The US pharmaceutical market’s valuation varies meaningfully across research houses, and those discrepancies are worth understanding rather than averaging away. Grand View Research pegged 2024 revenues at USD 634.32 billion. Nova One Advisor put 2023 at USD 602.19 billion. Magnet ABA cited USD 574.37 billion for the same year. These are not contradictory numbers so much as products of different definitional boundaries — some include OTC products, others restrict to prescription drugs only, and the treatment of compounding pharmacies and 340B transactions creates further divergence.
What all of the projections agree on is direction and magnitude. A compound annual growth rate in the 5.5%-6.2% range from 2024 to 2030 is consensus, yielding a 2030 market between USD 857 billion (Grand View, conservative case) and over USD 1 trillion (Magnet ABA). The most bullish model, from Nova One Advisor, targets USD 1,093.79 billion by 2033 on a 6.15% CAGR. For market entry planning purposes, the mid-case projection — roughly USD 885-900 billion by 2030 — is the operational assumption worth stress-testing.
What drives the growth is less interesting than the composition of that growth. Specialty drugs accounted for approximately 51% of total market revenue in 2023 and grew 11.7% year-over-year in the same period. Biologics took roughly 46% of total pharmaceutical spending in 2023 and are expanding as a share. That means the market is not growing uniformly: the specialty and biologic segments are compounding much faster than the generic small molecule base, which is deflationary by design. Any ROI analysis built on generic market share projections needs to account for that structural compression.
The Inflation Reduction Act introduces a significant variable that most pre-2022 market models did not price in. Beginning in 2026, CMS can directly negotiate prices for a defined set of Medicare Part D drugs, and that list expands substantially in 2028 and 2029. The Congressional Budget Office estimated cumulative Medicare savings of USD 98.5 billion over 10 years from the drug pricing provisions alone. Branded drug manufacturers with molecules at or near negotiation eligibility thresholds — nine or more years post-approval for small molecules, thirteen or more for biologics — face a material revision to their revenue forecasts.
The Structural Forces Driving Sustained Market Growth
Several demographic and epidemiological forces make the growth projections credible rather than speculative. The US population aged 65 and older will reach approximately 80 million by 2040, up from roughly 58 million in 2022. Per capita drug spending among seniors runs approximately three to four times the national average. That alone provides a structural tailwind that pricing reform cannot fully offset. The prevalence of chronic disease compounds it: approximately 60% of American adults have at least one chronic condition, and 40% have two or more. Cancer, type 2 diabetes, heart failure, COPD, and Alzheimer’s disease represent the dominant cost centers, and each is growing in patient volume.
R&D investment remains a secondary but real driver. Global pharmaceutical R&D spend reached USD 200 billion in 2023 and is growing, even if US-specific spend slipped just under USD 100 billion in the same year. The FDA approved 50 novel drugs in 2024, following 55 in 2023. That pace of innovation — roughly one new molecular entity per week — reflects both the maturation of the human genome as a target space and the productivity gains from AI-assisted target identification and compound screening. IQVIA’s 2025 Global R&D Trends report notes that the overall number of pipeline assets in active development passed 20,000 for the first time in 2024, a record. The pipeline is broad, and it is translating into approvals at a historically high rate.
AI in healthcare spending is projected to reach USD 188 billion by 2030. That number captures a category that ranges from chatbot patient scheduling to genuine computational biology, and the quality of those investments varies enormously. What is demonstrably valuable is AI’s application to hit identification and lead optimization in early drug discovery: cycle times that once required 4-5 years of wet lab iteration can now compress to 18-24 months through in-silico screening, structure prediction (AlphaFold-class tools), and generative molecular design. The firms that have built proprietary AI-guided discovery engines — Recursion Pharmaceuticals, Insilico Medicine, and a number of internal platforms at large pharma — have not yet produced approved drugs at scale from pure AI pipelines, but their Phase I/II read-outs in 2025-2027 will be the real test.
The Pharma 4.0 market — defined broadly as the integration of AI, IoT, and automation into pharmaceutical manufacturing and supply chains — sat at USD 13.7 billion in 2024 and is projected to reach USD 40.3 billion by 2030. Connected drug delivery devices are an especially active sub-segment: the US leads globally with over 5,000 active patents in the space, emphasizing Bluetooth and NFC integration. That matters for market entry because combination products — a drug paired with a delivery device — can generate fresh IP protection independent of the underlying molecule’s patent status, a tactic now widely used in respiratory disease (inhalers), diabetes (insulin pens), and rare disease (enzyme replacement infusion devices).
The Macro Policy Risk: IRA, Drug Pricing Negotiation, and the IRA’s Second-Order Effects
No market size analysis is complete without a detailed accounting of the IRA’s mechanics, because it is not a uniform tax on the pharmaceutical industry. It hits specific molecules in specific reimbursement channels at specific lifecycle stages.
The IRA’s Medicare price negotiation authority applies to drugs meeting three criteria: the drug accounts for substantial Medicare expenditure, it lacks a generic or biosimilar competitor, and it has been on the market long enough to exceed the exclusivity threshold (9 years for small molecules, 13 years for biologics). The first ten negotiated drugs, announced in August 2023, include Eliquis (apixaban, Bristol Myers Squibb and Pfizer), Jardiance (empagliflozin, Eli Lilly and Boehringer Ingelheim), Xarelto (rivaroxaban, Johnson & Johnson), and seven others. Negotiated prices take effect in 2026, and the list expands to 15 drugs in 2027 and 20 in 2028.
The second-order effects are what interest portfolio strategists. Drugs that fall just outside the eligibility window in 2026 may be on the negotiation list by 2028 or 2029. That creates an incentive to accelerate patent expiration management (more below), pursue indication extensions that reset commercial timelines, and invest in successor molecules early enough to capture market share before the originator faces negotiated pricing. It also creates a pricing umbrella effect: the IRA suppresses prices at the high end of the Medicare channel, potentially widening the commercial attractiveness of drugs priced below the negotiation threshold or targeting smaller populations.
For institutional investors, the IRA risk is most concentrated in drugs with high Medicare utilization among patients aged 65+, long post-approval histories without biosimilar entry, and high gross revenue relative to total therapy area revenue. Eliquis and Jardiance exemplify this profile. For drugs targeting pediatric or rare disease populations, or those with Medicare utilization below the threshold, the IRA’s direct impact is limited.
Key Takeaways — Part I
The US pharmaceutical market grows at roughly 5.5%-6.2% CAGR through 2030, but that aggregate figure masks a bifurcated reality: the specialty and biologics segments grow at double the rate of the small molecule generic base. Market entry strategies built on specialty drug innovation or biosimilar entry against high-revenue biologics capture the high-growth portion of the market. The IRA does not kill pharmaceutical ROI, but it compresses the late-lifecycle revenue of specific blockbuster molecules in the Medicare channel — a manageable risk for companies with diversified portfolios and a critical variable for firms with concentrated exposure to a single large-market small molecule.
Part II: Patent Architecture and IP Valuation — The Core Asset in Any Market Entry Decision
How Drug Patents Are Structured and Why the Layering Matters
A pharmaceutical patent is not a monolithic protection. It is a portfolio layered across multiple dimensions of a drug’s commercial life. Understanding how those layers interact — and where the cracks appear — is the foundation of any serious market entry analysis.
The primary patent on a small molecule drug covers the compound itself: its chemical structure, its synthesis, and its basic pharmacological activity. This is the composition-of-matter (COM) patent, and it carries the highest legal weight in litigation because it is the broadest and earliest-filed claim. For drugs approved in the early part of their patent term, the COM patent can provide 12-15 years of effective market exclusivity after FDA approval, accounting for patent term restoration (PTR) under the Hatch-Waxman Act, which restores up to 5 years of patent life lost to the regulatory review process, with a cap of 14 years of remaining term post-approval.
Layered on top of the COM patent, brand-name manufacturers typically file a series of secondary patents. These include formulation patents (covering specific dosage forms, extended-release mechanisms, or salt forms), method-of-use patents (covering specific therapeutic indications), process patents (covering manufacturing methods), and polymorph patents (covering specific crystalline forms of the drug substance). Each can be listed in the FDA’s Orange Book if it meets the statutory requirement — the patent must claim the drug or a method of using the drug as approved. Secondary patents typically expire later than the COM patent, and their Orange Book listing triggers Hatch-Waxman’s 30-month stay of generic approval upon a Paragraph IV certification challenge.
For biologics, the structure is different. A biologic’s composition-of-matter protection comes not from a single compound patent but from a combination of patents covering the molecular structure, the cell line that produces it, the manufacturing process, and the formulation. The Biologics Price Competition and Innovation Act (BPCIA) — the biologic analog of Hatch-Waxman — grants 12 years of data exclusivity to reference biologics regardless of patent status, followed by 4 years of exclusivity for the first biosimilar applicant to file. The ‘patent dance’ under the BPCIA requires biosimilar applicants to share their abbreviated Biologics License Application (aBLA) with the reference product sponsor, triggering a two-round process for identifying and litigating patents.
The Orange Book: Your Primary Intelligence Tool for Small Molecule Entry
The FDA’s Orange Book (formally: Approved Drug Products with Therapeutic Equivalence Evaluations) is the canonical source for patent term information on small molecule drugs. Every patent listed in the Orange Book, along with its expiration date, is publicly accessible. The Drug Patent Watch platform aggregates Orange Book data and enriches it with patent term extension (PTE) calculations, market exclusivity tracker data, and competitive filing histories.
Reading the Orange Book correctly requires understanding what is and is not listed there. Only patents that the NDA holder certifies meet the statutory criteria are listed. Patents that NDA holders choose not to list — or that do not qualify because they claim only manufacturing processes or inactive ingredients — do not appear and cannot trigger the 30-month stay. Generic filers frequently challenge the legitimacy of Orange Book listings: the FTC has brought multiple actions against companies for listing patents that do not claim an approved drug or method of use, and the 2021 CREATES Act gave the FTC new enforcement tools in this area.
The Orange Book patent expiration date for any given drug is rarely a single date. Most large-market branded drugs carry five to twelve listed patents with staggered expiration dates spanning a decade or more. Lipitor (atorvastatin, Pfizer) at peak had eleven Orange Book patents. Humira (adalimumab, AbbVie) built a patent portfolio exceeding 100 granted patents globally, with US Orange Book listings extending effective exclusivity well beyond the compound patent expiry. That history illustrates why IP teams cannot limit their analysis to the earliest or most prominent patent: the full stack matters.
