlement agreements with all major biosimilar developers, specifying authorized entry dates between July 2023 (U.S.) and early 2024. The agreements included royalty structures that gave AbbVie a royalty stream from biosimilar sales during the early entry period, providing partial revenue continuity beyond LOE.
The first wave of Humira biosimilars, including Hadlima (Samsung Bioepis/Organon), Hyrimoz (Sandoz), Cyltezo (Boehringer Ingelheim), and Amjevita (Amgen), entered the U.S. market in 2023. Amjevita was the first, launching in January 2023 under its authorized entry date. AbbVie responded with its own lower-cost version of Humira, marketed at a substantial discount from the original list price, a form of authorized generic strategy that moderated formulary substitution pressure.
The net result for AbbVie’s U.S. Humira revenue was a decline from approximately $14.4 billion in 2022 to $8.9 billion in 2023, a 38% decline, with further erosion projected through 2025 and 2026 as formulary changes and interchangeability designations accelerate substitution.
For Keytruda (pembrolizumab), Merck’s PD-1 checkpoint inhibitor generating over $25 billion annually, biosimilar development timelines are actively running. Multiple developers including Fresenius Kabi, Samsung Bioepis, and Celltrion have biosimilar programs at various clinical stages. The effective exclusivity cliff will be determined by the interaction between Merck’s patents, any settlement-authorized entry dates negotiated with biosimilar developers, and the completion of regulatory requirements for biosimilar approval.
How Competitors Benefit from Paragraph IV Outcomes: Company-Level Analysis
Which Generic Companies Are Best Positioned to Capture LOE Revenue Through 2030
The generic companies with the strongest Paragraph IV track records through 2024 are Teva Pharmaceuticals, Viatris (formerly Mylan and Upjohn), Sandoz, Sun Pharmaceutical Industries, and Aurobindo Pharma. Each maintains active Paragraph IV pipelines with first-filer positions on multiple pending challenges.
Teva’s Paragraph IV pipeline includes filings against drugs in the cardiovascular, central nervous system, and anti-inflammatory categories. Despite its own financial restructuring through the 2017-2022 period, Teva has maintained ANDA filing volume and has active exclusivity positions that will generate revenue through the LOE windows on several major drugs. Its settlement with GSK in the Coreg litigation remains the defining litigation in its recent history given the $235 million verdict that was ultimately reinstated.
Viatris, following the Mylan-Pfizer Upjohn merger in 2020, holds a large generic portfolio with multiple first-filer positions inherited from Mylan’s historically aggressive PIV filing strategy. Mylan’s challenge to Pfizer’s atorvastatin (Lipitor) patents, which led to a settlement and authorized entry date that produced one of the most profitable 180-day exclusivities in generic history, remains the benchmark for how first-filer economics work at scale.
For the biosimilar segment, the most commercially active developers include Samsung Bioepis, Sandoz, Celltrion, Amgen Biosimilars, and Fresenius Kabi. Each has products in various stages of FDA review or commercial launch across the adalimumab, infliximab, bevacizumab, trastuzumab, and rituximab categories.
How Bristol Myers Squibb and Pfizer Are Managing the Eliquis LOE
Eliquis (apixaban), co-promoted by BMS and Pfizer, generated approximately $12 billion in 2023 U.S. revenue. Multiple Paragraph IV challengers filed ANDAs and were met with litigation. Settlements with the principal challengers, including Mylan, Teva, and others, established authorized entry dates generally in the 2026-2028 range, depending on the challenge and the specific patents at issue.
BMS and Pfizer’s response to the approaching LOE has included preparing for the inevitable authorized generic launch, seeking patent term extension where available, and maximizing the drug’s formulary presence ahead of the transition. The commercial reality is that Eliquis generates a combined contribution of several billion dollars annually to both companies, and the settlement-authorized entry dates represent commitments from which neither company can deviate without breaching the settlement agreements.
For BMS, the Eliquis LOE arrives against a backdrop of continued revenue from Opdivo (nivolumab), which faces its own biosimilar development timeline, and the ramp-up of newer assets like Reblozyl and Camzyos. For Pfizer, which holds co-promotion rights rather than the NDA, the revenue impact is substantial but partially offset by the company’s diversified portfolio across vaccines, oncology, and rare disease.
Investment Strategy: How Paragraph IV Intelligence Translates to Portfolio Decisions
How to Use LOE Timelines and Settlement Entry Dates in Pharma Equity Valuation
Equity analysts and portfolio managers who model pharmaceutical company valuations without granular Paragraph IV intelligence are working with structural blind spots. The nominal patent expiry date shown in most financial models is often not the operative commercial variable. Settlement-authorized entry dates, which are typically negotiated well in advance of the nominal expiry and often precede it by years, define the actual revenue cliff.
