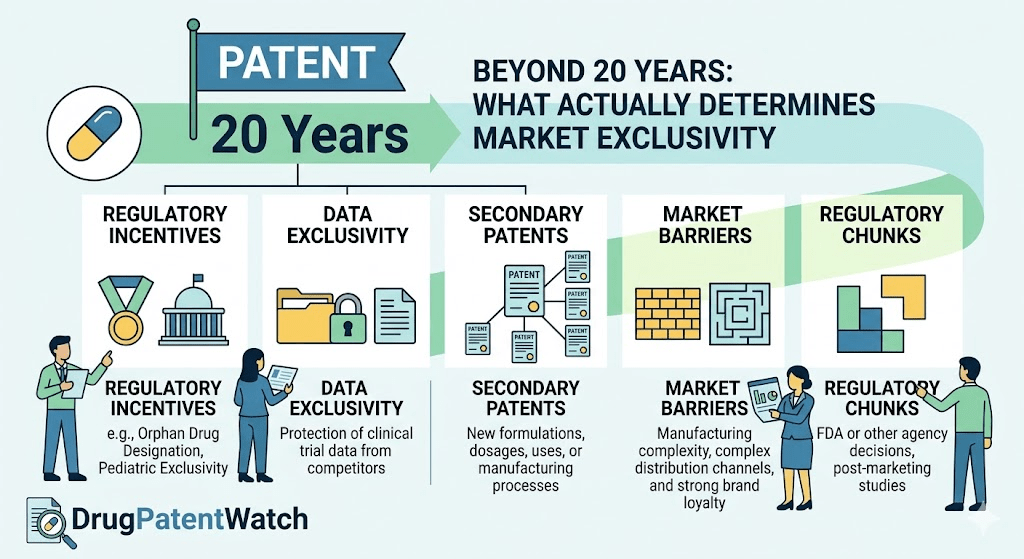
Ask anyone in pharma how long a drug patent lasts and they’ll say 20 years. The number is technically correct and commercially useless. By the time a new molecule reaches a pharmacy shelf, much of that statutory term has already burned off in lab notebooks, animal studies, and FDA review queues. The Tufts Center for the Study of Drug Development pegs the average time from compound synthesis to approval at 14.4 years [1]. Do the math on a patent filed at synthesis: there is roughly five and a half years of nominal patent life left on the day a sales rep first walks into a clinic.
The real number that matters to a portfolio manager, a generic challenger, or a hedge fund running a binary-event playbook is effective market exclusivity. That figure is the product of patent expiration math, FDA-granted regulatory exclusivities, lifecycle management tactics, litigation outcomes, the politics of price negotiation, and a half-dozen other factors that have nothing to do with the 20-year clock most outside observers fixate on. A 2024 analysis from George Mason’s Center for Intellectual Property and Innovation Policy found the average effective patent life across a representative sample of branded drugs was 13.35 years, with a median of 14.01 years [2]. That is a wide gap between the printed statute and the lived commercial reality.
This piece is for people who already know the basics of Hatch-Waxman and want the underlying mechanics, the case-study evidence, and the strategic levers. We will walk through every meaningful factor that compresses or extends a drug’s protected revenue window, with real companies, real molecules, and real numbers. Where the data tracking gets dense, platforms like DrugPatentWatch surface the Orange Book filings, terminal disclaimers, and exclusivity codes that determine when generic competition can actually file, get approved, and launch.
The 20-Year Patent Term Is a Ceiling, Not a Floor
Under 35 U.S.C. §154, a U.S. utility patent runs for 20 years from the earliest non-provisional filing date. For pharmaceuticals, the clock starts at the composition-of-matter filing, which typically happens shortly after a medicinal chemist identifies a promising lead. That filing usually predates the first human dose by several years and precedes FDA approval by closer to a decade.
The Tufts numbers cited above are not outliers. Independent analyses converge on a similar range. A pharmaceutical company typically identifies a promising compound, files a patent application covering it, and then spends the next 8 to 12 years running preclinical studies, Phase I through Phase III clinical trials, and navigating FDA review before the drug reaches patients [3]. The total clock from patent filing to approval frequently runs 10 to 14 years for new molecular entities, leaving anywhere from 6 to 10 years of remaining nominal patent term at launch.
Why the Filing-to-Approval Gap Keeps Widening
The development timeline has not compressed despite FDA process improvements. Clinical trial complexity has increased. Adaptive designs, biomarker-stratified enrollment, and combination-therapy pivots all add operational time. The diseases being targeted are also harder: oncology, neurology, and rare disease programs run longer than the cardiovascular and metabolic blockbusters of the 1990s. A 2024 Tufts update found median Phase I-to-approval times for oncology programs exceeded 10 years, compared with under 8 years for cardiovascular drugs in the same dataset [4].
The Compound Patent Versus the Method Patent
Companies often file multiple patents on a single asset. The composition-of-matter or compound patent is the strongest, covering the molecule itself. Method-of-use patents cover specific indications. Formulation patents cover delivery systems, salt forms, polymorphs, and excipient combinations. Process patents cover manufacturing methods. Each has a different filing date and therefore a different expiration date.
When industry analysts talk about a ‘patent cliff,’ they usually mean the day the compound patent expires. That is the strongest piece of IP, and its expiration removes the broadest legal barrier to generic entry. But it is rarely the last patent to expire. Pfizer’s Lipitor compound patent (U.S. 4,681,893) expired November 30, 2011 [5]. The enantiomer patent ran to June 2011 (with pediatric exclusivity), and various process and crystalline form patents ran to 2016 and 2017 [6]. The compound expiration triggered the cliff. The later patents did not stop generic entry but did affect formulation choice and litigation posture for the first few entrants.