Evergreening Tactics: A Comprehensive Technology Roadmap
Evergreening is the set of strategies by which an originator pharmaceutical company extends effective market exclusivity beyond the expiry of the original composition-of-matter patent. Calling it ‘evergreening’ implies a pejorative framing, which is analytically unhelpful. Some of these strategies represent genuine innovation and benefit patients; others are legal tactics with minimal clinical upside. Both matter to competitive intelligence.
The most technically grounded evergreening approach is the development of extended-release or modified-release formulations. When a company reformulates a drug from immediate-release to once-daily extended-release, it legitimately earns a new formulation patent and, if it files a new NDA or supplemental NDA for the new form, up to three additional years of Waxman-Hatch new clinical study exclusivity. Concerta (methylphenidate ER, originally marketed by Alza/Johnson & Johnson) and OxyContin (oxycodone ER, Purdue) both operated under this model. For market entry teams, the critical question is whether the branded extended-release formulation has been listed in the Orange Book and whether the clinical differentiation justifies payer coverage at a premium — because a generic of the IR form may already exist and capture most of the volume.
Pediatric exclusivity is a distinctly American mechanism that requires serious attention. Under the Best Pharmaceuticals for Children Act (BPCA), the FDA can request that a drug sponsor conduct pediatric studies. If the sponsor conducts those studies and submits a pediatric study report — regardless of whether the studies show efficacy in children — the sponsor receives a six-month exclusivity extension that attaches to every Orange Book patent on the drug. That six months can translate to hundreds of millions of dollars in additional revenue on a blockbuster molecule. Merck received pediatric exclusivity on Zocor (simvastatin); Abbott received it on Depakote (valproic acid). Generics firms routinely factor pediatric exclusivity expiry dates into their ANDA filing timelines.
Indication expansion generates fresh method-of-use patents and can restart data exclusivity periods for distinct indications. When a drug receives approval for a second indication, the new indication patent is listable in the Orange Book and triggers Paragraph IV rights for any generic filer who certifies non-infringement. If the new indication represents a genuine clinical advance (as with several oncology drugs repurposed to rare diseases), the NDA supplement warrants its own market analysis. If the indication expansion is primarily defensive, the litigation risk against a well-funded Paragraph IV challenger is high.
New chemical entity status based on enantiomers or active metabolites has been deployed selectively. The classic case is Nexium (esomeprazole, AstraZeneca), the S-enantiomer of omeprazole, which AstraZeneca launched as Prilosec (omeprazole) was approaching LOE. AstraZeneca secured a composition-of-matter patent on esomeprazole specifically, supported by manufacturing process patents that the company defended vigorously in litigation. Forest Laboratories used the same playbook with Lexapro (escitalopram), the S-enantiomer of citalopram (Celexa). These strategies work when the new entity can demonstrate clinically meaningful differentiation — better PK profile, improved tolerability, lower dose requirement — that supports payer coverage. When the differentiation is marginal or demonstrably pharmacologically equivalent, Paragraph IV challengers succeed at higher rates.
Salt form and polymorph patents protect specific physical forms of a drug compound. A drug can exist as a free acid, as a sodium salt, as a calcium salt, or as dozens of crystalline polymorphs — each with potentially distinct dissolution profiles, stability characteristics, and bioavailability. The original Hatch-Waxman filing for many drugs was based on a specific salt or polymorph, and the brand’s Orange Book listing protects that specific form. Generic filers routinely exploit this by developing a different acceptable salt form that is bioequivalent to the reference listed drug (RLD) but does not infringe the listed patents. 505(b)(2) applications built on alternative salts or polymorphs with improved characteristics can simultaneously challenge the originator’s market position and capture premium pricing.
The most aggressive evergreening mechanism from a litigation standpoint is the patent thicket: a dense web of overlapping patents covering different aspects of a drug, filed with the deliberate goal of making any challenger face simultaneous litigation on a dozen or more counts. AbbVie’s Humira portfolio — estimated at over 100 US patents — is the most studied example. The core composition-of-matter patent on adalimumab expired in 2016, but AbbVie’s thicket of formulation, manufacturing, method-of-use, and device patents kept biosimilar competitors off the US market until 2023, seven years after the core patent expiry. Amgen, Samsung Bioepis, and others had biosimilars approved by the FDA before AbbVie entered patent settlements that controlled biosimilar launch timing. The eventual biosimilar launches in 2023 — with 9 products entering the market within 12 months — demonstrate both the delayed-but-real opportunity in dense biologic thickets and the settlement structure companies use to manage entry timing.
Paragraph IV Filings: Anatomy of a Generic Entry Strategy
A Paragraph IV certification is a generic manufacturer’s formal assertion, filed in an ANDA with the FDA, that one or more patents listed in the Orange Book for the reference listed drug are either invalid, unenforceable, or would not be infringed by the generic product. Filing a Paragraph IV certification is, in patent law terms, an act of infringement — a legal fiction created by Hatch-Waxman specifically to permit early legal challenge to pharmaceutical patents before market entry.
Upon receiving notice of a Paragraph IV certification, the brand manufacturer has 45 days to file a patent infringement lawsuit in federal district court. If it does, an automatic 30-month stay activates: the FDA cannot approve the ANDA for 30 months from the date the brand receives notice, regardless of the FDA’s review timeline. The brand wins the stay whether or not it ultimately wins the litigation. This is why patent litigation defense is itself a value-creation mechanism for brands, even when they eventually lose on the merits.
The first ANDA filer with a Paragraph IV certification is entitled to 180 days of generic marketing exclusivity. This is the most commercially powerful provision in Hatch-Waxman from a generic filer’s perspective: for 180 days after the first generic launches, no other ANDA filer can receive final approval regardless of the FDA’s review clock. On a branded drug with USD 2 billion or more in annual US sales, 180-day exclusivity can generate USD 500 million to USD 1 billion in operating profit for the first generic filer, depending on price erosion dynamics.
The decision to file Paragraph IV requires a detailed IP validity analysis, a non-infringement claim construction, and a litigation budget that can reach USD 5-25 million for a full trial. The probability of success varies with patent type: composition-of-matter patents issued before 2000 are successfully challenged at a higher rate than post-2000 COM patents, largely because pre-GATT (pre-1995) patents had 17-year terms from issue rather than 20-year terms from filing, and the art available at the time of filing was less thoroughly developed. Secondary patents — formulation, method of use, polymorph — are successfully challenged at higher rates than COM patents. The FTC’s 2002 generic drug study found that generic challengers prevailed in roughly 73% of fully litigated Paragraph IV cases; more recent studies suggest the rate is closer to 50-60% as brands have improved their patent prosecution strategies.
Pay-for-delay settlements — in which a brand manufacturer pays a generic challenger to delay launch — were significantly curtailed by the Supreme Court’s 2013 decision in FTC v. Actavis. The Court held that large reverse payments create a presumption of antitrust violation, requiring analysis under the rule of reason. The practical effect has been a shift from cash payments to value-transfer arrangements: co-promotion rights, supply agreements, license to a peripheral market, or manufacturing contracts. The FTC continues to challenge these settlements, and any market entry strategy that involves a contemplated settlement with a brand challenger requires careful antitrust counsel.
Biologics IP: The 12-Year Exclusivity Clock and Biosimilar Interchangeability
The BPCIA’s 12-year reference product exclusivity is the dominant feature of the biologic market entry landscape. Unlike patent protection, which can be challenged before it expires, the 12-year exclusivity period is a statutory bar: the FDA will not accept a biosimilar BLA until 4 years after the reference product’s approval, and it will not approve a biosimilar until 12 years have passed. No patent challenge changes that clock.
The clinical requirements for biosimilar approval are substantially more demanding than those for small molecule generic approval. A generic drug applicant must demonstrate bioequivalence — essentially, that the generic’s PK profile in healthy volunteers is statistically similar to the RLD’s. A biosimilar applicant must demonstrate that the proposed biosimilar is highly similar to the reference product in terms of structure, function, purity, and clinical pharmacology, and it must show that there are no clinically meaningful differences in safety, purity, and potency. That typically requires a full analytical characterization package, comparative PK/PD studies, and at least one comparative clinical trial with immunogenicity data.
Biosimilar interchangeability — the designation that permits a pharmacist to substitute a biosimilar for the reference product without prescriber intervention, analogous to the ‘AB’ designation for generic small molecules — requires an additional demonstration that switching between the reference product and the biosimilar does not produce greater risk than using the reference product alone. Cyltezo (adalimumab-adbm, Boehringer Ingelheim) was the first adalimumab biosimilar to receive interchangeability designation in the US, in 2021. As of the end of 2024, the FDA had approved 63 biosimilars across 17 reference products, with approximately 25-30 carrying interchangeability designation.
The commercial impact of interchangeability is significant but market-specific. In states with automatic substitution laws (the majority), an interchangeable biosimilar can be dispensed at pharmacy without prescriber action, mirroring the generics substitution model. In practice, PBM formulary positioning, rebate contracting, and specialty pharmacy channel management often matter more than interchangeability status for biologic market share. Humira biosimilars that launched in 2023 without interchangeability — including Hadlima (Samsung Bioepis) and Yusimry (Coherus) — captured market share primarily through formulary access, not automatic substitution. Interchangeability remains a differentiator in commoditized categories where formulary decisions are made primarily at the pharmacy benefit level.
IP Valuation Methodology for Core Drug Assets
Pharmaceutical IP assets are valued using a risk-adjusted net present value (rNPV) framework, which applies a probability-of-technical-success (PTS) discount to expected future cash flows and discounts them at a rate that reflects the cost of capital for pharmaceutical development.
The core rNPV formula is:
rNPV = Sum over all scenarios of [ (probability of scenario) x (NPV of cash flows in that scenario) ]
In practice, this requires estimating peak sales (based on prevalence, penetration, price, and competitive displacement), the revenue ramp from launch to peak (typically 3-6 years for specialty drugs, 2-4 years for primary care), the duration of revenue (patent-protected period minus development time remaining), the cash flow from revenue minus cost of goods, operating expenses, royalties, and milestones, and the discount rate (typically 8-12% for late-stage assets, 12-20% for early-stage assets).
Probability of success estimates should come from empirical base rates, not management guidance. For Phase I assets, the overall probability of reaching approval is approximately 10-13% (across all therapeutic areas). Phase II to approval: approximately 20-25%. Phase III to approval: approximately 50-60%. These rates vary substantially by therapeutic area: oncology Phase II to approval probability is lower (approximately 15-20%) than rare disease (approximately 35-45%), reflecting the higher clinical bar and competitive landscape in oncology.
