Generic drugs fill more than 90% of U.S. prescriptions yet capture only around 18% of total drug spending. That gap tells you almost everything about the structural economics of the generic industry — and why many manufacturers are exiting products, watching margins collapse, and staring at a regulatory and legal environment more hostile than at any point since the passage of Hatch-Waxman in 1984.
The business of making cheap drugs is, paradoxically, very hard. You compete on price, manufacture in a heavily regulated environment, depend on APIs often sourced from a handful of overseas facilities, face patent litigation from brand-name companies with nine-figure legal budgets, and then sell into a buyer market where three pharmacy benefit managers control access to 80% of U.S. prescriptions [1]. The reward for winning this game is often single-digit gross margins on products that can be knocked off by the next low bidder within months of launch.
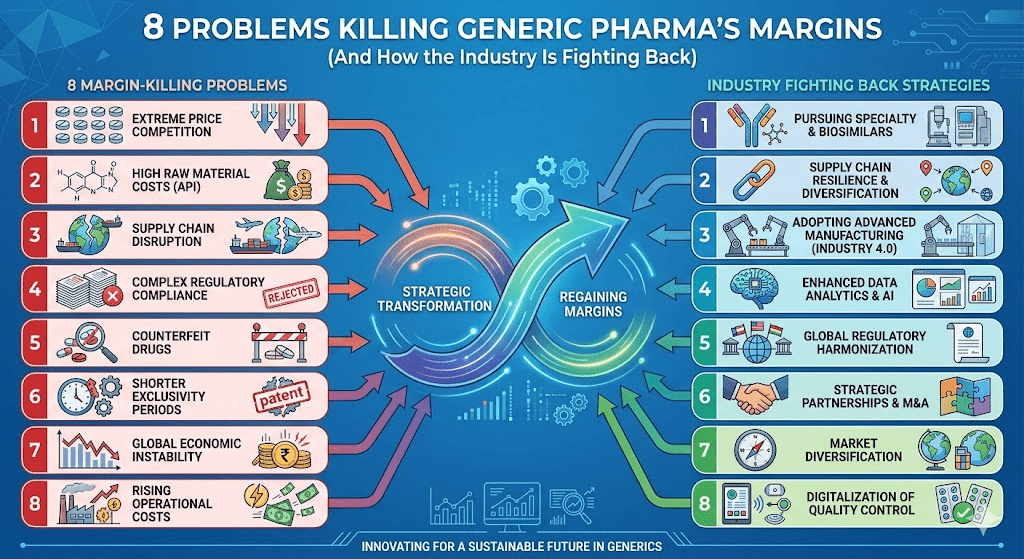
This article maps the eight structural problems that define the generic industry’s current condition. Each one is real, documented, and consequential for the companies that supply the backbone of American healthcare.
The Numbers That Frame Everything
Before getting to the problems, a few figures that put the stakes in context.
The U.S. generic drug market was valued at approximately $95.87 billion in 2024 and is projected to reach $131.8 billion by 2033 [2]. That growth sounds like good news until you recognize that it is driven almost entirely by volume — more prescriptions, more molecules coming off patent — not by price increases. Between 2025 and 2030, branded drugs generating an estimated $200 billion to $400 billion in annual sales will face patent expiry [3]. Generics manufacturers are supposed to be the beneficiaries of that expiry wave. In practice, for many of them, it will intensify the very problems already crushing their margins.
Drug shortages are already at historic levels. In the first quarter of 2024, active drug shortages in the U.S. hit 323 — an all-time record, surpassing the prior record of 320 set in 2014 [4]. These shortages are concentrated in generics, particularly older, low-margin products where manufacturers have exited rather than absorb losses.
These are not unrelated facts. They are the same story told from different angles.
PROBLEM 1 Price Erosion That Has No Floor
How Fierce Competition Destroys the Economics of Generic Entry
When a brand-name drug loses its patent and the first generic enters the market, the initial price is usually 20% to 30% below the branded price. That can still generate reasonable margins for the first mover. The problem starts roughly six months later, when the next ANDA approval comes through, and the one after that, and so on.
For small-molecule blockbusters with thin secondary patent protection, the initial year of multi-source generic competition typically produces an 80% to 90% price and volume decline from the branded baseline [3]. A drug that was generating $2 billion annually for its innovator can be reduced to a handful of commodity suppliers fighting over a $200 million generic market — at prices so compressed that any manufacturing hiccup makes the product economically unviable.
The price erosion mechanism is well understood: more ANDA filers means more competition; more competition means lower bids in GPO tender processes; lower bids set the new market price; every manufacturer either follows the price down or loses volume. In practice, the number of active competitors on any given generic product is the single most reliable predictor of the margin available to each participant.
“Generic drugs now account for over 90 percent of all prescriptions dispensed in the United States yet represent only about 18 percent of total U.S. drug spending.”— DrugPatentWatch, The Global Generic Drug Market: Trends, Opportunities, and Challenges, 2026 [5]
The industry’s theoretical response — file early, earn first-generic exclusivity, extract premium pricing for 180 days before competition arrives — is sound in theory. In practice, first-generic exclusivity under Hatch-Waxman is shared among all applicants who file on the same day, which for high-value products often means dozens of simultaneous filers. The 180-day exclusivity period has in many cases become a marginal advantage rather than a structural one.