Patent Term Extension: Restoring Time Lost to Regulatory Review
The Drug Price Competition and Patent Term Restoration Act of 1984, known almost universally as Hatch-Waxman, established the Patent Term Extension mechanism under 35 U.S.C. §156. The statute restores a portion of patent life consumed by FDA regulatory review, with two hard caps that pharmaceutical patent attorneys memorize on day one.
The Five-Year Maximum
The absolute maximum extension is five years, regardless of how long the regulatory review period was [7]. If the FDA took eight years to approve a drug, the maximum PTE is still five years. The statutory formula adds half of the Investigational New Drug (IND) phase to all of the FDA review phase, then subtracts any periods the applicant failed to act with due diligence. The result is the extension period, capped at five years.
The 14-Year Effective Patent Life Cap
The second limitation is more restrictive in practice. The 14-year effective patent life cap stipulates that the total remaining patent term after the extension is applied cannot exceed 14 years from the date of FDA approval [8]. If the patent would still have 13 years remaining as of the date of FDA approval even without any PTE, the extension is limited to one year. This 14-year ceiling reflects Congress’s judgment that 14 years of post-approval exclusivity is sufficient to justify the R&D investment.
The practical effect of the 14-year cap is asymmetric. Drugs with long development timelines (12 or more years from patent filing to approval) will have short remaining patent lives at approval, often 7 to 8 years, and will qualify for extensions approaching the 5-year maximum. Drugs approved quickly relative to their patent filing date face significant cap erosion. Speed to market can paradoxically reduce the PTE benefit [9].
The One-Patent Rule
Section 156 permits PTE for only one patent per approved product. The patent selected receives the extension, and the others run out on their original schedule. This makes patent selection a material financial decision. Picking the wrong patent for extension can cost years of protected revenue. The choice typically goes to the compound patent because it provides the broadest legal coverage, but a strategically valuable method-of-use or formulation patent can sometimes be the better selection if the compound patent has separate strength concerns or if its expiration is already far enough out that the cap binds.
Eligibility Conditions for PTE
Four conditions must be satisfied. The patent must claim the approved product, a method of using it, or a method of manufacturing it. The patent must not have previously been extended. The application for extension must be filed within 60 days of FDA approval. The product must be subject to a regulatory review period before commercial marketing. The 60-day filing deadline is absolute. Miss it and the entire extension is forfeit, no matter how meritorious the underlying case.
FDA Regulatory Exclusivities: A Parallel Fortress
Patents and FDA exclusivities are different legal instruments that run on different clocks and serve different purposes. Patents are property rights granted by the USPTO. Exclusivities are statutory periods during which the FDA cannot approve, or in some cases cannot even accept, a competing application. The two protections often overlap, but each can outlast the other depending on filing dates, approval dates, and the type of exclusivity granted.
New Chemical Entity Exclusivity
The five-year New Chemical Entity (NCE) exclusivity is granted upon approval of a drug containing an active moiety never previously approved by the FDA [10]. Under NCE exclusivity, an ANDA or 505(b)(2) new drug application cannot be filed by anyone until four years after the NDA’s approval. The FDA cannot approve an ANDA or 505(b)(2) application until five years after the NDA’s approval. The four-year filing bar can be shortened by one year if the generic applicant files a Paragraph IV certification challenging an Orange Book-listed patent.
New Clinical Investigation Exclusivity
The three-year exclusivity for new clinical investigations protects approvals that rely on new clinical studies essential to a change in a previously approved drug. The change can be a new indication, dosage form, or strength. The three-year period bars FDA approval of generics that rely on the new clinical data, but it does not bar generics referencing the original approval.
Orphan Drug Exclusivity
The Orphan Drug Act provides seven years of market exclusivity for drugs treating rare diseases (defined in the U.S. as those affecting fewer than 200,000 patients). During this period, the FDA cannot approve another sponsor’s application for the same drug for the same orphan indication. Orphan Drug Exclusivity (ODE) is indication-specific. A drug approved for multiple indications, only one of which is an orphan designation, will face generics with the protected indication ‘carved out’ of the label.
Abilify illustrates the carve-out mechanic. The atypical antipsychotic is indicated for multiple conditions, including schizophrenia and Tourette’s Disorder. In December 2014, the FDA granted ODE for the Tourette’s Disorder indication. The FDA could not approve generic versions of Abilify labeled for Tourette’s until the ODE expired in December 2021. Generic versions of Abilify were approved with the Tourette’s indication omitted from the label [11]. Pharmacists could still dispense those generics for other approved indications, and off-label use closed the rest of the gap.
Pediatric Exclusivity
Pediatric exclusivity is a six-month add-on that attaches to the end of all existing patents and exclusivities covering a drug, provided the sponsor conducts pediatric studies in response to an FDA Written Request and complies with the relevant requirements [12]. The mechanism is uniquely powerful for two reasons. It stacks on top of every other form of protection rather than running concurrently. It applies to all of the drug’s exclusivities and Orange Book-listed patents, not just one.
For a $5 billion-a-year product, six months of pediatric exclusivity is worth roughly $2.5 billion in protected revenue. The math on running pediatric studies almost always works out in the sponsor’s favor, even for indications where pediatric use is clinically marginal. The FDA’s Written Request system, the Best Pharmaceuticals for Children Act, and the Pediatric Research Equity Act collectively shape which drugs get studied and which qualify for the extension.