The IP asset’s exclusivity period is the single most sensitive variable in a pharmaceutical rNPV model. A one-year reduction in patent exclusivity on a blockbuster molecule with USD 5 billion in annual sales reduces the asset’s NPV by approximately USD 2-3 billion at a 10% discount rate, depending on the competitive landscape at the time of patent expiry and the rate of generic or biosimilar penetration. For large-market branded drugs, the 90% market share loss in year one following the first generic entry is the standard base case for US generic erosion modeling, driven by Medicaid, PBM, and VA formulary switches.
Key Takeaways — Part II
Every serious market entry analysis starts with an IP audit. The Orange Book tells you when and how to enter the small molecule generic market. The BPCIA data exclusivity clock and the Purple Book tell you when the biologic entry window opens. The layer of secondary patents — formulations, polymorph, method-of-use, pediatric exclusivity — determines how much litigation risk and timeline uncertainty attaches to that entry. IP valuation, done with discipline, converts patent expiry data into defensible NPV estimates that support capital allocation decisions.
Investment Strategy — Part II
For pharma IP and licensing teams, the immediate priority is mapping the secondary patent landscape for drugs within 3-5 years of primary composition-of-matter expiry. Those molecules represent the highest-value generic entry opportunities because the legal pathway is clearest and the first-mover advantage in Paragraph IV is still accessible. For biologics-focused investors, the interchangeability pipeline — drugs whose 12-year reference exclusivity expires before 2030 and which have not yet attracted multiple biosimilar applicants — represents the highest-concentration ROI opportunity. Adalimumab taught the market that a dense patent thicket plus settlement management can delay biosimilar entry by 7 years. The question for each high-revenue biologic is: how dense is the thicket, and what is the litigation risk profile for challenging it?
Part III: Therapeutic Areas With High Unmet Medical Needs — A Deep-Dive Analysis
Oncology: $110 Billion in Sales and a Pipeline That Has Not Peaked
Oncology generated approximately USD 110 billion in 2024 pharma sales among the top 50 products, making it the single largest therapeutic class by revenue. That concentration of revenue reflects a combination of high per-unit pricing (typically USD 100,000-300,000 per patient per year for approved antibody therapeutics), reasonable duration of therapy in maintenance settings, and a steady supply of new approvals. The FDA’s Oncology Center of Excellence has been one of the most active units in the agency, using accelerated approval, breakthrough therapy designation, and priority review to compress development timelines.
Precision oncology is the organizing principle of the current treatment paradigm. Genomic profiling of tumors — now standard of care at major cancer centers — produces biomarker data that stratifies patients into sub-indications, each of which can support a separate NDA or supplemental NDA. KRAS G12C, BRAF V600E, HER2 amplification, NTRK fusions, RET fusions, EGFR exon 20 insertions: each represents a targetable mutation with approved or in-development therapeutics. This biomarker-stratified model has several implications for market entry. The addressable patient populations are smaller — KRAS G12C accounts for approximately 13% of non-small cell lung adenocarcinoma, or roughly 25,000 US patients per year — which makes each program an orphan-scale opportunity from a prevalence standpoint, even though NSCLC itself is not a rare disease. Smaller populations can support premium pricing under value-based frameworks, and orphan drug designation may be available, which reduces FDA fees, provides tax credits for clinical development costs, and grants 7-year market exclusivity upon approval.
Antibody-drug conjugates (ADCs) are the current consensus ‘next wave’ in oncology. The class combines the targeting specificity of a monoclonal antibody with the cytotoxic potency of a small molecule payload, theoretically delivering higher drug concentrations to tumor cells while sparing normal tissue. The FDA approved Datopotamab deruxtecan-dlnk (Datroway, AstraZeneca/Daiichi Sankyo) in 2025 for HR+/HER2- metastatic breast cancer — a population of approximately 211,000 new cases per year in the US, with a market for therapeutics of approximately USD 7.5 billion in 2023. The ADC market broadly is expected to exceed USD 30 billion in annual sales by 2030 based on current pipeline progression rates, with multiple agents in Phase III trials across breast cancer, urothelial carcinoma, gastric cancer, and NSCLC.
From an IP perspective, ADCs present unusual complexity. The antibody component, the linker chemistry, the cytotoxic payload, and the drug-to-antibody ratio (DAR) each generate separate patent claims. Daiichi Sankyo holds dominant IP positions on the DXd payload and the tetrapeptide-based cleavable linker used in Enhertu (trastuzumab deruxtecan) and Datroway. Licensing agreements between Daiichi Sankyo and AstraZeneca, Merck, and others have defined the commercial structure of the ADC market. Any entrant into the ADC space that does not control its own payload or linker IP is dependent on licensing economics that can consume 10-15% of revenues in royalties.
Multiple myeloma is a specific oncology sub-market where unmet need, IP dynamics, and market size converge in a way that warrants dedicated attention. The condition’s standard of care has shifted to proteasome inhibitors (bortezomib, carfilzomib) and immunomodulatory agents (lenalidomide, pomalidomide) plus anti-CD38 antibodies (daratumumab). All of the backbone agents are now or soon will be subject to generic or biosimilar competition: lenalidomide (Revlimid) lost its primary patent protection and generic entry began in 2022 under a settlement with Bristol Myers Squibb; daratumumab (Darzalex) patents begin to expire around 2028-2030. The CAR-T segment in myeloma — ciltacabtagene autoleucel (Carvykti, Janssen/Legend Biotech) and idecabtagene vicleucel (Abecma, Bristol Myers Squibb) — represents the current high-value innovation space, with list prices exceeding USD 460,000 per infusion. The IP for CAR-T constructs targeting BCMA (the dominant myeloma antigen) is contested: Juno Therapeutics, Janssen, Kite Pharma, and Legend Biotech have all filed patents and engaged in licensing disputes. Any company entering CAR-T for myeloma needs a freedom-to-operate analysis on BCMA targeting, lentiviral manufacturing, 4-1BB vs. CD28 co-stimulatory domains, and allogeneic vs. autologous construct IP.
Immuno-oncology continues to generate revenue for PD-1/PD-L1 checkpoint inhibitors at a rate that no one predicted in 2010. Keytruda (pembrolizumab, Merck) generated approximately USD 25 billion in 2024 sales, making it the single largest-selling pharmaceutical product globally. Opdivo (nivolumab, Bristol Myers Squibb) and Tecentriq (atezolizumab, Roche/Genentech) follow at lower scales. Pembrolizumab’s core composition-of-matter patent expires in 2028 in the US (with PTR-extended term), and biosimilar applications are already in preparation. The competitive positioning of the checkpoint inhibitor biosimilar market will depend heavily on whether FDA grants interchangeability — which is technically challenging for monoclonal antibody biosimilars with complex glycosylation profiles — and whether PBMs are willing to manage formularies to drive substitution.
Neurology: The Alzheimer’s Bet, MSA, and the Under-Served Neurodegenerative Tier
Alzheimer’s disease is the most high-profile unsolved problem in neuropharmacology, with a clinical trial failure rate exceeding 99% over the past 25 years. The amyloid hypothesis dominated the field for decades, and the repeated failures of amyloid-clearing agents appeared to condemn the whole approach — until lecanemab (Leqembi, Eisai/Biogen) and donanemab (Kisunla, Eli Lilly) demonstrated statistically significant slowing of cognitive decline in Phase III trials targeting amyloid-beta.
The commercial launch of lecanemab in 2023 and donanemab’s approval in 2024 validate the anti-amyloid approach at the regulatory level, but the market access reality has been complicated. The requirement for amyloid PET imaging or CSF biomarker testing to establish eligibility, the IV infusion administration (biweekly for lecanemab, monthly for donanemab), and the risk of amyloid-related imaging abnormalities (ARIA) — serious neurological events affecting approximately 20-35% of patients — have slowed commercial uptake relative to peak-sales projections. CMS initially proposed covering lecanemab only in the context of clinical evidence development (CED), a coverage restriction that limited prescribing to Medicare beneficiaries enrolled in approved registries. That restriction was partially lifted in July 2023. By early 2025, lecanemab’s US uptake remained below initial projections, though a subcutaneous formulation in development by Eisai could substantially improve the convenience profile.
The IP landscape for Alzheimer’s disease-modifying therapies is currently Biogen/Eisai-dominated for anti-amyloid approaches, and Eli Lilly-dominated for donanemab’s specific N-truncated amyloid form targeting. Tau-targeting strategies — which address the neurofibrillary tangle pathology downstream of amyloid accumulation — represent the most scientifically motivated next wave. AbbVie, Roche, and UCB all have anti-tau programs in Phase II. If any tau-targeting agent demonstrates disease modification in a Phase III trial, the patent space will intensify rapidly. For companies entering now, the most defensible IP position is in novel tau targeting mechanisms (e.g., tau phosphorylation at specific residues, tau spreading inhibition) rather than amyloid, where the composition-of-matter space is crowded.
KarXT (xanomeline-trospium, Bristol Myers Squibb, acquired via the USD 14 billion acquisition of Karuna Therapeutics in 2024) is approved for schizophrenia and is in trials for Alzheimer’s-related psychosis. The acquisition price reflected both the drug’s commercial potential and the M1/M4 muscarinic agonism mechanism’s competitive differentiation from the dominant D2 dopamine antagonist framework. KarXT’s US patent protection extends to approximately 2035 on composition-of-matter, and the M1/M4 mechanism creates meaningful barriers to generic substitution because there are no structurally related alternatives with the same mechanism approved for psychotic disorders. The BMS acquisition illustrates the premium that differentiated CNS mechanisms command: a USD 14 billion acquisition for a drug that had not yet reached the market at the time of announcement.
Multiple System Atrophy (MSA) is a synucleinopathy with prevalence of 1-9 per 100,000 individuals and approximately 25,000-75,000 Americans living with the condition. Alterity Therapeutics received FDA Fast Track designation in 2025 for ATH434, a small molecule targeting iron dysregulation in MSA, a mechanism distinct from alpha-synuclein aggregation approaches. MSA has no approved disease-modifying treatment, and the FDA’s willingness to grant Fast Track designation signals regulatory openness to a surrogate endpoint strategy. The orphan drug designation pathway is available for MSA, providing the 7-year exclusivity, tax credits, and expedited review that make rare neurological diseases commercially viable despite their small patient populations.