The Commodity Trap on Solid Oral Dosage Forms
The worst erosion occurs in the most common generic category: solid oral dosage forms — tablets and capsules of older small molecules. These products have the lowest manufacturing barriers, the most ANDA filers per product, and the least differentiation. Companies that built their business on commodity solid orals have spent the past decade watching those revenues structurally deflate. Teva, once the world’s largest generic company by revenue, has been restructuring its portfolio for years partly in response to this dynamic, shedding products and headcount in an effort to right-size its cost structure to margin realities [6].
The KPMG analysis of the generics industry found that when consolidated buyers exert price pressure, generics can sell for only 20% of branded drug prices or less [7]. On a product with significant API costs, complex manufacturing requirements, and regulatory compliance overhead, selling at 20% of branded price frequently means selling below the true cost of quality manufacture.
What Sustainable Looks Like
The companies navigating price erosion most successfully have moved up the complexity curve — away from commodity solid orals toward complex dosage forms (injectables, inhalers, transdermals, microspheres) where the number of potential filers is smaller, development costs create a natural barrier, and the first-mover premium lasts longer. This is not accessible to all manufacturers; the capital and technical requirements for complex generics are substantially higher.
PROBLEM 2 Buyer Concentration That Functions as an Oligopsony
GPOs, PBMs, and the Structural Power to Set Prices Below Sustainable Levels
The U.S. generic market has a buyer concentration problem that most industries would recognize as structurally dangerous. On the supply side, hundreds of generic manufacturers compete aggressively for volume. On the demand side, a handful of entities control access to the market at terms they largely dictate.
Three pharmacy benefit managers — CVS Caremark, Express Scripts (Cigna), and Optum Rx (UnitedHealth Group) — collectively managed 80% of U.S. prescription claims as of 2024 [8]. The three largest wholesale buying consortia represent roughly 90% of all generic drug purchases by volume [7]. Ninety-eight percent of U.S. hospitals use at least one group purchasing organization [9].
This is not a competitive buyer market. It is an oligopsony — many sellers competing for the favor of a small number of buyers with enormous negotiating power. The consequence is that even when a generic manufacturer reduces its cost structure to competitive levels, the GPO or PBM can extract the majority of those cost savings as price concessions rather than allowing margin to accrue to the manufacturer [5].
The Lowest-Price-Wins Logic and Its Perverse Outcomes
GPO contracting is typically structured around lowest-price-wins tender logic. Manufacturers face a binary: bid aggressively and accept margin compression, or exit the contract and lose volume. Many have chosen to exit — not just from specific contracts, but from entire product categories.
Teva’s 2019 discontinuation of vincristine, a pediatric oncology drug, illustrates the endpoint of this logic. Low profitability made the product commercially unsustainable; Teva exited; a shortage followed [7]. The same pattern has repeated across dozens of products, particularly older generic injectables where manufacturing complexity is high and price pressure is severe.
The FTC’s interim staff reports from July 2024 and January 2025 revealed a parallel problem at the specialty generic level: PBM-affiliated pharmacies extracted over $7.3 billion in revenue above estimated acquisition costs for specialty generic drugs between 2017 and 2022, with that excess revenue growing at 42% annually [10]. The commission found that 63% of specialty generic drugs dispensed by PBM-affiliated pharmacies were marked up more than 100% over acquisition costs between 2020 and 2022, and 22% were marked up by more than 1,000% [10]. Manufacturers received commodity prices. PBMs captured the spread.
Private-Label PBMs and Vertical Integration
The most recent evolution — the “private label” PBM model — represents an additional layer of buyer power. CVS Caremark, through its Cordavis subsidiary, negotiated directly with AbbVie to become the preferred supplier of an authorized biosimilar version of Humira, effectively converting the world’s largest drug franchise into a white-label product. AbbVie retained manufacturing volume; CVS captured the margin differential [11]. Other PBMs have since pursued similar structures. For generic manufacturers competing in the biosimilar space, this trend signals that the value chain will increasingly be captured upstream, not at the manufacturing level.
PROBLEM 3 Patent Thickets and Branded Companies’ Litigation Playbook
How Innovators Use IP Strategy to Delay Generic Entry by Years
The Hatch-Waxman Act was designed to create a structured pathway for generic entry. Brand-name companies have spent the decades since its passage developing sophisticated counter-strategies that use the same legal framework to delay competition — legally, for as long as possible.
The core mechanism is the “patent thicket” — surrounding a blockbuster drug with dozens of secondary and tertiary patents covering formulations, dosing regimens, metabolites, manufacturing processes, and patient populations, staggered in expiration to create an effective exclusivity wall extending years or decades past the expiry of the original composition-of-matter patent. Orange Book patent listings for complex drugs can include 50 or more separate patents, each of which must be challenged individually by a Paragraph IV (PIV) filer, each of which triggers a potential 30-month automatic stay on FDA final approval.
Serial Litigation: The One-Bite Principle Under Assault
A 2025 study in a peer-reviewed pharmacology journal documented a pattern that practitioners call “serial patent litigation” — brand companies filing successive lawsuits built on new but substantively indistinguishable patents after earlier challenges have been resolved [12].
The mirabegron (Myrbetriq) case is instructive. After an initial Hatch-Waxman case settled in 2020 with generic entry expected in 2024, Astellas pursued four additional lawsuits, each built on newly filed patents. As of the study’s publication, only two firms had launched in 2024 under the threat of substantial damages [12]. Similar patterns appeared with bimatoprost (Latisse), aflibercept (Eylea), and tasimelteon (Hetlioz) [12].
