Biosimilars, as highly similar and clinically equivalent versions of originator biologics, are fundamentally reshaping the biopharmaceutical market. Their emergence is driven by the expiration of blockbuster biologic patents and the global imperative to reduce healthcare costs and expand patient access to life-saving therapies.1 This report provides a comprehensive analysis of the profound influence biosimilars exert across every stage of a biologic drug’s lifecycle, from initial research and development (R&D) investment decisions and complex manufacturing processes to intricate regulatory pathways, aggressive intellectual property (IP) strategies, dynamic market competition, and evolving post-market surveillance. The biosimilar era necessitates adaptive strategies for originator and biosimilar manufacturers, proactive policy interventions from governments and payers, and enhanced education for healthcare providers and patients to fully realize the cost-saving and access-enhancing potential of these critical medicines.
1. Introduction: The Evolving Biopharmaceutical Landscape
The pharmaceutical industry has undergone a significant transformation with the advent of biologic drugs, offering unprecedented therapeutic options for a myriad of complex diseases. However, the high costs associated with these innovative therapies have spurred the development of biosimilars, which are now fundamentally altering the landscape of drug lifecycle management.
1.1 Defining Biologics: Nature, Complexity, and Therapeutic Significance
Biologics represent a distinct class of medications derived from living organisms, such as animal cells, yeast, or bacteria.4 This biological origin sets them apart from conventional small-molecule drugs, which are typically synthesized from chemicals.4 The inherent complexity of biologics means that their structures are generally more intricate, and they cannot be exactly copied through chemical synthesis.4 Consequently, the manufacturing processes for biologics are significantly more intricate and sensitive to variations, with even minor changes in production potentially impacting their safety and efficacy.4
Biologics are indispensable for treating a wide array of serious and chronic conditions, including autoimmune diseases, various cancers, and diabetes.1 Their therapeutic significance is paramount, offering life-saving or life-altering treatments for millions of patients. However, the high cost of these therapies often presents a substantial barrier, limiting patient access and placing considerable strain on healthcare systems globally.1
A foundational concept in understanding biologics is that “the product is the process”.11 This principle arises directly from the living systems used in their production, where even minor batch-to-batch variations are normal and expected for both originator biologics and biosimilars.4 This means the specific manufacturing process, from cell line development and purification to formulation, intrinsically defines the final product’s characteristics, including its identity, purity, and potency.6 This fundamental characteristic of biologics is crucial because it dictates the entire regulatory approach for biosimilars, requiring them to be “highly similar” rather than “identical”.1 It also underpins the complexity and rigor of comparability studies 16 and explains why manufacturing patents are so prevalent in biologic litigation.19
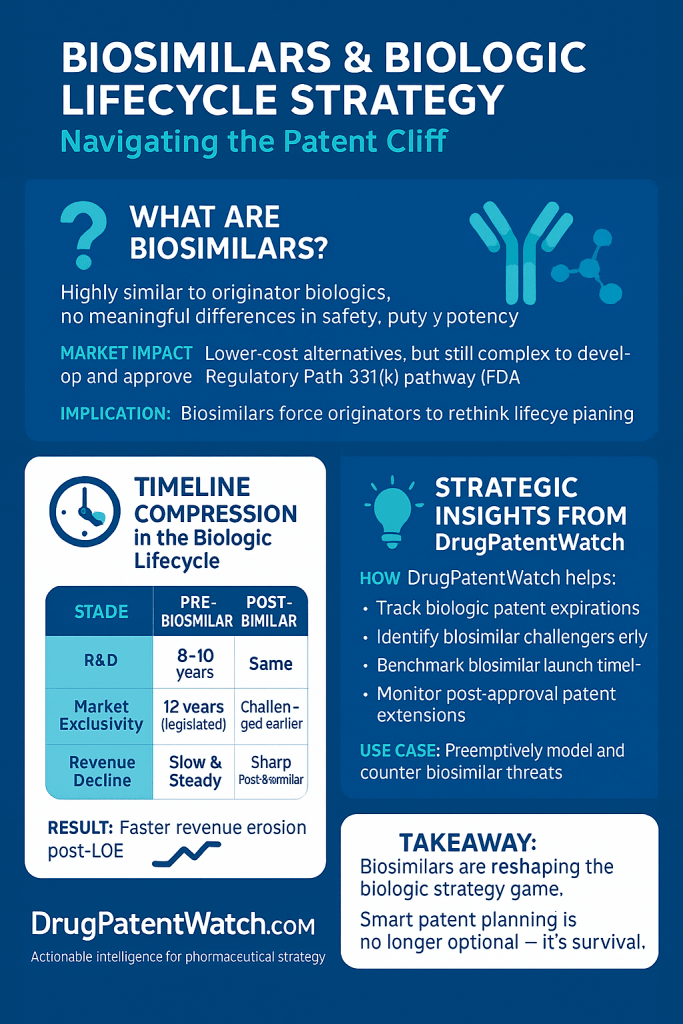
1.2 Defining Biosimilars: Scientific Basis of “High Similarity” and Interchangeability
A biosimilar is a biologic product that has been demonstrated to be “highly similar” to an already approved original biologic, referred to as the reference product.1 A critical requirement for biosimilar approval is the demonstration of “no clinically meaningful differences” from its reference product in terms of safety, purity, and potency.1 This ensures that biosimilars are administered in the same way, possess the same strength and dosage, and carry the same potential side effects and treatment benefits as the original biologic.4