Neurofibromatosis Type 1 (NF1) achieved a treatment milestone in 2025 with the approval of mirdametinib (Gomekli, SpringWorks Therapeutics) for symptomatic plexiform neurofibromas. NF1 has a prevalence of 1-5 per 10,000, or roughly 100,000 Americans. Mirdametinib’s approval followed that of selumetinib (Koselugo, AstraZeneca) in 2020 for the same population; both target the MEK pathway. The MEK inhibitor IP space for NF1 now has two approved products, meaning the primary opportunity for new entrants is differentiation from MEK: alternative RAS pathway targets, combination strategies, or patient sub-populations defined by co-occurring mutations.
Metabolic Disease: The GLP-1 Gold Rush and the Question of What Comes After
Glucagon-like peptide-1 receptor agonists have produced the fastest-growing pharmaceutical category in history. Wegovy (semaglutide, Novo Nordisk) and Zepbound (tirzepatide, Eli Lilly) generated approximately USD 6 billion and USD 4.9 billion in 2024 sales, respectively, in their first full year of commercialization. The GLP-1 market as a whole — combining obesity and type 2 diabetes indications — is projected to reach USD 130 billion by 2030 across all marketed agents. Novo Nordisk and Eli Lilly currently control roughly 90% of the addressable GLP-1 market, and both companies have multi-year manufacturing capacity constraints.
The IP landscape for semaglutide and tirzepatide is dense and well-defended. Novo Nordisk’s core composition-of-matter patents on semaglutide expire in the US around 2032 (with possible PTR), though a Paragraph IV campaign against peripheral formulation and method-of-use patents is already underway from at least one generic filer as of 2024. Tirzepatide’s core patents extend to approximately 2036. Both companies have invested heavily in device and formulation IP to extend commercial protection beyond the active pharmaceutical ingredient (API) patents.
The competitive response to GLP-1 dominance is coming on several fronts. Oral small molecule GLP-1 receptor agonists are in development by Structure Therapeutics, Pfizer (danuglipron, discontinued after tolerability issues), and Novo Nordisk (oral semaglutide is approved for diabetes but not yet obesity at the 50mg dose studied in Phase III). The oral route eliminates the injection burden that is the primary patient experience barrier for GLP-1 penetration in obesity. Any oral GLP-1 agonist that demonstrates comparable weight loss and tolerability to injectable semaglutide would command a significant premium and capture market share disproportionate to its weight loss efficacy advantage.
Triple agonists — simultaneous activation of GLP-1, GIP, and glucagon receptors — represent the pharmacological frontier. Retatrutide (Eli Lilly) demonstrated greater than 20% weight loss in Phase II trials, exceeding even tirzepatide’s Phase II performance. The glucagon component drives additional metabolic benefit through hepatic glucose output suppression and energy expenditure increase. Phase III data for retatrutide are expected in 2025-2026. If confirmed, retatrutide will face an immediate IP differentiation challenge: GLP-1/GIP dual agonism is Lilly’s established position (tirzepatide), and adding glucagon agonism is a logical extension that both Lilly and Novo Nordisk have IP claims on. Any entrant into the triple agonist space needs a novel molecular scaffold that does not infringe the GIP/glucagon receptor co-agonist claims held by these two companies.
Non-alcoholic steatohepatitis (NASH, now more formally called Metabolic Dysfunction-Associated Steatohepatitis or MASH) finally achieved regulatory validation with resmetirom’s (Rezdiffra, Madrigal Pharmaceuticals) FDA approval in March 2024 — the first approved drug for the condition. Resmetirom is a thyroid hormone receptor beta (THR-beta) selective agonist that reduces hepatic fat and improves fibrosis scores. Madrigal’s market exclusivity rests on a composition-of-matter patent expiring in approximately 2033 for the active enantiomer (MGL-3196), plus a method-of-use patent for THR-beta selective agonists in NASH. The addressable NASH/MASH market is estimated at 16-20 million US patients with moderate-to-severe fibrosis (F2-F4), the population most likely to respond to drug therapy and most likely to receive reimbursement. Peak sales projections for resmetirom have been revised upward to USD 4-6 billion annually as the market develops. Second-wave competitors include GLP-1-based approaches (semaglutide showed fibrosis improvement in a Phase II NASH trial), FXR agonists (obeticholic acid, Intercept, has a troubled safety record but the FXR pathway remains active), and FGF21 analogs (efruxifermin, Akero Therapeutics, in Phase III).
Rare Diseases: Orphan Designation, Premium Pricing, and the Genetic Revolution
Rare diseases collectively affect approximately 30 million Americans across 7,000-8,000 distinct conditions. Despite individual disease rarity, the aggregate commercial opportunity is substantial because orphan drug designation enables premium pricing, accelerated development timelines, and regulatory fee waivers that collectively generate ROI profiles comparable to primary care blockbusters, at a fraction of the development cost.
The FDA approved a record number of rare disease therapies with orphan drug designation in 2024 — approximately 60% of the novel drug approvals carried orphan drug designation. That reflects a deliberate regulatory strategy: orphan designation reduces the typical Phase III sample size requirement (100-400 patients in many cases vs. thousands in primary care), allows use of surrogate endpoints with accelerated approval, and creates a more predictable regulatory dialogue. For development-stage companies, the orphan drug tax credit — currently 25% of qualified clinical trial costs — provides a meaningful cash flow benefit.
Hemophilia represents one of the most commercially active rare disease markets. The US hemophilia market was approximately USD 9 billion in 2023, with hemophilia A (factor VIII deficiency) comprising roughly 80% of that and hemophilia B (factor IX deficiency) the remainder. Fitusiran (Qfitlia, Sanofi), approved in January 2025 for prophylaxis in both hemophilia A and B regardless of inhibitor status, operates via a subcutaneous small interfering RNA (siRNA) mechanism targeting antithrombin — a fundamentally different approach from factor replacement. The hemophilia B market alone is projected to reach USD 24.7 billion globally by 2035, driven by gene therapy entry: hemgenix (etranacogene dezaparvovec, CSL Behring), the first approved hemophilia B gene therapy, carries a list price of USD 3.5 million per treatment.
The hemophilia gene therapy IP landscape centers on adeno-associated virus (AAV) serotype selection, promoter sequences, transgene codon optimization, and manufacturing process. Spark Therapeutics, BioMarin, Takeda, and UniQure have all filed extensive patent portfolios in this space. Biosimilar entry for factor replacement products — historically treated as biologic-like in regulatory complexity — has been slow due to the product complexity and the shift of the market to longer-acting agents (extended half-life factor concentrates) and non-factor approaches like fitusiran and emicizumab (Hemlibra). For generic and biosimilar entrants, factor replacement is not an attractive market entry pathway; the real opportunity is in second-wave gene therapy constructs that improve on vector yield, immunogenicity, and durability.
Hereditary angioedema (HAE) affects approximately 6,000 people in the US, with global prevalence of 1 in 50,000. The current treatment standard includes plasma kallikrein inhibitors (lanadelumab, Takhzyro; berotralstat, Orladeyo) for prophylaxis and on-demand therapy with icatibant (Firazyr) or C1-esterase inhibitor concentrate. NTLA-2002 (Intellia Therapeutics) is a CRISPR/Cas9-based in vivo gene editing treatment in Phase III trials targeting the KLKB1 gene, which encodes plasma prekallikrein. If approved, it would represent the first approved CRISPR gene editing therapy for a non-oncologic indication and would command extraordinary pricing — Intellia has indicated Phase III interim data could support NDA filing in 2025-2026.
The IP on CRISPR gene editing therapies is concentrated in three foundational layers: the Cas9 endonuclease itself (where a licensing dispute between the Broad Institute and UC Berkeley was resolved in favor of the Broad for eukaryotic cell applications), the guide RNA design, and the delivery vehicle (lipid nanoparticles for in vivo editing, lentiviral vectors for ex vivo). Intellia licenses the Broad’s Cas9 IP and uses proprietary LNP delivery. Beam Therapeutics, which uses base editing rather than double-strand break editing, has differentiated IP. Prime editing (Broad Institute, commercialized via Prime Medicine) is earlier stage but avoids many of the double-strand break off-target concerns that limit Cas9 editing’s regulatory pathway.
Transthyretin (ATTR) amyloidosis has a US prevalence of 17.4-173 per million — the wide range reflecting diagnostic under-recognition rather than genuine prevalence uncertainty. Two approved RNA interference (RNAi) therapies target TTR gene expression: patisiran (Onpattro, Alnylam) and vutrisiran (Amvuttra, Alnylam). CRISPR-based treatment nexiguran ziclumeran (nex-z, Intellia), designed as a one-time in vivo gene editing treatment, is in Phase III trials. The TTR RNAi and gene silencing IP landscape is controlled primarily by Alnylam (GalNAc delivery conjugate chemistry, phosphorothioate chemistry for RNAi) and Ionis Pharmaceuticals (antisense oligonucleotide chemistry). The competition between RNAi (requiring quarterly or annual dosing) and one-time CRISPR gene editing is primarily a health economic argument: CRISPR’s one-time treatment requires a higher upfront cost but eliminates the cumulative cost of chronic maintenance dosing, which can run USD 300,000-450,000 per year. Value-based reimbursement models that allow installment payments or outcomes-based rebates will determine which modality captures more of the ATTR market.
Key Takeaways — Part III
The highest-ROI therapeutic areas in 2025-2030 are defined by the combination of large unmet need, receptive regulatory pathway, defensible IP exclusivity, and pricing environment that supports premium economics. Alzheimer’s disease, MASH, rare genetic diseases amenable to gene editing, and ADC-driven oncology each meet these criteria. GLP-1 in obesity and diabetes is real but increasingly competed; the entry opportunity is in differentiated mechanisms (triple agonism, oral formulations, combination approaches), not in additional GLP-1/GIP dual agonists.
Investment Strategy — Part III
For institutional investors, the three most actionable near-term signals are: Phase III data readouts for donanemab (cognitive endpoints in specific patient sub-groups by amyloid burden), CRISPR/gene editing Phase III completions (NTLA-2002 and nex-z both expected 2025-2026), and the evolution of the MASH market from single-agent resmetirom to combination regimens. Companies that control differentiated IP in these three intersections — Intellia Therapeutics, Madrigal Pharmaceuticals, and the companies with Phase III tau-targeting assets in Alzheimer’s — are the highest-conviction positions for therapeutic area specialization.