The intelligence gap has commercial consequences. At the drug level, a brand company that negotiates a 2026 entry date on a drug whose nominal patent doesn’t expire until 2030 will see LOE revenue erosion four years earlier than a model using only patent expiry dates would project. For drugs with $5-10 billion in annual revenue, that gap translates to $20-40 billion in valuation error if discounted cash flow models use the wrong baseline date.
Several data inputs are required for accurate LOE modeling. First, the identity and entry date of all settled first-filer generic challengers, as settlement terms are sometimes disclosed in litigation filings or SEC disclosures but must often be inferred from ANDA approval timing and generic launch announcements. Second, the status of any unsettled Paragraph IV challenges, which represent potential at-risk launches even before the negotiated entry date if a challenger obtains an early favorable ruling. Third, the list of secondary patents that may provide additional protection beyond the settlement date if they were not included in the settled challenge.
Common Investor Questions on Paragraph IV and Loss of Exclusivity
Q: Why don’t brand companies simply let every patent challenge go to trial and win?
Because winning costs money, takes time, and is not guaranteed. Average pharma patent litigation costs approximately $5 million per case. Cases with significant prior art disputes or prosecution history vulnerabilities carry substantial invalidity risk even for commercially successful drugs with apparently strong patents. The expected cost of litigation, weighted by the probability of losing, must be compared against the cost of settling on commercially tolerable terms. Settlements where the brand negotiates a favorable entry date, maintains royalty rights, or avoids an authorized generic commitment during the first-filer’s exclusivity window are often financially superior to litigating to conclusion.
Q: Can a brand company prevent a Paragraph IV challenge by not listing a patent in the Orange Book?
A brand company is required by statute to list any patent that claims the drug substance, drug product, or approved method of use. Failure to list a qualifying patent, or intentional misinformation in the listing, can result in removal from the Orange Book and loss of the Hatch-Waxman litigation protections, including the 30-month stay. Brands do, however, have discretion in how broadly they describe their use codes, and Caraco addressed the consequences of exploiting that discretion through inflation.
Q: What happens to the 180-day exclusivity if the first filer settles with the brand?
The settlement terms typically specify the authorized entry date, at which point the first filer launches its product and the 180-day exclusivity clock begins running. Subsequent filers cannot receive final ANDA approval until the first filer’s exclusivity either expires after 180 days or is forfeited. A first filer that enters a settlement agreement meeting the FTC’s definition of an anticompetitive agreement may trigger forfeiture, eliminating the exclusivity entirely. Modern settlements are structured to avoid this outcome, but the risk requires careful antitrust analysis of every settlement term.
Q: How do Paragraph IV outcomes affect drug pricing after generic entry?
The price trajectory after generic entry depends primarily on the number of competing generic manufacturers. During a 180-day exclusivity period with a single first-filer generic, prices typically fall 15-25% from the brand’s list price. When the exclusivity expires and additional generics enter, prices decline rapidly, often reaching 80-95% below brand list price within the first year. Brand products that survive this transition typically do so by retaining a narrow specialist segment, relying on payer contracts that maintain formulary position for the brand, or benefiting from patient loyalty programs that shift cost burden to the healthcare system rather than the patient.
Key Takeaways
The Hatch-Waxman Paragraph IV certification is not simply a regulatory filing mechanism. It is the primary driver of generic drug market economics, the central battleground for pharmaceutical patent strategy, and a major determinant of brand company valuations across a horizon extending fifteen years into the future.
Several high-value conclusions emerge from the case law and commercial analysis presented here.
Barr v. Lilly (2000) established that no drug is too large or too profitable to challenge. The 180-day exclusivity reward structure is designed to make that challenge economically rational, and it works. The result is a competitive dynamic where any drug with meaningful revenue and a challengeable patent faces a high probability of Paragraph IV attack.
FTC v. Actavis (2013) ended the era of straightforward pay-for-delay settlements and replaced it with a regime where every settlement term requires antitrust analysis. The “payment” concept has been extended by King Drug (2015) to cover non-cash value transfers, including no-authorized-generic agreements.
GSK v. Teva (2021) made skinny labeling an enterprise compliance problem, not just a regulatory filing exercise. The 36-percentage-point decline in skinny label approvals from 2021 to 2023 shows the chilling effect is already material, and it is likely producing delayed generic competition across a range of multi-indication drugs.