The development process for biosimilars is exceptionally rigorous, involving extensive analytical characterization, nonclinical studies, and human clinical testing to establish this high similarity. This comprehensive approach is often termed the “Totality of Evidence”.16 The process is considerably more demanding and costly than developing small-molecule generics, often taking 5 to 9 years and costing over $100 million, excluding regulatory fees.17
A subset of biosimilars may achieve an “interchangeable biosimilar” designation. This is an additional legal classification that permits pharmacy-level substitution for the reference product without prior consultation with the prescriber, akin to how generic drugs are routinely substituted for brand-name drugs.4 To earn this designation, manufacturers must conduct specific “switching studies” to demonstrate that alternating between the biosimilar and the reference product does not impact safety or efficacy.22
It is important to recognize that the interchangeability designation functions primarily as a market access lever, rather than a quality indicator. Regulatory agencies, such as the FDA, explicitly state that both biosimilars and interchangeable biosimilars are equally safe and effective as their reference products.4 The “interchangeable” designation is an
additional legal requirement, not an indication of superior safety or efficacy.23 Despite this, the absence of an interchangeability designation can impede biosimilar uptake.22 This situation suggests that the interchangeability designation, while designed to facilitate market access through pharmacy-level substitution 4, has inadvertently become a source of confusion among healthcare professionals and patients.23 This confusion can create an unnecessary barrier, potentially slowing adoption despite scientific evidence supporting the safety of switching between products.25 This highlights a disconnect between the regulatory intent and the market’s perception and behavior.
1.3 The Strategic Imperative: Driving Competition, Affordability, and Patient Access
Biosimilars serve as a critical tool for fostering competition within the biologics market, which has historically been characterized by high-priced originator products and limited competitive alternatives.1 The overarching goal of introducing biosimilars is to reduce healthcare expenditures and broaden patient access to essential, often life-saving, biologic therapies.1
The economic impact of biosimilars is substantial. They typically cost between 10% and 50% less than their reference products 4, leading to billions of dollars in savings for patients, insurers, and healthcare systems.29
However, a notable paradox exists between the inherent cost-effectiveness of biosimilars and the significant barriers they face in achieving widespread market entry and adoption. While biosimilars are explicitly designed to be more affordable alternatives 1 and have already generated substantial savings 29, their market entry is frequently delayed. These delays stem from a confluence of complex regulatory hurdles, extensive intellectual property disputes, and market access issues, such as the incentives driving Pharmacy Benefit Managers (PBMs).1 The substantial upfront investment required for biosimilar development, ranging from $100 million to $300 million 17, becomes a disincentive when market entry is prolonged by litigation or when intense pricing pressures threaten long-term sustainability.10 This situation suggests that the current ecosystem, while theoretically supportive of biosimilars, contains systemic flaws that impede their optimal market function, thus limiting the full realization of their cost-saving and patient access potential.