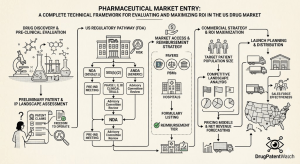
Part IV: Market Entry Vectors — A Technical Comparison of Pathways
The NDA (505(b)(1)) Route: Full Development Costs, High Exclusivity, Maximum Pricing Power
The standard new drug application under 21 CFR 314.50, commonly called the 505(b)(1) route, requires a complete pre-clinical and clinical dataset wholly owned by or licensed to the applicant. It is the pathway for first-in-class drugs and for any drug whose mechanism of action or molecule has not been previously approved by the FDA. The 505(b)(1) route confers 5 years of new chemical entity (NCE) data exclusivity upon approval — no ANDA referencing the drug can be submitted for 5 years, regardless of patent status — and the approval activates the Orange Book patent listing framework.
The average cost of a 505(b)(1) NDA, from IND-enabling studies through Phase III completion to NDA submission, is approximately USD 1.3-2 billion in direct R&D costs, with total development cost (including cost of capital) estimated at USD 2-2.6 billion per approved drug when Phase I/II failures in the same program are allocated. Phase III costs alone can reach USD 200-500 million for a registration trial in a large indication. Oncology Phase III trials, with their requirement for overall survival endpoints, specialized imaging endpoints, and centralized tumor assessment, run toward the top of that range.
Timeline from first-in-human to approval averages 10-15 years. Several FDA designations can compress that timeline materially. Breakthrough Therapy Designation (BTD), granted when preliminary clinical evidence shows substantial improvement over available therapy on a clinically meaningful endpoint, provides more intensive FDA guidance, rolling review, and organizational commitment of senior FDA staff. Drugs with BTD have averaged 5-6 years from IND to approval vs. 8-10 years for standard pathways. Priority Review, granted to drugs that offer significant improvement over available therapy, reduces the FDA’s review clock from 12 months to 6 months. Accelerated Approval allows drugs for serious conditions to receive approval based on a surrogate endpoint reasonably likely to predict clinical benefit, with post-approval confirmatory trials required. Fast Track Designation facilitates early and frequent communication with the FDA and allows rolling NDA submission.
The ANDA Route: Bioequivalence, Orange Book Navigation, and First-to-File Economics
An Abbreviated New Drug Application relies on the FDA’s prior finding of safety and effectiveness for the reference listed drug. The ANDA applicant must demonstrate that its generic drug contains the same active ingredient, dosage form, route of administration, strength, labeling, and conditions of use as the RLD. The critical scientific demonstration is bioequivalence: a pharmacokinetic study in a small number of healthy adult subjects showing that the generic’s 90% confidence interval for AUC and Cmax falls within 80-125% of the reference product’s value.
ANDA filing costs range from approximately USD 1-3 million for a straightforward tablet or capsule formulation without patent challenges, to USD 5-50 million for complex formulations (modified-release systems, nasal sprays, transdermal patches, ophthalmic solutions) or for drugs requiring a full Paragraph IV litigation campaign. The FDA’s ANDA approval timeline has been a source of significant frustration in the generic industry: as of early 2025, the FDA’s total pending ANDA queue exceeds 3,000 applications, with median total approval time from filing to final approval averaging 45-55 months for standard applications. Complex generics — the FDA’s category for products with documented formulation complexity or clinical endpoint bioequivalence requirements — average longer.
The 180-day first generic exclusivity provision creates enormous value concentration in the first ANDA approval. A generic manufacturer that achieves first-to-file status on a USD 2 billion annual revenue branded drug and successfully defeats or avoids the originator’s Paragraph IV litigation campaign can generate USD 500 million to USD 1.2 billion in operating profit during the 180-day exclusivity window, before the price collapses 70-90% when multiple generics enter.
The 505(b)(2) Pathway: The Most Underutilized Entry Strategy in the Market
The 505(b)(2) NDA pathway, defined at 21 CFR 314.54, allows applicants to rely on published literature or the FDA’s previous findings of safety and effectiveness for the listed drug, while providing their own data for the differences between their proposed product and the reference. It occupies the space between a full 505(b)(1) and an ANDA: the applicant gets the regulatory efficiency of relying on existing data but retains the ability to differentiate the product clinically and commercially.
The 505(b)(2) pathway is appropriate for reformulations (new dosage form, new route of administration, new dosing regimen), new combinations of previously approved drugs, new indications for approved molecules, enantiomers or active metabolites of approved drugs, and new salt or polymorph forms with different biopharmaceutical properties. Because the 505(b)(2) applicant is not simply copying the RLD but introducing clinical differences, the FDA can grant new clinical study exclusivity (3 years for a study supporting the change) or NCE exclusivity (5 years if the active moiety has never been approved), depending on the nature of the application.
The 505(b)(2) pathway is commercially attractive precisely because it combines lower development costs than a full 505(b)(1) NDA — by relying on the safety database of the reference compound — with pricing power higher than a generic. A 505(b)(2) drug enters the market as a branded or branded-generic product, with list pricing at a premium to the generic baseline but below the originator’s branded price. Suzetrigine (Journavx, Vertex), approved in January 2025 for moderate-to-severe acute pain, represents a novel NaV1.8 selective inhibitor that required a full 505(b)(1) development program; but many of the NaV1.8 early safety characterization studies could have relied on published literature on the target, illustrating how the 505(b)(2) pathway supports mechanistically novel but chemically derivative drug development.
Biosimilar Market Entry: The 63-Approved Baseline and the Interchangeability Premium
As of the end of 2024, the FDA had approved 63 biosimilars across 17 reference products. The commercial launches have been concentrated in adalimumab (Humira biosimilars), etanercept (Enbrel biosimilars), bevacizumab (Avastin biosimilars), trastuzumab (Herceptin biosimilars), and pegfilgrastim (Neulasta biosimilars). The penetration rates vary dramatically by reference product and market segment: trastuzumab and bevacizumab biosimilars have achieved 50-65% unit market share in some payer channels, while adalimumab biosimilars have achieved only 20-35% combined share despite nine products being available as of early 2025.
The biosimilar development cost is approximately USD 100-250 million per reference product, depending on the analytical complexity of the molecule and the scope of the required clinical program. That cost compares favorably to the USD 1.3-2 billion cost of a novel biologic NDA, but it is substantially higher than the USD 1-3 million cost of a simple small molecule ANDA. The higher development cost reflects the need for comprehensive comparative analytical characterization (structural similarity studies), comparative functional studies, comparative PK/PD studies in healthy volunteers or patients, and at least one comparative efficacy and safety trial. Manufacturing costs for biosimilars are also substantially higher than for small molecule generics, requiring cell culture facilities, purification trains, and cold-chain management.
The biosimilar interchangeability designation requires data from a switching study — a randomized study in which some patients are switched between the reference product and the biosimilar multiple times to demonstrate that the immunogenicity and clinical outcomes are not meaningfully worse than maintaining the reference product. As of 2025, approximately 15-20 biosimilars carry interchangeability designation, concentrated in adalimumab, ranibizumab, and insulin products. Interchangeability is most commercially meaningful in the retail pharmacy channel, where state automatic substitution laws allow pharmacists to dispense the interchangeable biosimilar without prescriber authorization. In the medical benefit channel (where most biologic infusions are administered), formulary management and acquisition cost contracts drive biosimilar utilization irrespective of interchangeability status.
Orphan Drug Designation: Seven Years of Exclusivity and the Financial Case
The Orphan Drug Act of 1983 provides three distinct financial incentives that, taken together, can transform the economic case for developing drugs for rare diseases with fewer than 200,000 US patients. The 7-year market exclusivity provision bars the FDA from approving a competing drug with the same active moiety for the same rare disease indication for 7 years from the date of the first approval. It is not patent-dependent: even if the compound’s patents have expired, orphan exclusivity prevents generic or biosimilar entry for 7 years. The tax credit for qualified clinical testing costs, currently 25% of qualifying expenses, provides a direct reduction in federal tax liability (not just a deduction) during the development years. The waiver of FDA application fees for the NDA or BLA — which can exceed USD 3 million for a standard application — reduces upfront cash outlay.
The economics of orphan drug development have been studied extensively. A 2020 analysis in Drug Discovery Today found that orphan drugs had a significantly higher probability of progression from Phase I to approval (approximately 25-30% for orphan drugs vs. 12% for all indications) and generated higher sales per approved drug relative to development costs. The higher success rate reflects several factors: smaller, faster trials, more direct FDA engagement, willingness to use surrogate endpoints, and patient communities that are actively supportive of clinical participation. The higher sales per drug reflects both premium pricing (average orphan drug list prices in the US exceeded USD 200,000 per patient per year by 2023) and the absence of generic competition during the 7-year exclusivity window.
Key Takeaways — Part IV
The five market entry pathways — 505(b)(1), ANDA/Paragraph IV, 505(b)(2), biosimilar, and orphan drug — are not mutually exclusive categories. A compound can qualify for both orphan designation and 505(b)(1) submission; a 505(b)(2) application can incorporate a Paragraph IV certification against a reference drug’s secondary patents; a biosimilar program can pursue interchangeability designation at the same time as seeking standard biosimilar approval. The optimal pathway is a function of the molecule type, the competitive patent landscape, the target patient population, and the company’s manufacturing and regulatory capabilities.
Part V: Competitive Intelligence and Late-Stage Pipeline Analysis
The Big Pharma Landscape: Revenue Concentration and Pipeline Stress
The top five US pharmaceutical companies by 2024 sales — Merck & Co., Pfizer, Johnson & Johnson, AbbVie, and AstraZeneca — collectively generate approximately USD 250-270 billion in annual revenues. Their market positions are built on patent-protected blockbusters, but each faces material patent cliff exposure between 2025 and 2030:
Merck faces LOE (loss of exclusivity) on pembrolizumab (Keytruda) around 2028, with US composition-of-matter protection expiring and biosimilar applications expected to be filed in 2025-2026. Keytruda accounts for approximately USD 25 billion of Merck’s revenues — roughly 40% of total company sales. The Keytruda cliff is the single largest patent expiry event in pharmaceutical industry history in terms of revenue at risk.
AbbVie’s post-Humira transition has been partially executed through the acquisitions of Allergan (USD 63 billion, 2020) and Cerevel Therapeutics (USD 8.7 billion, 2023). Skyrizi (risankizumab) and Rinvoq (upadacitinib) are growing to fill the Humira revenue gap, and both have patent protection extending into the mid-2030s. But AbbVie’s combined reliance on immunology means it is exposed to any structural shift in the PBM formulary landscape that drives biosimilar substitution at higher rates than projected.
Pfizer generated an extraordinary USD 100 billion in 2022-2023 revenues from COVID-19 vaccines and Paxlovid, which has now normalized dramatically. The company has invested in a post-COVID rebuild: the USD 43 billion acquisition of Seagen in 2023 brought in four ADC products and a deep ADC pipeline, making Pfizer one of the dominant ADC companies globally. The Seagen IP — particularly the ADC linker technology underlying Adcetris (brentuximab vedotin) and the pipeline — is the most commercially valuable element of the acquisition.