The practical effect is that a generic company can win a Paragraph IV challenge — establish that the brand’s patents are invalid or not infringed — and still face years of subsequent litigation delay. Each new lawsuit requires legal resources, creates uncertainty around launch timing, and forces the generic company to weigh the risk of launching at-risk (before litigation concludes) against waiting through potentially several more years of 30-month stays.
Pay-for-Delay Agreements Under Antitrust Scrutiny
The Federal Trade Commission has pursued “reverse payment” or “pay-for-delay” settlements for years — arrangements where a brand company pays a generic challenger to accept a delayed entry date rather than launch at risk. The FTC v. Actavis Supreme Court decision in 2013 established that such payments can violate antitrust law, but the settlements have continued in various forms. Teva paid $100 million to resolve reverse-payment litigation related to certain HIV medicines as part of a broader 2024 settlement package [13].
Tools for Navigating the Thicket
Competitive intelligence platforms like DrugPatentWatch have become core infrastructure for generic companies’ portfolio strategy. By aggregating FDA Orange Book data, ANDA filing records, Paragraph IV certification filings, litigation dockets, and patent expiration timelines, DrugPatentWatch allows analysts to model the realistic entry date for any target molecule — accounting for the full stack of secondary patents and the probability of litigation outcomes rather than just the nominal compound patent expiry [14]. This kind of patent landscape analysis has shifted from a nice-to-have to an operational necessity for companies deciding whether to file ANDAs on contested molecules.
PROBLEM 4 cGMP Compliance Failures and the Cost of Manufacturing Overseas
Data Integrity Violations, Warning Letters, and Import Alerts
The generic drug industry’s manufacturing base is heavily concentrated in India and China. India alone accounts for roughly 40% of the generic drugs approved for the U.S. market, and China supplies approximately 80% of the active pharmaceutical ingredients used globally [15]. This concentration creates two interrelated problems: supply chain fragility and persistent cGMP compliance failures at overseas facilities.
FDA warning letter data from 2025 reveals a pattern of significant regional disparity in compliance performance. A review of 85 warning letters issued through December 2025 found that Indian sites received warning letters with associated data integrity concerns at a 60% rate, compared to 21% for Chinese sites and 10% for U.S. sites [16]. Data integrity violations — which include deleted or overwritten raw analytical data, backdated records, and audit trail manipulation — represent the most serious category of cGMP deficiency because they undermine the entire quality system’s evidentiary foundation [17].
What the Warning Letters Actually Describe
A 2024 FDA inspection of one Indian manufacturer found live rodents and birds in storage areas for finished drug products and packaging materials, along with animal feces and feathers on walls, floors, and materials [18]. The company had not tested incoming materials for identity, relied solely on unverified certificates of analysis from suppliers, and had used a critical raw material known to have been supplied with incorrect labeling — without performing identity testing [18]. This is an extreme case, but it illustrates the gap between the regulatory expectations embedded in cGMP and the operational reality at some overseas facilities.
For generic companies with supply chains dependent on overseas API sources, a single warning letter to a key supplier can create a product supply gap that takes 12 to 24 months to resolve — long enough to lose formulary position and customer relationships that may not return. FDA import alerts, which ban products from affected facilities from entering the U.S. market, have affected a growing number of Indian and Chinese manufacturers in recent years [19].
The Compliance Investment Squeeze
The irony is that the same price pressure that compresses margins also limits the capital available for quality system upgrades. Building the data integrity infrastructure, validation documentation, and quality oversight systems that meet FDA expectations costs money — money that manufacturers bidding at the lowest possible price in GPO tenders are under pressure not to spend. The result is a compliance investment squeeze: the manufacturers most dependent on low-cost overseas production are often the ones least able to invest in the quality systems that would reduce their regulatory risk.
GDUFA and the Regulatory Fee Burden
The Generic Drug User Fee Amendments (GDUFA) added a layer of direct cost. For fiscal year 2025, the ANDA filing fee exceeded $320,000 per application [2]. For manufacturers filing multiple ANDAs annually across a broad product portfolio, these fees represent a meaningful overhead cost that falls before any revenue is generated. GDUFA was designed to fund faster FDA review — and it has improved review timelines — but it also raised the breakeven threshold for each product in development.
PROBLEM 5 Drug Shortages: When Low Margins Force Exit and Patients Pay
The Structural Economics of Shortage Creation
Drug shortages are not random events. They are the predictable output of a system that has priced generic manufacturing at or below the cost of reliable quality production for certain product categories.
When GPO contracts reward the lowest bidder and the market reaches a point where only one or two manufacturers remain on a product — often because others have exited as unprofitable — any disruption to those remaining manufacturers creates a shortage. The disruption can be a failed inspection, a facility damage event (as with the July 2023 tornado that severely damaged Pfizer’s sterile injectables facility in Rocky Mount, North Carolina), or a natural disaster (as with Hurricane Helene’s temporary closure of Baxter’s North Cove IV fluids facility in 2024) [20].
Drug shortage numbers reflect this structural fragility. The first quarter of 2024 saw a record 323 active shortages in the U.S. FDA shortage list. By 2025, that number had declined to approximately 270, but healthcare systems still describe shortages as “a major concern” at any given point in time [4]. Shortages disproportionately affect generic injectables — the category most dependent on specialized sterile manufacturing, most concentrated in overseas supply, and least able to generate margins that incentivize redundant domestic capacity.