GAIN and Other Specialty Exclusivities
The Generating Antibiotic Incentives Now (GAIN) Act adds five years to the applicable NCE, new clinical investigation, or orphan exclusivity for qualifying antibacterial and antifungal products that treat serious or life-threatening infections. Other narrow categories exist for tropical disease priority review vouchers and rare pediatric disease vouchers, though those are transferable assets that operate differently from exclusivities tied to a specific molecule.
The Biologic Exception: BPCIA and 12 Years of Reference Product Exclusivity
Biologics live under a separate statutory regime. The Biologics Price Competition and Innovation Act (BPCIA), enacted as part of the Affordable Care Act in 2010, provides 12 years of reference product exclusivity from the date of biologic license approval [13]. A biosimilar application cannot be approved during this period. An additional four-year bar at the front prevents the FDA from even accepting a biosimilar application until four years after reference product approval.
The 12-year figure was a political compromise. The branded industry pushed for 14 years. Biosimilar advocates and consumer groups pushed for 7. The commercial rationale for the longer term involves the genuine complexity of biologic manufacturing, the longer development timelines for large molecules, and the historically weaker effect of biosimilar competition on prices compared with small-molecule generic entry.
No Patent Linkage for Biologics
Unlike Hatch-Waxman’s Orange Book system, BPCIA does not link FDA biosimilar approval to patent expiration. The FDA can approve a biosimilar regardless of any biologic patents that may still be in force. Patent disputes between reference product sponsors and biosimilar applicants proceed through the BPCIA’s ‘patent dance,’ a structured exchange of information that ultimately leads to litigation, but the FDA does not stay biosimilar approval pending patent resolution. The absence of patent linkage explains why biologic exclusivity periods are systematically longer than the Hatch-Waxman cap: the FDA needs to give branded biologics direct statutory exclusivity because patent challenges cannot delay biosimilar approval the way they delay generic approval.
The Median Biologic Reaches Biosimilar Competition at 20+ Years
Historically, small molecule drugs reach generic entry at a median of 12.6 years post-approval, while biologics face biosimilar competition at a median of 20.3 years [14]. The biologic gap reflects the combined effect of the 12-year exclusivity, manufacturing complexity that deters biosimilar entrants, complex patent thickets, and the BPCIA litigation framework.
Patent Thickets: The Humira Playbook
The textbook example of patent layering as a market exclusivity tool is AbbVie’s Humira (adalimumab). The drug generated nearly $200 billion in cumulative sales between launch in 2002 and the start of biosimilar competition in the U.S. in January 2023 [15]. Its primary composition-of-matter patent expired in 2016. Biosimilar competition did not arrive in the United States for another seven years.
The gap was created by what academics, regulators, and biosimilar makers call a patent thicket. AbbVie applied for approximately 247 patents on Humira, of which 132 were granted [16]. The portfolio covered the adalimumab molecule itself, but most of the filings came after launch and covered formulations, dosing regimens, manufacturing methods, and treatment protocols. Roughly 90% of Humira’s patent filings came after the drug was already on the market, and almost half were filed in 2014 or later, in advance of the expiration of the primary patent [17].
How the Thicket Functioned
Biosimilar developers faced a strategic problem. Even after the compound patent expired, a biosimilar manufacturer could not market its product without potentially infringing dozens of secondary patents covering formulation, manufacturing, and method of use. Litigating each patent individually would take years and cost tens of millions per defendant. AbbVie asserted as many as 63 patents against one biosimilar entrant [18].
The economically rational response for biosimilar makers was to settle. Eight to ten manufacturers entered individual settlement agreements with AbbVie. Each agreement permitted the biosimilar to enter the U.S. market on a staggered schedule beginning in January 2023, with royalty payments to AbbVie continuing afterward. The same biosimilars launched in Europe in October 2018 because AbbVie’s European patent portfolio was substantially thinner [19].
The Antitrust Challenge Failed
A coalition of Humira buyers, including consumer groups, wholesalers, and union welfare funds, sued AbbVie under the Sherman Act, alleging that the patent thicket was an unlawful scheme to monopolize the adalimumab market. The Seventh Circuit affirmed dismissal of the antitrust claims in 2022, holding that filing patent applications protected under the Noerr-Pennington doctrine generally is not actionable antitrust conduct, even when done in volume. The plaintiffs would have needed to prove that the patents were objectively baseless, a high bar given that 132 of them were granted by the USPTO [20].
Why Humira Was the Exception, Not the Rule
One reason Humira’s strategy has not been replicated at scale is that it requires a combination of patentable subject matter (formulations that admit meaningful variation, manufacturing processes with multiple paths), regulatory complexity (biologic-class manufacturing review), and commercial scale that justifies the litigation budget. Most blockbuster drugs lack one or more of these elements. The Humira approach is an exception rather than the rule [21], but it has shaped policy debates about patent quality, terminal disclaimers, and obvious-variant filings ever since.
Terminal Disclaimers and Obviousness-Type Double Patenting
Patent thickets often rely on terminal disclaimers, a procedural device that allows an applicant to overcome a rejection for obviousness-type double patenting. A terminal disclaimer ties the disclaimed patent’s expiration to the expiration of an earlier-issued patent in the same family. Humira’s thicket included 436 terminal disclaimers [22]. Revlimid’s portfolio of 30 patents included 18 terminal disclaimers and protected Celgene (later Bristol Myers Squibb) from generic competition well past the primary patent’s expiration date [23].