Novo Nordisk and Eli Lilly’s GLP-1 dominance is covered in Part III. Their combined market capitalization, which peaked at approximately USD 700 billion and USD 750 billion respectively in late 2024, reflects the market’s assessment that the GLP-1 market will sustain growth through 2030 and that their manufacturing advantages — Novo Nordisk’s manufacturing scale at its Kalundborg facility, Lilly’s rapid facility expansion — are genuine competitive moats.
FDA Approvals 2024-2025: What the Recent Wave Tells You About Market Direction
The FDA approved 50 novel drugs in 2024 and has approved multiple products in the first months of 2025. The 2025 approvals through early in the year include:
Nipocalimab-aahu (Imaavy, Johnson & Johnson) for generalized myasthenia gravis — an FcRn antagonist that reduces IgG antibody titers including pathogenic acetylcholine receptor antibodies. The gMG market was approximately USD 2.7 billion in 2021 and is growing. The approved FcRn mechanism (also used by argenx’s efgartigimod/Vyvgart and UCB’s rozanolixizumab/Rystiggo) represents a crowded but expanding class in antibody-mediated autoimmune diseases.
Atrasentan (Vanrafia, Chinook Therapeutics/Novartis) for IgA nephropathy — an endothelin-A receptor antagonist that reduces proteinuria by improving mesangial cell function. IgA nephropathy has a US prevalence of approximately 200,000 patients. The prior approval of sparsentan (Filspari, Travere Therapeutics) and the endothelin/AT1 dual antagonism mechanism, plus atrasentan’s more selective endothelin-A mechanism, create a two-drug market with distinct mechanistic profiles. Novartis’ USD 3.5 billion acquisition of Chinook in 2023 valued atrasentan primarily on its IgA nephropathy potential, implying a peak sales estimate in the range of USD 1.5-2.5 billion.
Suzetrigine (Journavx, Vertex Pharmaceuticals) for moderate-to-severe acute pain — the first selective NaV1.8 inhibitor approved for pain, and the first non-opioid analgesic with a novel mechanism approved in over 20 years. The US acute pain therapeutics market was USD 17.2 billion in 2023 and is growing at approximately 3-4% annually. Vertex’s NaV1.8 composition-of-matter patents extend to approximately 2038. The competitive response from AstraZeneca (which has NaV1.8 programs), Pfizer (which partnered with Biohaven on Nav programs), and emerging biotech companies will define whether NaV1.8 becomes a multi-drug class or remains Vertex’s exclusive franchise.
Mirdametinib (Gomekli, SpringWorks Therapeutics) for NF1 plexiform neurofibromas — the second MEK inhibitor approved for this rare disease after selumetinib (Koselugo, AstraZeneca), and the first approved for adult patients. SpringWorks licensed mirdametinib from Pfizer, which had abandoned the compound after Phase II data in NF1 did not meet internal thresholds. The SpringWorks development program — targeting a specific, genetically defined patient population with a clear mechanism — is a case study in how compounds abandoned by large pharma can be rescued and developed to approval by focused rare disease companies with lower overhead and more targeted clinical programs.
Fitusiran (Qfitlia, Sanofi) for hemophilia A and B prophylaxis — a GalNAc-conjugated siRNA targeting antithrombin (AT), which restores the balance between procoagulant and anticoagulant factors. The RNAi mechanism and subcutaneous once-monthly dosing represent genuine clinical differentiation from factor replacement. Sanofi acquired fitusiran via the acquisition of Alnylam’s former partnership and paid significant milestones. The hemophilia market’s evolution toward non-factor approaches (fitusiran, emicizumab) and eventually gene therapy is compressing the revenue opportunity for factor replacement biosimilars — a reminder that market entry into a therapeutic area requires an understanding not just of patent timelines but of therapeutic modality shifts.
Late-Stage Pipeline Candidates: Where the 2026-2028 Market Is Being Built
The most closely watched late-stage pipeline assets in 2025 include:
Donanemab (Kisunla, Eli Lilly) is approved in the US for Alzheimer’s disease. Phase III data from the TRAILBLAZER-ALZ 2 trial showed 35% slowing of cognitive decline in patients with low-to-medium tau burden. The drug’s differentiation from lecanemab rests on monthly vs. biweekly dosing, a potentially finite treatment duration (Lilly’s labeling includes discontinuation criteria), and differences in the specific amyloid-beta species targeted (N-truncated pyroglutamate form). The commercial competition between donanemab and lecanemab will ultimately be decided by head-to-head trial data (none yet planned), ARIA rate comparisons in real-world settings, and payer coverage policies.
Retatrutide (Eli Lilly) showed greater than 22% mean weight reduction at 24 weeks in Phase II at the highest dose, the strongest efficacy signal observed in any obesity program. Phase III TRIUMPH trial results are anticipated in 2025-2026. If the efficacy holds and the tolerability profile is acceptable, retatrutide will likely be filed for regulatory approval by 2026. Its intellectual property, built on Lilly’s proprietary peptide engineering platform, represents the most commercially significant obesity IP position after tirzepatide itself.
Nexiguran ziclumeran (nex-z, Intellia Therapeutics) for ATTR-CM (transthyretin cardiac amyloidosis) is in Phase III with interim data expected in 2025. If successful, it would be the first CRISPR-based gene editing therapy approved for a cardiovascular indication and would compete with Alnylam’s patisiran and vutrisiran in the ATTR market. Intellia’s market capitalization — substantially below its all-time high as of early 2025 — reflects investor uncertainty about Phase III success, not about the commercial opportunity if the trial reads out positively.
NTLA-2002 (Intellia) for hereditary angioedema, targeting KLKB1 gene editing, is in Phase III. The HAE market currently generates approximately USD 3-4 billion in annual revenues from approved prophylactic agents. A one-time CRISPR treatment priced at USD 3-5 million per patient would be commercially transformative but would require a health economic argument that displaces USD 300,000-400,000 per year in chronic dosing costs — a straightforward NPV calculation for payers over a 15-20 year time horizon.
Deucravacitinib (Sotyktu, Bristol Myers Squibb) is approved for plaque psoriasis and is in Phase III (POETYK PsA-2) for psoriatic arthritis. The TYK2 inhibition mechanism differs fundamentally from JAK1/2/3 inhibitors (upadacitinib, tofacitinib, baricitinib) in its allosteric mechanism, which targets the regulatory rather than catalytic domain of TYK2 and produces a more selective kinase inhibition profile with lower off-target activity. If the PsA Phase III data support approval, Sotyktu will compete in an immunology market dominated by IL-17 inhibitors (secukinumab, ixekizumab) and IL-23 inhibitors (guselkumab, risankizumab), with differentiation based on mechanism and oral administration.
Key Takeaways — Part V
The current pipeline concentration in five areas — Alzheimer’s disease-modifying therapy, CRISPR gene editing for single-gene disorders, GLP-1 class extension into triple agonism and oral formulation, ADC expansion across oncology indications, and TYK2/JAK immunology — reflects where the probability-adjusted commercial opportunity was strongest when these programs were initiated 5-8 years ago. Market entry into any of these areas in 2025 requires a credible differentiation story: a mechanism, a patient sub-population, or a delivery approach that is not already represented by an approved or late-stage product.
Part VI: Financial Framework — Building a Defensible ROI Model
Clinical Development Costs: Phase-Level Reality, Not Headline Averages
The USD 2-2.6 billion average total development cost per approved drug is a useful headline but a dangerous single number. It is an average across all therapeutic areas and all development stages, and it includes the cost of failed programs attributable to each approved drug. The distribution is highly skewed: a straightforward rare disease drug with a defined biomarker, approved via accelerated approval on 150 patients, may cost USD 150-300 million to develop. A large cardiovascular outcomes trial enrolling 15,000 patients over 6 years may cost USD 500-700 million for Phase III alone.
Phase-level cost benchmarks from published literature (primarily Sofpromed and Johns Hopkins analyses) provide more useful guidance:
Phase I trials cost between USD 4 million and USD 22 million on average, with the wide range reflecting study design complexity — PK/PD characterization, SAD/MAD designs, drug-drug interaction studies, special populations studies. First-in-human studies in healthy volunteers for small molecules run USD 4-8 million. Phase I cancer studies in patients (where healthy volunteer studies are ethically impermissible for cytotoxics) run USD 8-20 million or more.
Phase II trials average USD 7-30 million, with oncology Phase IIa proof-of-concept trials in biomarker-defined populations often landing at the lower end. Phase IIb dose-finding studies with larger sample sizes run USD 15-40 million.
Phase III costs span the widest range: USD 20-500 million per pivotal trial. The median cost for all clinical trials supporting an FDA approval is approximately USD 19 million (Johns Hopkins), but this includes many approvals based on single, small confirmatory trials in rare diseases with accelerated approval endpoints. Phase III cardiovascular outcomes trials, oncology overall survival trials, and large respiratory or metabolic indication trials are all in the USD 100-500 million range.
FDA advisory committee preparation, NDA preparation and submission, and the FDA PDUFA user fee (USD 4.04 million for a standard FY2025 NDA application) add USD 50-100 million to total development costs for a standard drug program. Post-approval commitments — confirmatory trials required for accelerated approvals, pediatric studies under PREA, Phase IV safety studies as a condition of approval — add another USD 30-150 million.
Probability of Technical and Regulatory Success: Benchmarks by Phase and Therapy Area
The probability of regulatory success (PRS) is the most consequential variable in an rNPV model because it multiplicatively discounts all expected cash flows. The industry-standard benchmark, derived from aggregated clinical trial database analyses by BIO, Citeline, and IQVIA, is:
Phase I to approval: approximately 12-13% overall (all therapy areas, all drug types). Phase II to approval: approximately 22-24%. Phase III to approval: approximately 55-65% (with NDA filing assumed following successful Phase III). NDA submission to approval: approximately 85-90%.
These rates mask substantial therapeutic area variation. Hematological malignancies have a Phase I to approval rate of approximately 30-35% — the highest of any major indication — reflecting the use of biomarker-defined patient selection, established surrogate endpoints (complete response, minimal residual disease), and aggressive use of breakthrough therapy designation. Alzheimer’s disease has historically had a Phase III to approval rate below 10%, though the anti-amyloid approvals of 2023-2024 suggest a structural improvement in the category.