Concentrated Manufacturing and Single Points of Failure
According to the FDA, less than 30% of the active pharmaceutical ingredients used to manufacture drugs in the U.S. are actually produced in the U.S. [4]. For many critical generic drug categories, the API is manufactured at two or three facilities globally — sometimes only one. A quality problem at any of those facilities triggers a cascade: finished-goods manufacturers cannot source their API; production slows or stops; inventory runs down; shortages appear at the dispensing level.
The BIOSECURE Act of 2024 responded to this risk by restricting federal agencies from contracting with companies that use certain Chinese biotechnology manufacturers [14]. It is a national security response to what is also a public health problem — but it does not resolve the underlying economics that created single-source dependency in the first place.
Vincristine and the Political Failure Point
Teva’s 2019 discontinuation of vincristine — a drug used in pediatric cancer treatment — produced a shortage that reached Senate hearing rooms. The episode illustrated the political exposure that generic manufacturers face when low-margin product exits generate visible patient harm. It also illustrated the policy gap: no regulatory mechanism existed to require Teva to continue manufacturing a product at a loss, and no viable competitor existed to step in quickly. The FDA can facilitate voluntary supply measures and communicate with potential entrants, but it cannot compel commercial decisions [7].
PROBLEM 6 The Complexity Barrier: Biosimilars, Complex Generics, and the Cost of Moving Up-Market
Why the “Move to Complexity” Strategy Is Harder Than It Sounds
The standard prescription for generic companies facing commodity erosion is to move up the complexity curve — from solid oral dosage forms into injectables, inhalers, extended-release formulations, complex drug-device combinations, and eventually biosimilars. The logic is sound: these product categories have fewer ANDA filers, longer development timelines that serve as natural barriers to entry, and price erosion curves that are less severe than for simple tablets.
The execution challenge is formidable. Complex generics require specialized manufacturing capabilities, more extensive analytical characterization, and regulatory science expertise that most commodity generic manufacturers do not have on staff. A scoping review published in 2025 covering the period from 2014 to 2024 found that the significant startup and investment costs and specialized manufacturing knowledge required for complex generics create “oligopolistic market dynamics” — effectively limiting meaningful participation to large or well-capitalized companies [21].
Biosimilars: The PBM Rebate Problem That Slows Adoption
Biosimilars — the biologic equivalent of generic drugs — represent the highest-value opportunity in the generic space, and the most structurally complicated one. Biologics constitute 5% of U.S. prescriptions but 51% of total drug spending as of 2024 [22]. Capturing even a fraction of that market through biosimilar entry would generate substantial revenue. The problem is that biosimilar adoption has been much slower than anticipated.
Humira (adalimumab) provides the clearest case study. Multiple biosimilar adalimumab products launched in 2023 and 2024. By 2025, biosimilars had captured approximately 20% to 30% of adalimumab volume — a meaningful but underwhelming share given that equivalent situations with small-molecule generics typically result in 80% to 90% brand erosion within the first year [3].
The reason is the PBM rebate structure. AbbVie had built a formulary defense through massive rebate payments to PBMs — payments so large that PBMs preferred to keep Humira on formulary and collect the rebate rather than switch patients to cheaper biosimilars. The “net price” of Humira after rebates was already competitive with biosimilar list prices. Biosimilar developers found themselves competing against a drug whose true cost to payers had already been eroded by a private negotiation they could not see or replicate. The eventual erosion of Humira’s position has occurred in steps coinciding with PBM formulary contracting cycles (typically January 1 and July 1), not as a competitive market response [3].
The Interchangeability Barrier and FDA’s October 2025 Response
An additional challenge for biosimilar adoption is the U.S. regulatory distinction between “biosimilar” and “interchangeable” status. An interchangeable biosimilar can be substituted at the pharmacy level without a new prescription — the equivalent of generic substitution for small molecules. Obtaining interchangeable status previously required “switching studies” demonstrating safety through alternating courses of treatment, adding cost and time to development.
In October 2025, the FDA announced draft guidance eliminating the routine requirement for switching studies for biosimilar approval, and signaled plans to make interchangeable status effectively the default for all approved biosimilars [22]. This regulatory shift should, over time, unlock pharmacy-level substitution and accelerate the biosimilar adoption curve. It does not, however, change the PBM rebate dynamics that have been the primary barrier to adoption for the highest-revenue biologic targets.
PROBLEM 7 Antitrust Liability: The Price-Fixing Legacy and Its Ongoing Legal Costs
The Department of Justice Investigation and Its Aftermath
The generic pharmaceutical industry has spent the better part of a decade dealing with the fallout from a DOJ investigation into price-fixing and market allocation that began around 2014. The investigation revealed a pattern of collusion among sales representatives at competing generic companies — coordination on pricing and customer allocation that reduced competition in ways that cost payers and patients billions of dollars.
Teva Pharmaceuticals reached a deferred prosecution agreement with the DOJ in 2023, agreeing to pay $225 million over five years — with annual payments of $22.5 million from 2024 through 2027 and a final lump sum of $135 million in 2028 [23]. Separately, Teva agreed to pay $450 million to resolve civil False Claims Act allegations related to the same conduct, as well as kickback violations involving the multiple sclerosis drug Copaxone [24].
Sandoz reached a $195 million criminal settlement with the DOJ in 2020, and a separate $275 million civil class action settlement received preliminary court approval in February 2025 [25]. The civil litigation against remaining non-settling defendants is ongoing.