A proposed USPTO rule (now withdrawn) would have required that terminally disclaimed patents share validity standing with their parent patent, meaning that a successful invalidity challenge to the parent would automatically invalidate all of the children. Modelers found that applying such a rule retroactively could have cut Humira’s thicket significantly and reduced Revlimid’s thicket by approximately 70% [24]. The rule’s withdrawal in 2024 ended the immediate policy push, but the underlying terminal-disclaimer mechanic remains a frequent feature of pharmaceutical patent strategy.
The Orange Book and Paragraph IV: Generic Challenges as a Compression Mechanism
Hatch-Waxman set up an information-sharing system that requires NDA holders to list patents covering an approved drug in the FDA’s Orange Book. Generic applicants filing ANDAs must certify with respect to each listed patent. The four certification options are: (I) no patent information has been submitted; (II) the patent has expired; (III) the generic will not launch until the patent expires; or (IV) the patent is invalid, unenforceable, or not infringed.
The Paragraph IV Mechanism
A Paragraph IV certification is the generic industry’s primary tool for compressing branded exclusivity. By certifying that an Orange Book patent is invalid or not infringed, the generic applicant triggers a 45-day window during which the NDA holder may sue for patent infringement. If suit is filed within 45 days, the FDA imposes a 30-month stay on generic approval to allow the litigation to proceed. If suit is not filed, the stay does not apply.
180-Day First-Filer Exclusivity
To incentivize Paragraph IV challenges, the first generic to file a substantially complete ANDA with a Paragraph IV certification receives 180 days of generic market exclusivity. During that period, no other ANDA referencing the same listed drug can be approved. The first-filer position is commercially valuable. Ranbaxy’s first-filer status on atorvastatin was estimated to be worth roughly $600 million in earnings during its 180-day window [25].
Plavix and the Apotex Disaster
Apotex’s clopidogrel (Plavix) challenge in 2006 illustrates how Paragraph IV gambles can go wrong. Apotex launched at risk after a failed reverse-payment settlement with Bristol-Myers Squibb and Sanofi. The company shipped enough product to flood the U.S. market for months before the litigation resolved against it. The damages exposure ended Apotex’s first-filer exclusivity and forced subsequent generics to compete on commodity-like terms when the patent finally expired in 2012 [26].
The Patent Cliff: What Happens When Protection Ends
The financial consequences of loss of exclusivity (LOE) are documented in painful detail across two decades of branded drug histories. Generic entry typically drops branded drug prices by 80% to 95% and shifts market share to generics within 6 to 12 months [27].
Lipitor: The Reference Case
Pfizer’s Lipitor (atorvastatin) lost its compound patent on November 30, 2011. Within a few years, annual revenue dropped from approximately $13 billion to under $3 billion, with reports indicating a 71% sales drop in a single quarter [28]. Generic atorvastatin entered priced 90% to 95% below brand. Pfizer ran a 180-day defense strategy that preserved roughly 30% market share through May 2012, but the long-run trajectory was set on patent-expiration day.
Nexium and Plavix
AstraZeneca’s Nexium (esomeprazole) faced generic competition in 2014, with generic versions reaching an estimated 80% price reduction. Plavix sales declined precipitously upon patent expiration in 2012, with generics leading to a 90% price reduction [29]. Both cases tracked the Lipitor template, with the brand retaining a residual share of patients on chronic therapy who resisted switching for non-clinical reasons.
Humira: The Slow Cliff
Humira’s loss-of-exclusivity dynamic looks different from a small-molecule cliff. Biosimilar entry has been slower and more price-stable than analysts predicted in 2022. The eight biosimilars launched in 2023 generally took a 5% to 30% list-price discount in the first 12 months, with substantial rebate negotiations playing out behind the formularies. By mid-2024, biosimilars held roughly 5% of U.S. adalimumab volume, far below the 60%-plus penetration biosimilars achieved in European markets over comparable time horizons. The mechanism is partly PBM economics, partly interchangeability designations, and partly AbbVie’s contracting strategy.
The Inflation Reduction Act: A New Compression Variable
The Inflation Reduction Act of 2022 introduced a different kind of exclusivity compression. Medicare can now negotiate prices for certain high-spend drugs through the Drug Price Negotiation Program (DPNP). The eligibility timeline is the new variable that changes how pharma companies think about effective exclusivity.
The Pill Penalty
Small molecule drugs become eligible for Medicare price negotiation seven years after FDA approval, with the negotiated price taking effect in year nine. Biologics become eligible for negotiation 11 years post-approval, with the negotiated price taking effect in year 13 [30]. The industry calls the four-year gap the ‘pill penalty.’ It functionally creates a different effective exclusivity ceiling for small molecules than for biologics, separate from patent or FDA exclusivity considerations.
What the Pill Penalty Does to Investment Decisions
A 2022 PhRMA survey found 63% of responding member companies planned to shift R&D focus away from small molecules in response to the IRA, and 95% planned to develop fewer new uses for existing medicines [31]. Whether the shift will persist at scale is contested, but early evidence is consistent with the trend. A 2025 analysis published in Health Affairs Scholar found that following the IRA, small molecule oncology drugs experienced a disproportionately greater reduction in post-approval clinical trial initiations compared with biologics [32].
Why the Asymmetry Exists
Some analysts argue the longer biologic window reflects manufacturing complexity and longer development timelines for large molecules. Others point to the political influence of the biologics industry or to a misunderstanding of the historical patent landscape. Whatever the origin, the asymmetry matters financially. Historically, small molecule drugs reached generic entry at a median of 12.6 years post-approval. The IRA effectively moves the revenue cliff forward to year nine for selected drugs, capturing the asset just as it typically reaches commercial peak [33].