2. Regulatory Pathways and Global Harmonization
The regulatory landscape for biologics and biosimilars is complex and varies significantly across major global markets, directly influencing their development, approval, and market entry.
2.1 The US Framework: Biologics Price Competition and Innovation Act (BPCIA) and FDA Approval (351(a) vs. 351(k))
In the United States, the Biologics Price Competition and Innovation Act (BPCIA), enacted on March 23, 2010, fundamentally transformed the regulatory environment by creating the first dedicated pathway for biosimilar approval.1 Prior to this legislation, no such mechanism existed for follow-on biologics.1 The BPCIA established a framework enabling the FDA to approve biosimilars that are “highly similar” to an already-approved reference biologic, with the critical stipulation that there are “no clinically meaningful differences” in terms of safety, purity, and potency.1
The approval processes for originator biologics and biosimilars differ significantly. Reference products undergo a comprehensive review via the traditional 351(a) pathway, which necessitates extensive clinical trials to demonstrate their efficacy, safety, and purity.24 In contrast, biosimilars are approved through the abbreviated 351(k) pathway. This pathway leverages a “totality of the evidence” approach, relying on the FDA’s prior determination that the original biologic is safe and effective.15 This abbreviated process is designed to reduce the need for unnecessary duplication of human testing, thereby accelerating development and reducing costs.1
A key provision of the BPCIA is the grant of a 12-year period of market exclusivity for reference products, commencing from the date of first licensure. During this timeframe, biosimilar applications cannot be approved by the FDA.1 Additionally, a 4-year data exclusivity period prohibits biosimilar manufacturers from even submitting an application that relies on the originator’s data.39 Since the first FDA approval of a biosimilar in 2015, the US market has seen significant growth, with nearly 70 FDA-approved biosimilars for 19 different reference products, including a record 18 approvals in 2024.1
The BPCIA was intended to strike a balance between encouraging innovation in the biologics industry through exclusivity and fostering competition through the timely entry of biosimilars.1 However, the implementation of the 12-year exclusivity, coupled with the potential for extensive and protracted patent disputes, often leads to significant delays in biosimilar market entry well beyond the statutory exclusivity period.1 A notable example is Humira, where biosimilar launch was delayed for years after its exclusivity expired due to an extensive “patent thicket”.36 This situation suggests that while the BPCIA successfully established a regulatory pathway, its practical application has allowed originator companies to leverage intellectual property strategies to prolong their market monopolies, thereby undermining the act’s secondary goal of promoting rapid competition and reducing healthcare costs for patients and the system.9
2.2 The European Model: European Medicines Agency (EMA) and Centralized Procedure
Europe has been a global leader in biosimilar regulation, with the European Medicines Agency (EMA) establishing the first comprehensive regulatory framework in the early 2000s and approving the first biosimilar as early as 2006.2 The EMA employs a centralized procedure for evaluating biosimilar applications, which, upon approval, grants a single marketing authorization valid across the entire European Union market.21
The European framework for market protection follows an “8+2+1” exclusivity model. This includes an 8-year period of data exclusivity, during which generic or biosimilar manufacturers cannot reference the original product’s data in their applications.39 This is followed by an additional 2-year period of market exclusivity, meaning that even if a biosimilar is approved, it cannot be marketed until this period concludes.39 Furthermore, this exclusivity can be extended by one additional year if the original product receives approval for new therapeutic indications that offer significant clinical benefit over existing therapies, potentially leading to a total of 11 years of protection.39 The EMA’s approach to biosimilarity is rooted in comprehensive comparability studies, rigorously designed to rule out any clinically meaningful differences between the biosimilar and its reference product.21