The Scale of the Conspiracy
DOJ filings describe coordination across dozens of generic drugs — oral contraceptives, cholesterol medications, antibiotics, diabetes drugs — with the “standard” being that competitors would divide market share according to a polite industry norm: “playing nice in the sandbox.” Representatives from competing companies communicated directly about which company would win which customer, what price increases were coming, and which markets each company “owned.” The conspiracy was not sophisticated; it was pervasive.
The reputational damage has been substantial, and the compliance costs of remediation — enhanced monitoring, antitrust training, revised communication protocols, ongoing DOJ oversight under the deferred prosecution agreement — add to operating costs for the companies involved. For Teva, whose financial position was already strained by portfolio erosion and opioid-related litigation, the DOJ settlement added another multi-year payment obligation to a balance sheet under structural pressure.
Ongoing Civil Litigation and State AG Actions
Beyond the DOJ settlements, the generic price-fixing scandal spawned a wave of civil class action litigation and state attorney general cases that remain active. The state AG litigation covers a broader set of products and defendants than the DOJ cases and is proceeding on a timeline measured in years, not quarters. Companies still facing unresolved liability must carry legal reserves that constrain capital allocation — funds that might otherwise go to R&D, manufacturing investment, or market entry on complex products.
PROBLEM 8 Tariffs, Geopolitics, and the Reshoring Pressure Generic Companies Cannot Easily Meet
A Policy Environment Designed for Brands That Catches Generics in the Crossfire
The Trump administration’s pharmaceutical tariff framework, as finalized in the April 2, 2026 executive order, targets patented pharmaceutical products and their APIs with a 100% ad valorem tariff, effective July 31, 2026 for large companies and September 29, 2026 for smaller manufacturers [26]. Generic pharmaceuticals and biosimilars are explicitly exempt from the tariffs as structured — an acknowledgment of the public health risk of raising the cost of commodity drugs that fill the vast majority of prescriptions [27].
But the “generics are exempt” framing understates the indirect exposure. Generic manufacturers source APIs from many of the same overseas supply chains that the tariff framework is designed to pressure. The White House’s April 2026 proclamation mandates a formal review of generic drug tariff policy within one year — meaning the current exemption is a starting position, not a settled policy [26].
The API Dependency That Cannot Be Resolved Quickly
The U.S. pharmaceutical trade deficit reached $139 billion in 2024 [28]. Approximately 53% of patented pharmaceutical products distributed domestically are produced outside the U.S.; for APIs, the dependence is even more concentrated, with China and India supplying the majority of global API production for most small-molecule drug categories [26].
Reshoring API production to the U.S. is not a two- or three-year project. It requires facility construction, regulatory filing and inspection, technology transfer from overseas suppliers, and sustained price parity — or a regulatory and contractual structure that makes the higher cost of domestic production commercially viable. The May 2025 executive order directing FDA and EPA to streamline permitting for domestic pharmaceutical facilities and the August 2025 order to fill the strategic API reserve are steps in that direction, but the capital investment cycle for pharmaceutical manufacturing runs on decade-scale timelines [29].
Geopolitical Risk and the BIOSECURE Act’s Downstream Effects
The BIOSECURE Act of 2024 restricts federal agencies from contracting with companies that use certain designated Chinese biotechnology manufacturers. For generic companies that rely on Chinese API suppliers or contract development and manufacturing organizations (CDMOs), this creates a compliance requirement that may necessitate supplier qualification of alternative sources — a process that can take 18 to 36 months and requires regulatory filing updates with the FDA.
China’s 14th Five-Year Plan explicitly identified pharmaceutical manufacturing as a strategic industry, which has driven investment in domestic capacity and pricing strategies designed to maintain market share even at margins that would be commercially unsustainable for Western competitors [20]. The practical effect is that Chinese API suppliers can price at levels that are difficult for potential domestic or European competitors to match — reinforcing the very dependency that BIOSECURE and the tariff framework are designed to reduce.
Investment Capital Is Tightening
The capital markets constraint compounds the geopolitical one. According to GlobalData, venture funding for biopharmaceutical companies fell by 20% in Q1 2025 compared to the same period in 2024 [20]. Private equity activity in pharmaceutical supply chain services has dropped by approximately 15% annually since 2021 [20]. For generic manufacturers seeking to invest in domestic manufacturing capacity or complex product development, the financing environment is less favorable than it was three years ago — precisely when the strategic argument for such investment is strongest.
How the Industry’s Strongest Players Are Responding
The first response is portfolio rationalization. Both Teva and Viatris have publicly committed to exiting hundreds of low-margin generic products and concentrating resources on complex generics, biosimilars, and branded generics in international markets. Teva’s “Pivot to Growth” strategy, announced in 2023, targets a smaller but higher-margin portfolio oriented around injectables, respiratory, and specialty generics [6]. The strategy acknowledges that the company cannot compete sustainably in commodity solid orals at current market prices.
Complex Generics as the Competitive Moat
Companies with established track records in complex dosage forms — Hikma in injectables, Amneal in extended-release and inhalers, Lannett in topicals — have achieved marginally better pricing power by operating in product categories where the ANDA barrier to entry is higher. Analysts track these companies’ ANDA pipelines against the complexity profile of their submissions: a pipeline weighted toward paragraph IV certifications on complex molecules, with limited head-to-head competition from other filers, implies substantially better economics than a pipeline of commodity tablet filings.