The EPIC Act Proposal
The bipartisan Ensuring Pathways to Innovative Cures (EPIC) Act would give small molecules the same 13-year market window before price setting that biologics receive. The bill has not passed as of this writing, but it represents the most serious legislative response to the pill penalty and tracks closely with PhRMA’s policy ask.
According to IQVIA Institute analysis, biosimilars and unbranded biologics generated $36 billion in cumulative U.S. savings between 2015 and 2023, with the bulk of those savings concentrated in the post-2019 wave of competition. Despite this, biologics still face biosimilar competition at a median of 20 years post-approval — roughly eight years later than small molecules face generics — making biologic franchises structurally more durable than chemical drugs even before factoring in IRA price negotiation effects [34].
Lifecycle Management Tactics That Extend Effective Exclusivity
Beyond patents and exclusivities, branded manufacturers use a set of well-worn tactics to preserve revenue past the nominal cliff. Some are clinically meaningful. Others are largely defensive.
Reformulation and Line Extensions
Switching patients to a new formulation before the compound patent expires moves prescription volume to a product with fresh patent and exclusivity coverage. Extended-release versions, fixed-dose combinations, and reformulated salt forms all qualify. AstraZeneca’s switch from Prilosec (omeprazole) to Nexium (esomeprazole, the active enantiomer) is the canonical example. Forest Laboratories’ switch from Celexa (citalopram) to Lexapro (escitalopram) used the same enantiomer-isolation playbook. The legal mechanic relies on three-year new clinical investigation exclusivity for the new product plus method-of-use patents on the reformulation.
Authorized Generics
The NDA holder can license a generic version to a marketing partner or a wholly-owned subsidiary, who launches it during the first-filer’s 180-day exclusivity window. The authorized generic shares the market with the first-filer and compresses generic pricing during the period of theoretically protected first-filer pricing. Pfizer used this strategy with Lipitor through its partnership with Watson Pharmaceuticals.
Product Hopping
Product hopping involves removing the original formulation from the market shortly before patent expiration and replacing it with a reformulated version. Pharmacies are then unable to substitute generics of the original formulation because the reference listed drug is no longer commercially available. The tactic has drawn antitrust scrutiny. In New York v. Actavis, the Second Circuit ruled in 2015 that Actavis’s discontinuation of Namenda IR in favor of Namenda XR before generic Namenda IR could launch was an exclusionary act under Section 2 of the Sherman Act [35].
Citizen Petitions
NDA holders sometimes file citizen petitions with the FDA raising scientific or regulatory questions about pending generic applications. The FDA must respond to citizen petitions, and the response process can delay generic approval. The FDA Safety and Innovation Act of 2012 tightened the rules to prevent citizen petitions from triggering automatic stays of generic approval, but the petitions remain a friction-inducing tool.
Tracking the Real Exclusivity Map: Why Orange Book Reading Is a Skill
For analysts, business development teams, and generic strategists, the practical question is not ‘when does the patent expire’ but ‘when can a competitor actually launch.’ The answer requires reading the Orange Book listings, the FDA exclusivity codes, any pending pediatric studies, terminal disclaimer records, and pending PTE applications, then layering on litigation status and any settlements with prior filers.
DrugPatentWatch aggregates this data across thousands of approved drugs, surfacing the Orange Book filings, exclusivity expirations, and patent challenges that determine when generic competition can actually file, get tentative approval, and launch. For a single asset, the work can be done by hand. For a portfolio, the aggregation matters. The same applies to biosimilar tracking through the Purple Book, where pending interchangeability designations and reference product exclusivity dates drive a different set of timing calculations.
What Sophisticated Users Track
Working through a single drug, the questions a portfolio analyst will want answered include the following. What is the compound patent expiration date, and has PTE been applied for or granted? What method-of-use and formulation patents are Orange Book-listed, and what are their nominal expirations? Are any pending pediatric studies likely to add six months? What is the NCE, ODE, or new clinical investigation exclusivity status? Are there any pending Paragraph IV ANDAs, and if so, when did the 45-day window close and is the 30-month stay running? Has the manufacturer entered any settlements with first-filers that fix a specific launch date? For biologics, when did the 12-year BPCIA exclusivity end, and are biosimilar applications pending or approved? For IRA-eligible drugs, where does the asset sit in the selection cycle?
Each of these has a specific data source. The Orange Book lists patents and exclusivities by drug product. The FDA’s regulatory exclusivity page tracks NCE and ODE codes. The USPTO’s patent term extension records show pending and granted PTEs. The PACER docket system tracks Paragraph IV litigation. CMS publishes IRA selection lists and negotiated price effective dates. Pulling these together for a portfolio of 50 to 100 assets is the work of a dedicated analyst team or a specialized platform.
Case Study: Revlimid and the Orphan Drug Extension Strategy
Celgene’s Revlimid (lenalidomide) is a small molecule oncology drug approved in 2005 for multiple myeloma and other hematologic malignancies. Its compound patent expired in 2019. Generic competition did not begin until 2022, with full unrestricted generic entry delayed to 2026 under volume-limited settlement agreements [36].
How Celgene Extended Exclusivity
Revlimid’s protection came from a stacked combination of orphan drug exclusivity (granted for multiple indications), method-of-use patents covering specific dosing regimens and patient populations, and a Risk Evaluation and Mitigation Strategy (REMS) program that gated distribution. The REMS, which was clinically justified by lenalidomide’s teratogenicity, also functioned as a barrier to generic entry because generic applicants needed access to the REMS to demonstrate bioequivalence and obtain approval.