Regarding interchangeability, the EMA’s stance differs from that of the US. While interchangeability determinations are ultimately made at the Member-State level within the EU, and most EU countries have rules prohibiting automatic substitution without explicit physician consent 47, the EMA and Heads of Medicines Agencies generally consider all biosimilars approved in the EU to be interchangeable with their reference products and other biosimilars equivalent to the same reference product.24
Europe’s pragmatic approach to interchangeability appears to foster faster uptake of biosimilars. Unlike the US, the EU does not require a separate “interchangeable” designation that necessitates additional switching studies for biosimilars; rather, the scientific and regulatory stance is that all approved biosimilars are inherently interchangeable at the EU level.24 While national policies on automatic substitution may vary 47, the unified underlying scientific and regulatory position reduces market confusion and the development burden for manufacturers. This approach is linked to Europe’s demonstrably faster biosimilar uptake and greater cumulative cost savings compared to the US.2 This suggests that a streamlined and consistent regulatory perspective on interchangeability, even with variations in national dispensing laws, can significantly accelerate biosimilar adoption and the realization of associated cost savings. Conversely, the US’s more stringent and separate interchangeability pathway 22 appears to be an unnecessary barrier that hinders market penetration.22
2.3 Comparative Analysis of Global Regulatory Frameworks (US, EU, Canada, Japan)
Major global regulatory bodies, including the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), Health Canada, and Japan’s Pharmaceuticals and Medical Devices Agency (PMDA), have all established biosimilar pathways. These frameworks share the fundamental principle of requiring “high similarity” and “no clinically meaningful differences” between a biosimilar and its reference product.1 All these agencies rely on a “totality of evidence” approach, emphasizing comprehensive analytical and functional characterization as the foundational step in demonstrating biosimilarity.15
However, significant differences exist in their implementation, which can profoundly impact the global development and market entry of biosimilars.
Exclusivity Periods: The US framework provides a straightforward 12-year market exclusivity for reference biologics.39 In contrast, the EU employs an “8+2+1” model, offering 8 years of data exclusivity followed by 2 years of market exclusivity, with a potential 1-year extension for new indications.39 Exclusivity periods in Canada and Japan are not explicitly defined in the same segmented manner as the EU, often relying on a combination of patent protection and regulatory data protection.
Interchangeability Policy: The US has a unique, optional “interchangeable” designation that requires additional switching studies to permit pharmacy-level substitution.4 In the EU, all approved biosimilars are generally considered interchangeable, with specific substitution policies varying by individual member states.24 Canada and Japan do not have a formal federal interchangeability designation, with substitution decisions often left to provincial/prefectural regulations or physician discretion.47
Mandatory Clinical Efficacy Trials: Historically, both the US and EU often required comparative Phase III clinical efficacy studies as part of the biosimilar approval process.24 However, there is a clear trend towards streamlining these requirements. Recent guidelines from bodies like the UK, WHO, and a new Canadian draft guidance suggest that these extensive trials may not be routinely necessary if robust analytical and pharmacokinetic/pharmacodynamic (PK/PD) data adequately support similarity.27 Japan’s PMDA has also re-evaluated the necessity of clinical efficacy studies.55
Local Reference Product Requirement: While the US and EU generally do not require analytical or clinical comparability studies against locally sourced reference products if the originator is globally approved, some countries, particularly in Asia (e.g., South Korea, China, Japan), may still mandate such studies.56 This can necessitate redundant testing for biosimilar developers.