Rare diseases overall have Phase I to approval probabilities approximately 2-2.5x higher than the all-indication average, because of smaller trials, surrogate endpoints, and regulatory accommodation. This makes the orphan drug pathway not just an exclusivity mechanism but a genuine probability-of-success multiplier.
NPV and IRR Modeling for Drug Development Portfolios
A defensible rNPV model for a drug development asset incorporates at minimum six quantitative inputs: peak annual sales (USD), time to peak from approval (years), duration of revenue above threshold (years of patent/exclusivity protection), net revenue margin (revenue minus COGS, royalties, and direct commercial spend, as a percentage), capital expenditures required (manufacturing buildout, if applicable), and a discount rate appropriate to the stage and risk profile.
Peak annual sales estimation is the highest-variance input. The standard methodology uses an epidemiological foundation (total eligible patients x addressable fraction x treatment initiation rate x price per patient-year) and adjusts for competitive displacement over time. A drug entering a market with two approved agents can typically capture 15-30% of incident patients in year 3-5, assuming comparable efficacy and pricing. A first-in-class drug in a condition with no prior therapy can capture 40-60% of eligible patients at peak, subject to diagnosis rates and treatment access.
Discount rates for pharmaceutical assets should reflect the risk profile: typically 8-12% for assets post-Phase III positive data with an NDA filed, 12-15% for assets in Phase III with mature data, 15-20% for Phase II assets, and 20-25% for Phase I assets. Higher discount rates are appropriate for assets in therapeutic areas with historically poor clinical translation (CNS, fibrosis) or in competitive markets where the pricing assumption is inherently uncertain.
IRR calculations for drug development partnerships and acquisitions require modeling the deal cash flows: upfront payment, development milestone payments, regulatory milestone payments, sales milestones, and royalty rates on net sales. Standard deal structures in pharma licensing typically feature upfront payments in the range of USD 25-250 million (depending on stage), development milestones totaling USD 200-1,500 million contingent on Phase III and NDA events, and royalty rates of 8-20% on net sales depending on the stage at licensing, the strategic value to the licensee, and the therapeutic area.
Drug Pricing Strategy Under the IRA: The New Commercial Math
The IRA’s direct price negotiation provision applies to 10 Medicare Part D drugs in 2026, expanding to 15 in 2027, 20 in 2028, and further in subsequent years. The negotiated prices represent an average discount of approximately 38-79% from current list prices (based on CMS’s released negotiated prices for the first 10 drugs). Eliquis’ negotiated price represents a 56% discount from list; Januvia’s represents a 79% discount. These are the prices paid by Medicare, not the commercial (private payer) price — commercial pricing remains unconstrained.
The mechanism by which the IRA influences commercial pricing is indirect: brand manufacturers set prices in part to defend against reference pricing claims and international comparisons. If Medicare pays substantially less than the commercial price, political and media pressure for commercial price alignment intensifies. Manufacturers facing IRA negotiation on their lead products have several strategic options. They can invest in successor compounds that reset the timeline, seek additional indications that expand the commercial patient population beyond the Medicare-dominant segment, pursue combination product strategies that create new IP, or argue on clinical grounds for the highest possible negotiated price.
The IRA also includes a drug pricing negotiation out-of-pocket cap for Medicare beneficiaries (USD 2,000 per year in out-of-pocket drug costs beginning in 2025) and an insulin price cap (USD 35 per month for Medicare beneficiaries). The out-of-pocket cap substantially improves medication adherence and access for high-cost specialty drugs, which may partially offset revenue reductions from negotiated pricing by expanding the actively treated patient population.
For new drugs entering the market post-2025, the pricing strategy must account for the IRA’s designation mechanisms from the beginning. Drugs that can demonstrate substantial clinical benefit relative to alternatives, serve younger patient populations with lower Medicare exposure, or target conditions that the IRA’s unmet medical need criteria would consider underserved, are better positioned commercially.
Barriers to Entry: The Capital and Regulatory Reality
The structural barriers to pharmaceutical market entry are high and rising. The capital barrier is self-evident: USD 1.3-2 billion in average development costs is beyond the reach of most single-asset startups without venture or strategic partnership support. The regulatory barrier — navigating a 10-15 year FDA review process with repeated interaction across Pre-IND meetings, End-of-Phase-2 meetings, Type B and Type C meetings, and the NDA review itself — requires regulatory affairs expertise that takes years to develop internally.
The manufacturing barrier is often underestimated. GMP manufacturing compliance for biologics and complex small molecules requires controlled facilities, validated processes, and a quality system that can survive pre-approval inspections. The FDA’s Good Manufacturing Practice regulations are enforced through pre-approval inspections (PAIs) that can delay NDA approvals by 6-24 months if deficiencies are found. CDMOs (contract development and manufacturing organizations) have partially democratized access to manufacturing capacity, but CDMO dependence creates supply chain risk and limits a company’s control over manufacturing cost and quality.
The competitive barrier is most acute for second-entrant drugs in established therapeutic areas. A drug targeting TNF-alpha (the same mechanism as adalimumab and etanercept) faces a saturated market and cannot price above the biosimilar baseline. A drug targeting IL-17 (the mechanism of secukinumab and ixekizumab) in psoriasis faces two established agents with long safety records and strong label differentiation data. New entrants into established mechanism classes need a meaningful clinical advantage — better efficacy, better safety, more convenient dosing, or a combination — to command market share.
Key Takeaways — Part VI
The financial framework for pharmaceutical market entry has four non-negotiable elements: a credible epidemiological basis for peak sales, empirically grounded probability-of-success assumptions by phase, a realistic discount rate for the asset’s stage, and an IRA-adjusted revenue model for products with significant Medicare exposure. Companies that build rNPV models with optimistic penetration assumptions, above-average phase transition probabilities, and pre-IRA pricing will systematically overestimate asset values. The reverse is equally true: companies that apply the wrong (too high) discount rate to a late-stage rare disease asset with orphan designation and clear clinical differentiation will systematically undervalue it.
Investment Strategy — Part VI
For institutional investors, the single most useful metric is the gap between a drug company’s traded enterprise value and its rNPV-based sum-of-parts valuation, adjusted for pipeline risk. Companies trading at significant discounts to rNPV often reflect either market mispricing of a specific clinical catalyst (approaching trial readout, underappreciated mechanism) or rational skepticism about commercial execution capabilities. Distinguishing between the two requires interrogating not just the trial design and endpoint but the company’s market access infrastructure, payer relationships, and manufacturing readiness.
Part VII: Targeted Entry Opportunities and Strategic Recommendations
The approval of anti-amyloid therapies for Alzheimer’s has opened regulatory space for disease-modifying approaches without solving the disease. The next clinical generation — tau-targeting agents, alpha-synuclein therapies for Parkinson’s/Lewy body dementia, TDP-43 approaches for ALS/FTLD, and neuroinflammation suppressors — represents the current Phase I/II opportunity set. The IP space in tau targeting is contested but not concentrated: small molecule tau aggregation inhibitors (TauRx’s LMTM, in Phase III), antisense oligonucleotides targeting tau expression (Ionis/Biogen), and tau PET-guided patient stratification approaches create multiple entry points.
For companies with RNA platform capabilities, the most defensible new IP position in CNS is in GalNAc-conjugated siRNA or ASO targeting CNS gene expression, specifically in conditions with defined genetic mutations or expression abnormalities. Huntington’s disease (HTT gene), spinocerebellar ataxia (various CAG repeat expansions), and genetic forms of ALS (SOD1, FUS, TDP-43 mutations) are genetically validated targets with defined patient populations and regulatory precedent (tominersen, an ASO for Huntington’s disease, completed Phase III though with mixed results that provide dose and timing guidance for follow-on programs).
The GLP-1 market rewards both innovation and manufacturing scale. Neither is easily replicated by new entrants in the short term. The actionable opportunity for companies not already at scale in GLP-1/GIP is one level removed: the drug products, devices, and monitoring technologies that support GLP-1 therapy.
GLP-1 injectable delivery devices — autoinjectors with connectivity features, dose titration tracking, and integration with continuous glucose monitors — are an active IP space (the US leads in connected drug delivery device patents with over 5,000 active patents). A combination product strategy that pairs a differentiated delivery device with a GLP-1 active ingredient can generate independent IP protection and a user experience premium that sustains brand loyalty post-generic entry of the GLP-1 molecule itself.
In MASH, the first-to-market advantage of resmetirom will not last beyond 2-3 additional approvals. The most technically differentiated next-wave candidates are FGF21 analogs (efruxifermin, Akero Therapeutics; pegozafermin, 89bio, in Phase III), which target a different biological pathway (fatty acid oxidation and lipid metabolism through FGFR1c/beta-klotho receptor activation) and may combine well with resmetirom in a combination regimen addressing different fibrotic pathways simultaneously. Combination MASH therapy — analogous to the multi-drug regimens that transformed HIV and hepatitis C — is the commercial model most likely to generate durable premium pricing.
Priority Opportunity 3: Genetically Defined Rare Diseases With CRISPR-Addressable Targets
The CRISPR gene editing opportunity is not limited to the Intellia/Beam/Prime Medicine companies that hold the foundational IP. The delivery technology — lipid nanoparticles for hepatic targeting, AAV for CNS and retinal targeting, ex vivo lentiviral editing for hematopoietic cells — is itself a differentiable IP layer. Companies that own proprietary LNP formulations with improved hepatic targeting efficiency, or AAV capsid variants with improved CNS penetration and lower immunogenicity, are positioned to out-license their delivery technology to gene editing companies that control the editing component. The royalty economics on a USD 3 million single-treatment gene editing therapy are extraordinary — a 5% royalty on delivery technology generates USD 150,000 per patient treated.
For companies with clinical capabilities rather than platform technology focus, the most actionable rare disease opportunities are in conditions meeting four criteria: single-gene disorder with identified mutation, existing natural history data supporting trial design, patient advocacy organization with research infrastructure, and sufficient US patient prevalence (at least 5,000 patients) to support NDA approval with credible sales projections. Conditions that meet these criteria and do not yet have an approved therapy as of 2025 include several specific mucopolysaccharidoses, alpha-1 antitrypsin deficiency (where gene therapy and RNAi approaches are in Phase II), and specific forms of primary hyperoxaluria beyond the two conditions addressed by Alnylam’s approved products.
Priority Opportunity 4: Biosimilar Entry Against the 2026-2030 Biologic Patent Cliff
Seven of the top fifteen best-selling biologics in the US will face initial composition-of-matter patent expiry between 2026 and 2030. These include pembrolizumab (Keytruda), nivolumab (Opdivo), dupilumab (Dupixent, Sanofi/Regeneron), and multiple others. The biosimilar development programs for these compounds should be in active development now — the 3-4 year development timeline from aBLA filing to approval means that a biosimilar developer beginning development in 2025 can realistically target a 2028-2029 launch date.