The Settlement Cascade
Celgene entered settlement agreements with multiple ANDA filers that permitted volume-limited generic entry beginning in March 2022, with progressively larger volume allowances each year through 2025 and unrestricted entry in January 2026. The agreements were structured to preserve Revlimid revenue during the highest-margin years while avoiding the antitrust exposure of a flat exclusion deal.
Revenue Trajectory
Revlimid revenue peaked at $12.8 billion in 2021. The 2022 figure dropped to roughly $10 billion under the volume-limited generic entry. Bristol Myers Squibb’s 10-K disclosures show progressive erosion through 2024, with the drug expected to lose meaningful revenue contribution as generic volume caps lift. The case demonstrates that orphan exclusivity plus method-of-use patents plus REMS-based access control can extend effective exclusivity well past the compound patent expiration, even for a small molecule.
Case Study: Eliquis and the Patent Cliff in Slow Motion
Bristol Myers Squibb and Pfizer’s Eliquis (apixaban) was approved in 2012 for stroke prevention in atrial fibrillation. Its compound patent expires in 2026. The drug was selected for the first cycle of IRA Medicare price negotiation, with the negotiated price taking effect in January 2026. Eliquis is the canonical case of an asset where IRA price negotiation, not patent expiration, defines the binding exclusivity constraint.
The IRA Timing
Eliquis was selected by CMS in August 2023, and the negotiated Maximum Fair Price (MFP) takes effect in January 2026. Generic apixaban is not expected before April 2028 under existing settlement agreements with Mylan, Micro Labs, and Sigmapharm. The branded drug’s effective exclusivity is bounded by IRA negotiation, not patent expiration, for the two years between January 2026 and April 2028.
What This Tells Us About the New Math
For drugs that reach blockbuster status quickly and have long remaining patent life, IRA negotiation now defines the binding constraint on revenue rather than patent expiration. The implications for new drug development math are significant. Discount rates applied to peak-year revenue must account for the year-nine MFP, not the year-12 or year-14 patent cliff.
What This Means for Different Stakeholders
The mechanics above produce different consequences depending on where you sit in the value chain.
For Branded Manufacturers
The pre-launch IP strategy is a multi-year exercise in maximizing effective exclusivity through patent filing sequence, PTE patent selection, regulatory exclusivity stacking, and lifecycle management planning. The cost of getting any one of these wrong runs into billions of dollars for blockbuster assets. A 24-month error in PTE calculation on an $8-billion-a-year product is worth $16 billion in protected revenue.
For Generic and Biosimilar Companies
The challenge is identifying assets where the gap between nominal exclusivity expiration and binding exclusivity expiration is largest. Drugs with weak secondary patents, limited regulatory exclusivity layering, and clean Orange Book listings are the most attractive Paragraph IV targets. The risk-adjusted value of a successful first-filer position can justify litigation budgets in the tens of millions.
For Investors and Analysts
The work is in pricing the right discount rate to revenue in the years between nominal patent expiration and binding exclusivity expiration. For biologics, this means understanding biosimilar launch economics, formulary contracting dynamics, and interchangeability designations. For small molecules under IRA scrutiny, it means understanding the selection criteria, the negotiation timeline, and the likely MFP discount range based on therapeutic alternatives.
For Policy Watchers
The terminal disclaimer rule, the pill penalty, the EPIC Act, the patent thicket debate, and the ongoing FTC scrutiny of Orange Book listings are all live issues. Each could materially change the effective exclusivity math for substantial swaths of the branded pharmaceutical market. The IRA selection process itself is iterating, with the second cycle of drugs already named and the third cycle in development.
The Effective Exclusivity Math, Restated
The 20-year patent term remains the starting point, but it is the wrong number for any commercial decision. The right number is the binding exclusivity expiration, calculated by working through the following layers in order:
The compound patent expiration date, plus any PTE granted under Section 156 (capped at five years of extension and 14 years of post-approval life). The expiration dates of secondary patents (method, formulation, process) listed in the Orange Book. Any FDA regulatory exclusivity (NCE, ODE, new clinical investigation, GAIN) that runs past the patent dates. Any pediatric exclusivity that adds six months to all patents and exclusivities. For biologics, the 12-year BPCIA reference product exclusivity. Any settlement agreements with generic or biosimilar applicants that fix specific launch dates. For IRA-eligible drugs, the year-nine (small molecule) or year-13 (biologic) MFP effective date.
The binding constraint is whichever of these expires last, with the IRA MFP date now superseding patent expiration as the binding constraint for an increasing share of high-value assets. Working through this math for a single product takes hours. Doing it across a portfolio of 100+ assets requires either a dedicated patent intelligence team or a platform like DrugPatentWatch that aggregates the underlying data and surfaces the binding constraint dates.
Key Takeaways
The 20-year statutory patent term has little bearing on commercial reality for pharmaceutical products. Effective patent life averages 13 to 14 years, with a wide range driven by development timelines, regulatory review duration, and IP strategy.
Patent Term Extension under Hatch-Waxman restores up to five years of regulatory review time, capped at 14 years of post-approval patent life. The one-patent rule makes PTE selection a financially material decision.
FDA regulatory exclusivities (NCE, ODE, new clinical investigation, pediatric, GAIN) run independently of patents and can extend effective exclusivity past patent expiration. Orphan drug exclusivity is indication-specific and subject to label carve-outs.