Animal Toxicology Studies: Although many health authorities have moved away from requiring comparative animal toxicology studies due to scientific advancements, a few countries still mandate them, adding an additional burden to biosimilar development.56
The persistence of regulatory divergence across these major markets significantly exacerbates development costs and delays global access to biosimilars. Requirements for local reference products, mandatory animal studies, and varied clinical study designs 56 compel biosimilar developers to conduct redundant or unnecessary studies. This fragmentation directly increases development costs 17 and prolongs market entry 38, effectively limiting the number and variety of biosimilars that can be brought to market.27 This undermines the global public health objective of expanding access to affordable biologics, as manufacturers may prioritize markets with less burdensome pathways. This situation highlights that regulatory policy is not merely about ensuring safety, but functions as a critical determinant of market dynamics and investment decisions.
Table 1: Key Regulatory Frameworks for Biosimilar Approval: A Comparative Analysis
Feature
United States (US)
European Union (EU)
Canada
Japan
Primary Regulatory Authority
FDA (Food and Drug Administration)
EMA (European Medicines Agency)
Health Canada
PMDA (Pharmaceuticals and Medical Devices Agency)
Enabling Legislation/Guidance
Biologics Price Competition and Innovation Act (BPCIA)
Regulation (EC) No 726/2004, EMA Guidelines
Food and Drugs Act, Health Canada Guidance Document
Japanese Guidelines for Biosimilars
Approval Pathway
Abbreviated (351(k))
Centralized Procedure
New Drug Submission
Case-by-case approach
Core Data Requirements
Totality of Evidence (Analytical, Non-clinical, Clinical)
High Degree of Similarity (Quality, Clinical, Non-clinical)
Equivalence (Quality, Safety, Efficacy)
Reference Product Exclusivity
12 years (market), 4 years (data)
8+2+1 years (data + market + extension)
Varies (combination of patent & data protection)
Varies (combination of patent & data protection)
Interchangeability Policy
Separate optional designation; state-level substitution laws
Integrated (all approved biosimilars are interchangeable); member-state substitution laws vary
No formal federal designation; provincial discretion
No formal designation
Mandatory Clinical Efficacy Trials
Historically often required; trend towards risk-based evaluation
Historically often required; trend towards risk-based evaluation
Historically often required; recent draft guidance suggests not always necessary
Re-evaluated; trend towards less emphasis if analytical data is strong
Local Reference Product Requirement
No (generally)
No (generally)
No (generally, but bridging data for non-Canadian source)
Yes (for analytical/clinical comparability)
2.4 Challenges and Opportunities in Global Regulatory Convergence
The path towards global regulatory harmonization for biosimilars is fraught with challenges. Inconsistent interchangeability policies across jurisdictions, coupled with fundamental variations in regulatory frameworks and disparities in resource allocation among nations, create significant hurdles.38 For instance, the requirement in some regions for analytical or clinical comparability studies against locally sourced reference products, even when a globally approved originator exists, forces biosimilar developers to conduct redundant and costly studies.56 Similarly, the persistence of mandatory animal toxicology studies in certain countries, despite global scientific consensus on their diminishing necessity, further complicates and delays development.56 The limited exchange of data and information between health authorities also impedes the convergence of regulatory standards.56
Despite these challenges, opportunities for global regulatory convergence are substantial. Harmonizing approval processes can streamline development, reduce redundant testing, and accelerate market access for biosimilars worldwide.38 Such convergence would foster market competition and stimulate innovation within the biopharmaceutical industry.38 Strategies to achieve this include promoting international collaboration, facilitating robust information sharing among regulatory bodies, and investing in comprehensive education for healthcare providers and regulators.38 Furthermore, streamlining development by eliminating routine comparative clinical efficacy studies and adopting a risk-based approach for immunogenicity testing can significantly reduce both development costs and timelines.27
The economic imperative for harmonization is clear. Biosimilar development is inherently costly, with investments ranging from $100 million to $300 million.17 Regulatory disparities add significantly to this financial burden, delaying the recouping of investments by manufacturers.38 Conversely, successful harmonization can substantially reduce these costs and expedite market entry.27 This indicates that the economic viability of biosimilar development is directly linked to the efficiency and consistency of the regulatory process. Unnecessary regulatory hurdles, stemming from a lack of global convergence, act as a significant disincentive for manufacturers to invest in biosimilars for certain markets or product types, thereby limiting the potential for global cost savings and expanded patient access.