The Dupixent (dupilumab) biosimilar opportunity deserves specific attention. Dupilumab generated approximately USD 14 billion in 2024 sales across atopic dermatitis, asthma, COPD, eosinophilic esophagitis, and other IL-4/IL-13 inhibition indications. Its core composition-of-matter patents begin expiring around 2027-2028 in the US, though the secondary patent landscape requires careful mapping. A biosimilar developer with anti-IL-4/IL-13 monoclonal antibody manufacturing capability and a robust analytical comparability package is positioned for one of the most commercially significant biosimilar launches since adalimumab.
Priority Opportunity 5: The 505(b)(2) Reformulation Premium for Off-Patent Specialty Molecules
The off-patent specialty drug reformulation opportunity is systematically underserved by both large pharma (too small to move the revenue needle) and generic manufacturers (insufficiently differentiated to justify the development cost). It is well-suited to mid-size specialty pharmaceutical companies with 505(b)(2) regulatory expertise.
The architecture of a successful 505(b)(2) reformulation program involves: identifying an off-patent specialty molecule with documented clinical limitations (suboptimal pharmacokinetics, tolerability burden, administration inconvenience, or need for therapeutic drug monitoring), developing a novel formulation that addresses the specific limitation with clinical differentiation data, filing a 505(b)(2) application that cites the original NDA for safety/efficacy and adds proprietary clinical data for the formulation change, earning 3-year Waxman-Hatch new clinical study exclusivity upon approval, and pricing the reformulated product at a substantial premium to the generic baseline.
Suzetrigine’s NaV1.8 mechanism aside, the recently approved non-opioid acute pain market illustrates this approach: VX-548, Vertex’s compound, was developed as a differentiated mechanism rather than a reformulation, but the commercial infrastructure for NaV-targeted pain drugs — specialty pharmacy, pain management KOL networks, payer coverage development — applies equally to any second-entrant in the mechanistic class.
Strategic Recommendations for IP, R&D, and Business Development Teams
IP teams should maintain a rolling Orange Book surveillance protocol covering all drugs within 5 years of primary COM patent expiry in the top 20 therapeutic categories by revenue. For any molecule with greater than USD 500 million in US annual revenues, the secondary patent landscape requires a complete freedom-to-operate analysis and a litigation risk assessment. Paragraph IV campaigns against secondary patents should be evaluated using empirical success rate benchmarks by patent type, not by the strength of the brand’s Orange Book listing.
For biologics-focused IP teams, the Purple Book surveillance equivalent is the biosimilar BLA tracker maintained by the FDA. Any biologic with greater than USD 1 billion in US revenues, reference product exclusivity expiring before 2030, and fewer than three active biosimilar development programs represents a white space on the biosimilar development map. The biosimilar development decision should include a manufacturing capability assessment: companies without an existing mAb production platform will spend USD 50-80 million on manufacturing development before filing a single clinical study.
R&D leadership teams evaluating therapeutic area strategy should weight three factors above all others: the probability-adjusted commercial value of the opportunity, the strength of the IP position the company can build (composition-of-matter vs. method-of-use vs. secondary patent), and the company’s differentiated scientific capability relative to the current pipeline. A company with best-in-class RNA chemistry capabilities is not well-positioned to compete in small molecule CNS, regardless of the market size.
Business development teams evaluating in-licensing and acquisition opportunities should apply rNPV analysis consistently across therapeutic areas without allowing revenue size alone to drive valuation. A USD 200 million rare disease asset with 7-year orphan exclusivity, a 40% probability of approval, and USD 500 million peak sales may generate a higher risk-adjusted IRR than a USD 500 million cardiovascular asset with 15% approval probability, USD 3 billion peak sales, and IRA negotiation eligibility by year 8.
Market Data Reference Tables
Table 1: US Pharmaceutical Market Size and Growth Projections, 2023-2033
Year
Market Size (USD Billion)
CAGR (Source Basis)
2023
574-602
Baseline (multiple sources)
2024
634
Base year (Grand View Research)
2030
857-884
5.7% CAGR from 2025
2030
>1,000
5.5% CAGR from 2024 (Magnet ABA)
2033
1,094
6.15% CAGR from 2024 (Nova One)
Table 2: High-Unmet-Need Therapeutic Areas — Prevalence, Market Size, and Key IP Dynamics
Therapeutic Area
Condition
US Prevalence
2023-2024 Market (USD)
Key IP Holder(s)
Patent Cliff Timing
Oncology
HR+/HER2- Breast Cancer
211,000 new cases/yr
USD 7.5B
AstraZeneca/DSP (ADC)
Datopotamab: ~2038
Neurology
Alzheimer’s Disease
6.5M (age 65+)
USD 3-5B (growing)
Eisai/Biogen; Eli Lilly
Lecanemab: ~2032; Donanemab: ~2034
Neurology
Multiple System Atrophy
25,000-75,000
<USD 100M (unmet)
Alterity (ATH434, FT)
Phase II/III
Metabolic
GLP-1 Obesity/Diabetes
110M+ eligible US adults
USD 25B+ (2024)
Novo Nordisk; Eli Lilly
Semaglutide: ~2032; Tirzepatide: ~2036
Metabolic
MASH
16-20M (F2-F4)
USD 1B (nascent)
Madrigal (Rezdiffra)
~2033
Rare Disease
Hemophilia B
~5,000 US patients
USD 2B+
CSL Behring (gene therapy)
Hemgenix: long exclusivity
Rare Disease
HAE
~6,000 US patients
USD 3-4B
Takeda; KalVista; Intellia
NTLA-2002: Phase III
Rare Disease
ATTR Amyloidosis
17-173 per million
USD 2-3B
Alnylam (RNAi); Intellia (CRISPR)
Patisiran: ~2033
Pain
Acute Pain (non-opioid)
50M acute episodes/yr
USD 17.2B
Vertex (Journavx)
~2038
Autoimmune
Psoriatic Arthritis
0.06-0.25% US adults
USD 8B+
BMS (Sotyktu); Janssen; UCB
Varied, 2028-2034
Table 3: 2025 Novel Drug Approvals and IP Status
Drug (Brand)
INN
Company
Indication
Mechanism
Approval Year
Primary Patent Expiry (Est.)
Imaavy
Nipocalimab-aahu
Johnson & Johnson
Generalized MG
FcRn antagonist
2025
~2034
Vanrafia
Atrasentan
Novartis/Chinook
IgA Nephropathy
ET-A receptor antagonist
2025
~2033
Qfitlia
Fitusiran
Sanofi
Hemophilia A/B
siRNA (antithrombin)
2025
~2035
Blujepa
Gepotidacin
GSK
Uncomplicated UTI
Topoisomerase II inhibitor
2025
~2036
Romvimza
Vimseltinib
Deciphera/Daiichi
TGCT
CSF1R inhibitor
2025
~2035
Gomekli
Mirdametinib
SpringWorks
NF1 plexiform
MEK inhibitor
2025
~2033
Journavx
Suzetrigine
Vertex
Acute pain
NaV1.8 selective inhibitor
2025
~2038
Datroway
Datopotamab deruxtecan-dlnk
AstraZeneca/DSP
HR+/HER2- mBC
ADC (TROP2-DXd)
2025
~2037
Table 4: Biosimilar Market Development Status as of End of 2024
Reference Product
INN
Originator
US Reference Sales (2024 est.)
Biosimilars Approved
With Interchangeability
Reference Exclusivity Status
Humira
Adalimumab
AbbVie
~USD 9B (declining)
10
2
Post-exclusivity
Keytruda
Pembrolizumab
Merck
~USD 25B
0
0
COM patent ~2028
Dupixent
Dupilumab
Sanofi/Regeneron
~USD 14B
0 (in dev.)
0
COM patent ~2027-2028
Opdivo
Nivolumab
BMS
~USD 9B
0 (in dev.)
0
COM patent ~2027
Herceptin
Trastuzumab
Roche/Genentech
~USD 1.5B (generic-eroded)
10+
Multiple
Post-exclusivity
Avastin
Bevacizumab
Roche/Genentech
~USD 1.5B (eroded)
8+
Some
Post-exclusivity
Stelara
Ustekinumab
J&J
~USD 7B
4
0
COM patent expired Jan 2024
Overall Key Takeaways
The US pharmaceutical market’s growth trajectory is real and durable through 2030, but the growth is not distributed uniformly across the market. The specialty drug and biologics segments grow at two to three times the rate of the generic small molecule base. Market entry strategies calibrated to that bifurcation — focusing on precision oncology, rare genetic diseases, CNS neurodegeneration, and next-generation metabolic therapies — capture the high-CAGR portion of the market. Strategies built on primary-care small molecule generic entry remain viable but operate in a structurally deflationary segment where first-mover advantage on 180-day exclusivity is the primary value creation event.
Patent analysis is not a compliance exercise. It is the primary source of competitive intelligence for market entry timing, litigation risk quantification, and IP valuation. Every company considering market entry into any therapeutic area should maintain a systematic Orange Book and Purple Book surveillance protocol, apply Paragraph IV probability benchmarks with empirical rigor, and value orphan exclusivity separately from patent protection in its financial models.
The IRA does not end pharmaceutical profitability; it concentrates the revenue risk in specific categories — long-duration, high-Medicare-utilization, large-market branded drugs. New market entries post-2025 that are designed from the outset with IRA eligibility in mind — targeting populations with lower Medicare concentration, pursuing combinations that maintain clinical differentiation, securing value-based reimbursement contracts — will outperform those that use pre-IRA commercial assumptions.
Clinical probability of success is the most controllable variable in pharmaceutical ROI, and it is controlled primarily through target validation quality, patient population precision, and trial design discipline. A biomarker-selected Phase II trial costs more per patient than an unselected trial, but it generates data of higher predictive value for Phase III success. Every percentage point improvement in Phase III probability of success adds approximately USD 150-400 million to the rNPV of a mid-market drug asset. That arithmetic makes upfront investment in biomarker development and patient stratification among the highest-returning capital allocation decisions in drug development.
Sources and methodology available on request. All market projections are sourced from third-party research and are subject to the definitional limitations noted in Part I. IP expiry estimates are based on publicly available Orange Book and USPTO data and may be affected by patent term extension applications, pediatric exclusivity grants, and litigation outcomes not reflected in current filings.