DrugPatentWatch’s ANDA data and patent expiration tracking tools have become standard infrastructure for this kind of competitive intelligence work. By mapping how many Paragraph IV filers have submitted for a given target molecule, and what the litigation history suggests about the realistic entry date, companies can model not just whether to file, but what margin profile they are actually signing up for [14].
Vertical Integration and Supply Chain Ownership
A smaller number of companies have pursued backward integration — acquiring or building API manufacturing capacity to reduce dependence on external suppliers and improve margin control. Sun Pharmaceutical’s investment in API manufacturing and Aurobindo’s integrated API-to-finished-dosage-form model represent versions of this approach. The trade-off is capital intensity: owning the supply chain requires investment that can only be justified by volume commitments and a long-term view of the market.
M&A: Scale as a Negotiating Tool
Consolidation has been the industry’s structural response to buyer concentration for a decade. The logic is straightforward: a larger generic company with a broader product portfolio has more leverage in GPO negotiations because it is harder to replace. The three largest wholesale buying consortia control 90% of generic purchasing volume [7]; a generic company with a large enough share of that volume becomes indispensable rather than interchangeable.
The 2025-2030 patent cliff — with an estimated $236 billion in branded revenues at risk — is expected to accelerate M&A activity as both branded companies seeking to acquire generic/biosimilar capabilities and well-capitalized generic companies seeking to acquire first-to-file positions pursue deals [3]. DrugPatentWatch’s analysis of mergers in the generic drug space suggests that “bolt-on” acquisitions in the $1 billion to $15 billion range, targeting specific assets rather than full-company mergers, are the preferred structure in the current antitrust enforcement environment [30].
The Regulatory Calendar That Shapes the Next Five Years
Several regulatory events will materially affect the generic industry’s competitive landscape between now and 2030.
The Inflation Reduction Act’s drug pricing negotiation framework for Medicare — applied initially to high-cost branded drugs with no generic competition — is restructuring the brand-to-generic transition economics for some of the highest-value molecules. When a brand drug’s Medicare price is negotiated downward, the economic gap between brand and generic narrows, which can reduce the revenue available to generic filers entering after negotiated price takes effect [31].
The FDA’s October 2025 biosimilar guidance, eliminating routine switching studies and moving toward automatic interchangeability designation, should materially improve biosimilar market uptake over the 2026-2030 period [22]. For generic companies that have invested in biosimilar development, this guidance reduces the regulatory cost of achieving interchangeable status and should unlock pharmacy-level substitution in more states.
The April 2026 pharmaceutical tariff order mandates a formal review of generic drug tariff policy within one year [26]. The outcome of that review will determine whether the current exemption for generics remains in place or whether domestic manufacturing pressure is applied to the commodity generic sector as well. That review is the single largest regulatory wildcard for the industry in the near term.
The DOJ’s ongoing antitrust investigation into generic drug pricing — the civil cases against non-settling defendants continue — will generate additional settlement disclosures, potentially expanding the documented scope of collusion beyond what is currently in the public record.
The Outlook: Structural Pressure, Selective Opportunity
The eight problems described here are not temporary. Price erosion from competition is the design intent of the Hatch-Waxman system; it is not a malfunction. Buyer concentration in GPOs and PBMs reflects rational consolidation in a cost-conscious healthcare system. Patent thickets are the product of innovator companies rationally protecting their most valuable assets. Manufacturing quality failures are real but remediable, at a price. Drug shortages are the visible endpoint of margin erosion that was predictable. Biosimilar adoption challenges reflect both PBM incentive misalignment and, now partly, a regulatory correction in progress. Antitrust liability is a consequence of conduct that occurred — and that has cost the industry severely. Geopolitical and tariff risk reflects a broader realignment of pharmaceutical supply chain policy that will not reverse.
None of this means the generic industry is in secular decline. The patent cliff that runs through 2030 will create entry opportunities of historic scale. The biosimilar market’s structural underdevelopment — FDA-approved biosimilars have captured less than 20% of the biologic market despite comparable safety and efficacy — represents a recoverable economic opportunity as regulatory and formulary barriers come down. Companies with the manufacturing capability, patent intelligence infrastructure, and regulatory expertise to file credibly on the right molecules at the right time will generate returns that commodity manufacturers cannot.
The industry’s central challenge is identifying those opportunities before the competition does, building the supply chain to serve them reliably, and pricing in a way that sustains the manufacturing quality that shortages have demonstrated the system desperately needs. That is not a simple optimization problem, but it is a solvable one for companies with the analytical discipline and capital to execute it.
Key Takeaways
Generic drugs fill 90%+ of U.S. prescriptions but capture only ~18% of drug spending. The gap between volume share and revenue share is the central fact of the industry’s economics.
Price erosion on commodity solid oral dosage forms has driven many manufacturers to exit products entirely, contributing directly to drug shortages that hit a record 323 in Q1 2024.
Three PBMs control 80% of U.S. prescription claims. Three wholesale buying consortia control 90% of generic purchases by volume. This oligopsony structure extracts margin from manufacturers structurally, not through negotiation failures.
Brand-name companies have developed serial patent litigation strategies that can delay generic entry by years after initial Paragraph IV challenges succeed. Patent landscape analysis — using tools like DrugPatentWatch — has become essential for realistic entry timing models.