Biologics receive 12 years of reference product exclusivity under BPCIA, with no patent linkage to biosimilar approval. The median biologic faces biosimilar competition at 20-plus years post-approval, compared with under 13 years for small molecules.
Patent thickets, terminal disclaimers, lifecycle management tactics, and settlement agreements can extend effective exclusivity well past nominal patent expiration. Humira’s 132-patent thicket extended effective U.S. exclusivity by approximately seven years past its compound patent expiration.
The Inflation Reduction Act has introduced a new exclusivity ceiling for high-spend Medicare drugs, with negotiated prices taking effect in year nine for small molecules and year 13 for biologics. The asymmetry is reshaping R&D investment allocation between molecule classes.
Effective exclusivity tracking requires aggregating Orange Book listings, FDA exclusivity codes, USPTO patent records, PACER litigation data, and IRA selection records. Platforms like DrugPatentWatch surface this data at portfolio scale.
FAQ
Why do drug patents last 20 years on paper but only 12 to 14 years in practice?
The 20-year clock starts at patent filing, which typically happens shortly after a medicinal chemist identifies a promising compound. FDA approval usually comes 10 to 14 years later, after preclinical work, three phases of clinical trials, and NDA review. By the time the drug is approved and can generate revenue, much of the 20-year term has elapsed. PTE under Section 156 can restore some of that lost time, but the 14-year post-approval cap limits the total restoration.
Can a drug have FDA exclusivity but no patents, or vice versa?
Yes, both situations occur. A drug can receive five-year NCE exclusivity even if its underlying patents have expired, providing standalone protection. A drug with strong patents may have minimal FDA exclusivity if its active moiety was previously approved (which precludes NCE eligibility). The two systems run on separate clocks and operate independently, which is why effective exclusivity calculations must consider both.
Why are biologics protected for so much longer than small molecules?
Biologics receive 12 years of statutory reference product exclusivity under BPCIA, compared with five years of NCE exclusivity for small molecules. The longer biologic window reflects manufacturing complexity, longer development timelines, and the absence of patent linkage between FDA approval and patent status. Biosimilar entry is also slower and produces smaller price reductions than small molecule generic entry, which compounds the effective exclusivity advantage.
How does the Inflation Reduction Act change effective exclusivity for affected drugs?
For drugs selected for Medicare price negotiation, the IRA imposes a new ceiling on effective exclusivity that operates independently of patent and FDA exclusivity. Small molecules become eligible for negotiation seven years after approval, with the negotiated price taking effect in year nine. Biologics become eligible at 11 years, with prices taking effect in year 13. For high-spend assets, IRA negotiation now defines the binding exclusivity constraint rather than patent expiration.
What is the most reliable way to track when a specific drug will face generic or biosimilar competition?
The starting point is the FDA Orange Book (for small molecules) or Purple Book (for biologics), which list patents and exclusivities. PACER provides docket information for any pending Paragraph IV litigation. USPTO records show pending and granted PTEs. CMS publishes IRA selection lists. For portfolio-scale tracking, DrugPatentWatch aggregates these data sources and surfaces the binding constraint dates across thousands of approved drugs, including settlement agreement terms that govern launch timing.
References
[1] DiMasi, J. A., Grabowski, H. G., & Hansen, R. W. (2016). Innovation in the pharmaceutical industry: New estimates of R&D costs. Journal of Health Economics, 47, 20-33.
[2] Center for Intellectual Property and Innovation Policy. (2024). Pharmaceutical ‘nominal patent life’ versus ‘effective patent life,’ revisited. George Mason University. https://cip2.gmu.edu/2024/05/20/pharmaceutical-nominal-patent-life-versus-effective-patent-life-revisited/
[3] DrugPatentWatch. (2026). Drug patents don’t last as long as you think. https://www.drugpatentwatch.com/blog/drug-patents-dont-last-as-long-as-you-think/
[4] DrugPatentWatch. (2026). How long does a drug patent actually last? The definitive analyst’s guide. https://www.drugpatentwatch.com/blog/how-long-does-a-patent-last-for-drugs/
[5] Pfizer Inc. (2010). Form CORRESP, FY2010. U.S. Securities and Exchange Commission. https://www.sec.gov/Archives/edgar/data/0000078003/000115752310001924/filename1.htm
[7] U.S. Food and Drug Administration. (2023). Small business assistance: Frequently asked questions on the Patent Term Restoration Program. https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/small-business-assistance-frequently-asked-questions-patent-term-restoration-program
[8] Azami Global. (2025). Patent term extension (PTE) under the Hatch-Waxman Act. https://azamiglobal.com/blog/patent-term-extension-pte-under-the-hatch-waxman-act/
[10] U.S. Food and Drug Administration. (2025). Frequently asked questions on patents and exclusivity. https://www.fda.gov/drugs/development-approval-process-drugs/frequently-asked-questions-patents-and-exclusivity