Pfizer v. Teva on celecoxib and Sunovion v. Teva on eszopiclone together define the two primary technical battlegrounds of Paragraph IV litigation: the prosecution history of the target patent and the specification of the ANDA itself. Both require pre-filing diligence that most generic companies treat as an afterthought rather than a strategic foundation.
The IPR pathway at PTAB creates genuine second-front leverage but requires careful management of estoppel risk and PTAB timing discretion. Patent thickets, deployed aggressively by companies like AbbVie, mean that clearing a single composition-of-matter patent is rarely sufficient for market entry. And the biosimilar competitive dynamic differs enough from small-molecule generics that the Hatch-Waxman playbook cannot be applied to BPCIA cases without significant adaptation.
For investors, the actionable intelligence is: know the settlement-authorized entry dates, not just the nominal patent expiry dates; assess the density and quality of secondary patent coverage; track IPR institution and outcome data for pending PTAB challenges; and model biosimilar market share erosion on a per-drug basis using indication-level prescription data, not aggregate revenue projections.
The generic companies that will generate disproportionate returns from the current LOE cycle are those that combine rigorous patent portfolio analysis with disciplined ANDA preparation, multi-forum litigation coordination, and antitrust-aware settlement strategy. The brands that will manage LOE transitions most effectively are those that deploy patent thickets with defensible secondary claims, negotiate settlement terms early enough to secure favorable entry dates, and invest in post-LOE brand defense strategies calibrated to the specific physician and payer dynamics of their therapeutic category.
Frequently Asked Questions
Is the skinny label strategy still viable after GSK v. Teva?
Yes, with substantially more compliance infrastructure. The legal pathway remains open. The Caraco precedent preserves the ability to challenge overbroad use codes. The GSK v. Teva risk is not that skinny labels have been banned but that marketing conduct can substitute for label language in an induced infringement analysis. A generic that limits all commercial communications to the approved non-patented indications, avoids broad equivalence claims, and implements enterprise-wide legal review of promotional materials can manage the risk. The 20% approval rate for skinny label generics in 2023 compared to 56% in 2021 shows that many companies are choosing not to manage the risk rather than investing in the compliance infrastructure to do so responsibly.
How does an IPR petition affect pending Paragraph IV district court litigation?
Filing a parallel IPR creates a second front that applies settlement pressure and provides an alternative path to patent invalidation. The PTAB’s lower burden of proof and broader claim construction can make invalidity arguments more successful than in district court. However, if the PTAB institutes the IPR and ultimately upholds the patent claims, the estoppel provisions bar the filer from raising any invalidity arguments in district court that were raised or could have been raised in the IPR. A lost IPR can be more damaging to a generic’s litigation position than never having filed at all. The decision requires an honest assessment of the prior art strength and the consequences of losing the estoppel arguments in the parallel proceeding.
What is the financial model for a generic company during the 180-day exclusivity period?
During the 180-day exclusivity, the first-filer generic competes in a duopoly with the brand. Typical pricing discounts run 15-25% below the brand list price. The first-filer typically captures 40-60% of total prescriptions during this period while the brand retains the remainder. For drugs generating $1 billion or more in annual revenue, the first-filer’s six-month revenue from exclusivity often falls in the $100-500 million range depending on the drug’s market size, the pace of conversion, and whether the brand launches an authorized generic. Companies typically earn 60-80% of their total lifetime revenue on a given product during this window.
How do settlement-authorized entry dates work when multiple generics have filed Paragraph IV certifications against the same drug?
Each generic challenger negotiates its own settlement with the brand company. Settlement terms are typically confidential, but they often include authorized entry dates that may differ among the various filers. The first-to-file generic’s authorized entry date typically arrives first, and its 180-day exclusivity blocks subsequent filers from receiving final approval until the exclusivity expires. Subsequent settlers often negotiate entry dates that begin shortly after the first-filer’s exclusivity ends, creating a structured wave of generic entry rather than a simultaneous multi-party launch.
What is the investor significance of a brand company receiving a Paragraph IV notice letter?
Receipt of a Paragraph IV notice letter signals that at least one generic challenger believes the drug’s patent protection is vulnerable and is prepared to fund multi-year litigation to prove it. The financial significance depends on the drug’s revenue scale, the remaining nominal patent life, the strength of the challenged patents, and whether the company has secondary patent protection the challenge does not address. For large-cap pharma companies, a single PIV notice letter on a drug generating under $500 million in annual revenue may represent a rounding error in the equity valuation. For mid-cap or small-cap companies where one or two drugs represent the majority of revenue, the same letter can trigger material equity re-pricing.


















")