FDA warning letter data from 2025 shows Indian manufacturing sites receiving data integrity-related enforcement at a 60% rate — a persistent compliance liability for the industry’s largest API and finished-goods source.
The DOJ generic price-fixing investigation resulted in Teva paying a total of $450 million across criminal and civil settlements, with Sandoz paying a combined $470 million. Civil litigation against remaining defendants is ongoing.
Biosimilar adoption remains far below the small-molecule generic equivalent: ~20-30% volume share for adalimumab (Humira) after two years of competition, versus 80-90% for small molecules within six months. PBM rebate economics are the primary structural barrier, not patient or prescriber resistance.
The April 2026 pharmaceutical tariff order exempts generics and biosimilars but mandates a review of generics policy within one year. The outcome of that review is the industry’s most significant near-term regulatory wildcard.
Frequently Asked Questions
Q: Why are drug shortages concentrated in generic drugs rather than branded ones?
A: Generic drugs — particularly older injectable generics — are manufactured by a small number of suppliers at margins that leave little room for redundant capacity or supply chain buffers. When a GPO contract is structured around the lowest-price bidder, manufacturers that invest in backup capacity are at a cost disadvantage and lose contracts. Over time, the market selects for the leanest possible manufacturing structure — which is also the most fragile. Branded drugs, by contrast, typically have higher margins that fund more resilient supply arrangements, and their manufacturers have greater leverage with payers to maintain pricing that covers quality investment.
Q: What is a Paragraph IV certification and why does it trigger litigation automatically?
A: A Paragraph IV (PIV) certification is a declaration by an ANDA filer that one or more patents listed in the FDA’s Orange Book for the reference drug is either invalid, unenforceable, or will not be infringed by the generic product. Under Hatch-Waxman, filing a PIV certification constitutes an artificial act of patent infringement — the generic company is challenging the patent’s validity before it has even manufactured a single commercial tablet. The brand company then has 45 days to file suit. If it does, the FDA cannot grant final approval to the generic for 30 months, regardless of the merits of the litigation. This 30-month automatic stay is the core mechanism through which patent litigation delays generic market entry.
Q: If biosimilars are as safe and effective as branded biologics, why do PBMs keep branded drugs on formulary?
A: The answer is the rebate structure. Brand biologic companies pay PBMs substantial rebates — confidential percentage discounts off list price — in exchange for favorable formulary placement. The “net price” of the brand drug (after rebates) can be competitive with or lower than the biosimilar’s list price, which means the PBM has no financial incentive to prefer the biosimilar. In fact, switching patients to the biosimilar can reduce the total rebate the PBM collects. The savings from biosimilar use accrue to the health plan or patient (through lower premiums or cost-sharing) — not to the PBM. The FDA’s October 2025 guidance on interchangeability addresses part of the regulatory barrier to substitution, but the rebate economics require contractual or regulatory restructuring to fully resolve.
Q: How do generic companies use patent intelligence tools in practice?
A: The decision about whether to file an ANDA on a target molecule involves several layers of analysis that go well beyond noting when the principal compound patent expires. A generic company’s business development team needs to know: how many other companies have already filed Paragraph IV certifications on the same molecule; what the litigation history suggests about the brand company’s willingness to sue; whether pediatric exclusivity or other regulatory exclusivities extend the effective entry date past patent expiry; and how many competitors are likely to be on the market at first launch (which determines the margin available). Platforms like DrugPatentWatch aggregate FDA Orange Book listings, ANDA filing records, PIV certification notices, and litigation dockets into a searchable database that allows analysts to model these variables systematically rather than relying on fragmented public sources. The platform is used by generic manufacturers for ANDA portfolio planning, by branded companies for competitive threat assessment, and by investors modeling the revenue risk associated with specific patent expiration events.
Q: Are the Trump administration’s pharmaceutical tariffs a significant risk for generic drug prices?
A: The April 2026 tariff order explicitly exempts generic pharmaceuticals and biosimilars — so as currently structured, the tariffs apply to patented branded products, not generics. However, the order mandates a review of generic drug tariff policy within one year, which means the exemption is a policy position under active reconsideration, not a permanent carve-out. The indirect risks to generics are more immediate: many generic manufacturers source APIs from China and India, which are subject to broader tariff and trade policy pressure. If API sourcing costs increase through tariffs on chemical precursors or materials (which are not categorically exempt), finished generic drug production costs increase even if the finished product itself is tariff-free. For manufacturers with predominantly overseas supply chains — which is most of the industry — the tariff and reshoring policy environment is a material cost uncertainty for the next three to five years.