[11] IPD Analytics. (2025). Potential market exclusivity granted during U.S. regulatory approval process. https://www.ipdanalytics.com/post/potential-exclusivity-granted-during-us-regulatory-approval-process
[12] DrugPatentWatch. (2026). The six-month windfall: Transforming pediatric exclusivity from regulatory hurdle to strategic asset. https://www.drugpatentwatch.com/blog/the-six-month-windfall-transforming-pediatric-exclusivity-from-regulatory-hurdle-to-strategic-asset/
[13] DrugPatentWatch. (2026). When do drug patents expire: Understanding the lifecycle of pharmaceutical innovations. https://www.drugpatentwatch.com/blog/when-do-drug-patents-expire/
[14] DrugPatentWatch. (2026). The Inflation Reduction Act pill penalty: Structural distortions in pharmaceutical R&D. https://www.drugpatentwatch.com/blog/the-inflation-reduction-act-pill-penalty-structural-distortions-in-pharmaceutical-rd-and-strategic-pricing-adaptations/
[15] BioPharma Dive. (2023, January 27). Two decades and $200 billion: AbbVie’s Humira monopoly nears its end. https://www.biopharmadive.com/news/humira-abbvie-biosimilar-competition-monopoly/620516/
[16] Knox, R., & Curfman, G. (2022). The Humira patent thicket, the Noerr-Pennington doctrine, and antitrust’s patent problem. SSRN. https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4215822
[17] Storz, U. (2017). Feeling evergreen: A case study of Humira’s patent extension strategies. Harvard DASH Repository. https://dash.harvard.edu/bitstreams/0b2cd634-f60c-422f-8861-74725c0c940b/download
[18] Tu, S. S., & Lemley, M. A. (2022). Biological patent thickets and delayed access to biosimilars, an American problem. Journal of Law and the Biosciences. https://pmc.ncbi.nlm.nih.gov/articles/PMC9439849/
[19] Moorkens, E., Vulto, A. G., & Huys, I. (2021). The expiry of Humira market exclusivity and the entry of adalimumab biosimilars in Europe. Frontiers in Pharmacology. https://pmc.ncbi.nlm.nih.gov/articles/PMC7839249/
[20] Biosimilars Law Bulletin. (2021, April 15). 7th Circuit hears oral arguments in Humira ‘patent thicket’ antitrust case. https://www.biosimilarsip.com/2021/04/15/7th-circuit-hears-oral-arguments-in-humira-patent-thicket-antitrust-case/
[21] Pharmaceutical Technology. (2021, February 19). AbbVie’s successful hard-ball with Humira legal strategy unlikely to spawn imitators. https://www.pharmaceutical-technology.com/comment/abbvies-successful-hard-ball-with-humira/
[22] BioSpace. (2024, December 18). Lessons from Humira on how to tackle unjust extensions of drug monopolies with policy. https://www.biospace.com/policy/opinion-lessons-from-humira-on-how-to-tackle-unjust-extensions-of-drug-monopolies-with-policy
[23] DrugPatentWatch. (2025). REVLIMID patent and exclusivity profile. https://www.drugpatentwatch.com/p/tradename/REVLIMID
[24] BioSpace. (2024, December 18). Lessons from Humira. https://www.biospace.com/policy/opinion-lessons-from-humira-on-how-to-tackle-unjust-extensions-of-drug-monopolies-with-policy
[25] PM360. (2012). Pfizer’s 180-day war for Lipitor. https://pm360online.com/pfizers-180-day-war-for-lipitor/
[26] PharmaTimes. (2012). FDA opens gates for generic Plavix as patent expires. https://pharmatimes.com/news/fda_opens_gates_for_generic_plavix_as_patent_expires_977292/
[27] UpCounsel. (2025). Drug patents: Essential guide to pharmaceutical patent protection. https://www.upcounsel.com/how-long-does-a-drug-patent-last
[28] DrugPatentWatch. (2026). The patent cliff’s shadow. https://www.drugpatentwatch.com/blog/the-effect-of-patent-expiration-on-sales-of-branded-competitor-drugs-in-a-therapeutic-class/
[29] DrugPatentWatch. (2026). The patent cliff’s shadow. https://www.drugpatentwatch.com/blog/the-effect-of-patent-expiration-on-sales-of-branded-competitor-drugs-in-a-therapeutic-class/
[30] Finnegan. (2023, May 1). Potential implications of Inflation Reduction Act on pharmaceutical patent litigation. https://www.finnegan.com/en/insights/articles/potential-implications-of-inflation-reduction-act-on-pharmaceutical-patent-litigation.html
[31] Congressional Research Service. (2024). Medicare drug price negotiation under the Inflation Reduction Act: Industry responses and potential effects (Report R47872). https://www.congress.gov/crs-product/R47872
[32] Zheng, H., Patterson, J. A., & Campbell, J. D. (2025). Early impact of the Inflation Reduction Act on small molecule vs biologic post-approval oncology trials. Health Affairs Scholar. https://doi.org/10.1093/haschl/qxaf152
[33] Brownstein Hyatt Farber Schreck. (2025, June 24). Reflections on the Inflation Reduction Act’s pill penalty. https://www.bhfs.com/insight/reflections-on-the-inflation-reduction-acts-pill-penalty/
[34] DrugPatentWatch. (2026). The Inflation Reduction Act pill penalty. https://www.drugpatentwatch.com/blog/the-inflation-reduction-act-pill-penalty-structural-distortions-in-pharmaceutical-rd-and-strategic-pricing-adaptations/
[35] New York v. Actavis, 787 F.3d 638 (2d Cir. 2015).
[36] DrugPatentWatch. (2025). REVLIMID patent and exclusivity profile. https://www.drugpatentwatch.com/p/tradename/REVLIMID










(2) Pathway: Unlocking a Hybrid Strategy for Drug Innovation")






(2) Beats the Branded Generic Every Time — Unless It Doesn't")


(2) Approval Strategy: The Complete Step-by-Step Guide to a Successful NDA Submission")