References
DrugPatentWatch. (2026). PBMs, formularies, and rebates: What investors should know. https://www.drugpatentwatch.com/blog/pbms-formularies-and-rebates-what-investors-should-know/
DrugPatentWatch. (2026). The definitive guide to generic drug approval in the U.S.: From ANDA to market dominance. https://www.drugpatentwatch.com/blog/obtaining-generic-drug-approval-in-the-united-states/
DrugPatentWatch. (2026). The patent cliff and beyond: A definitive guide to generic and biosimilar market entry. https://www.drugpatentwatch.com/blog/generic-drug-entry-timeline-predicting-market-dynamics-after-patent-loss/
University of Wisconsin School of Pharmacy. (2025, June 11). The future of generics. https://pharmacy.wisc.edu/2025/06/11/the-future-of-generics/
DrugPatentWatch. (2026). The global generic drug market: Trends, opportunities, and challenges. https://www.drugpatentwatch.com/blog/the-global-generic-drug-market-trends-opportunities-and-challenges/
Teva Pharmaceutical Industries Ltd. (2025). Annual report FY2024. U.S. Securities and Exchange Commission. https://www.sec.gov/Archives/edgar/data/0000818686/000117184325000493/exh_991.htm
KPMG. (2023). Generics 2030: Three strategies to curb the downward spiral. https://kpmg.com/kpmg-us/content/dam/kpmg/pdf/2023/generics-2030.pdf
Drug Channels Institute. (2024). The 2024 economic report on U.S. pharmacies and pharmacy benefit managers. https://drugchannelsinstitute.com/files/2024-PharmacyPBM-DCI-Overview.pdf
Drug Topics. (n.d.). The evolution of group purchasing organizations. https://www.drugtopics.com/view/evolution-group-purchasing-organizations
HIV-HCV Co-Infection Watch. (2025, January 27). FTC exposes PBM price gouging of specialty generic drugs. https://www.hiv-hcv-watch.com/blog/jan-27-2025
DrugPatentWatch. (2026). Decoding drug pricing models: A strategic guide to market domination. https://www.drugpatentwatch.com/blog/decoding-drug-pricing-models-a-strategic-guide-to-market-domination/
Sarpatwari, A., et al. (2025). Serial patent litigation: An emerging strategy to delay entry of generic competition. PubMed Central. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12757684/
Teva Pharmaceutical Industries Ltd. (2023, August 21). Teva settles price fixing charges with U.S. DOJ. https://www.tevapharm.com/news-and-media/latest-news/teva-settles-price-fixing-charges-with-u.s.-doj
DrugPatentWatch. (2026). Win the generic drug market: Patents, ANDAs, IP valuation, and the tactics that separate first-movers from also-rans. https://www.drugpatentwatch.com/blog/how-to-succeed-in-generic-drug-market-entry/
Center for Strategic and International Studies. (2026, February 13). Rebuilding resilience in U.S. pharmaceutical manufacturing. https://www.csis.org/analysis/rebuilding-resilience-us-pharmaceutical-manufacturing
Scilife. (2025). 2025 FDA warning letter trends: What pharma can learn from this year’s top citations. https://www.scilife.io/blog/worst-fda-warning-letters-pharma
DrugPatentWatch. (2025). Generic drug approval: The complete pharma playbook (FDA, EMA, patents, IP valuation). https://www.drugpatentwatch.com/blog/navigating-the-generic-drug-approval-process-a-comprehensive-guide/
GMP Compliance. (2025). Shocking conditions at Indian pharmaceutical manufacturer. https://www.gmp-compliance.org/gmp-news/shocking-conditions-at-indian-pharmaceutical-manufacturer
Pharmaceutical Online. (2025). What 2025 FDA warning letters tell us about GMP compliance. https://www.pharmaceuticalonline.com/doc/what-fda-warning-letters-tell-us-about-gmp-compliance-0001
Center for Strategic and International Studies. (2026). Rebuilding resilience in U.S. pharmaceutical manufacturing. https://www.csis.org/analysis/rebuilding-resilience-us-pharmaceutical-manufacturing
Sreedevi, A., et al. (2025). Exploring the challenges faced by generic version of complex drugs: A scoping review. PubMed Central. https://pmc.ncbi.nlm.nih.gov/articles/PMC12482112/
U.S. Department of Health and Human Services. (2025, October 29). FDA moves to accelerate biosimilar development and lower drug costs. https://www.hhs.gov/press-room/fda-accelerates-biosimilar-development-and-lowers-drug-costs.html
BioSpace. (2023). Teva to pay $225M in DOJ generic drug price-fixing settlement. https://www.biospace.com/teva-to-pay-225m-in-doj-drug-price-fixing-settlement
ArentFox Schiff. (2024, October 18). Teva Pharmaceuticals agrees to pay $450 million to resolve FCA claims. https://www.afslaw.com/perspectives/investigations-blog/teva-pharmaceuticals-agrees-pay-450-million-resolve-fca-claims
ClassAction.org. (2025, March 18). $275 million Sandoz settlement partially resolves generic drug price-fixing litigation. https://www.classaction.org/news/275-million-sandoz-settlement-partially-resolves-generic-drug-price-fixing-litigation
The White House. (2026, April 2). Adjusting imports of pharmaceuticals and pharmaceutical ingredients into the United States. https://www.whitehouse.gov/presidential-actions/2026/04/adjusting-imports-of-pharmaceuticals-and-pharmaceutical-ingredients-into-the-united-states/
PMC. (2025). The consequences of pharmaceutical tariffs in the United States. https://pmc.ncbi.nlm.nih.gov/articles/PMC12123191/
The White House. (2026, April 2). Fact sheet: President Donald J. Trump bolsters national security and strengthens U.S. supply chains by imposing tariffs on patented pharmaceutical products. https://www.whitehouse.gov/fact-sheets/2026/04/fact-sheet-president-donald-j-trump-bolsters-national-security-and-strengthens-u-s-supply-chains-by-imposing-tariffs-on-patented-pharmaceutical-products/
DrugPatentWatch. (2026). A strategic analysis of mergers and acquisitions in generic drug development. https://www.drugpatentwatch.com/blog/mergers-and-acquisitions-opportunities-and-challenges-in-generic-drug-development/












")

")