3. Intellectual Property and Market Exclusivity Strategies
Intellectual property (IP) rights and market exclusivity provisions are critical components of the biologic drug lifecycle, profoundly influencing competition and the pace of biosimilar market entry.
3.1 Patent Protection vs. Data Exclusivity: Mechanisms and Interplay for Biologics (US 12-year, EU “8+2+1” model)
Patents are a form of intellectual property right granted by national patent offices, such as the United States Patent and Trademark Office (USPTO), protecting an invention for a specified period, typically 20 years from the patent application filing date.57 Patents can encompass a broad range of claims, including the core molecule itself, specific manufacturing processes, drug formulations, and methods of use.57 They can be issued or expire at any point during a drug’s development or market life, irrespective of its regulatory approval status.57
Exclusivity, distinct from patents, refers to statutory delays or prohibitions on the approval or marketing of competitor drugs. These are granted by regulatory bodies like the FDA or EMA upon the approval of a drug, provided specific statutory requirements are met.41 Unlike patents, exclusivity is not based on intellectual property rights but on regulatory approval.41
The interplay between patents and exclusivity is complex. Patent terms and exclusivity periods may or may not run concurrently, and they may not necessarily cover the same aspects of the drug product.57 Importantly, exclusivity is not added to the patent life; rather, they are distinct protections.41
In the US, the BPCIA grants original biologics a 12-year period of market exclusivity from the date of first licensure, during which no biosimilar application can be approved.1 Within this 12-year window, there is also a 4-year data exclusivity period that prohibits biosimilar manufacturers from even
submitting an application that relies on the originator’s data.39
In the EU, the EMA framework offers an “8+2+1” exclusivity model. This comprises an 8-year data exclusivity period, preventing generic or biosimilar manufacturers from referencing the original product’s data in their applications.39 This is followed by an additional 2 years of market exclusivity, during which a biosimilar cannot be sold even if approved.39 An optional 1-year extension can be granted if the original product receives approval for new therapeutic indications that offer significant clinical benefit.39
The strategic layering of IP and exclusivity creates a formidable barrier to biosimilar entry. Originator companies leverage both patent protection (granted by patent offices) and data/market exclusivity (granted by regulatory bodies) to extend their market monopoly.41 While regulatory exclusivity is a fixed statutory period, patents can be continuously filed and litigated throughout a product’s lifecycle.9 This continuous expansion of patent protection means that even after the initial regulatory exclusivity period expires, a dense web of patents can effectively prolong market protection, leading to protracted litigation and delayed patient access to more affordable options.1 This highlights that IP strategy for biologics is a dynamic, ongoing process throughout the product’s lifecycle, rather than a one-time event at launch.
3.2 Originator Strategies: Patent Thickets and Evergreening Tactics (New Formulations, Indications, Combination Therapies, Device Patents)
Evergreening refers to a range of legal, business, and technological strategies employed by originator pharmaceutical companies to extend the commercial lifetime of their patents that are nearing expiration, thereby retaining significant revenues.58 This practice often involves securing new patents on minor variations or associated aspects of the original product.62
A prominent evergreening tactic is the creation of patent thickets. This involves a pharmaceutical company acquiring a “dense web” or “fortress” of numerous patents and other intellectual property rights surrounding a single product.9 This intricate network of overlapping IP rights makes it exceptionally challenging for biosimilar competitors to navigate the legal landscape and enter the market.9
Common evergreening tactics include:
New Formulations and Dosage Forms: Patenting minor modifications to a drug’s dosage form, such as extended-release versions, different crystalline forms (polymorphs), or novel delivery methods.58
New Indications and Methods of Use: Obtaining patents for newly discovered therapeutic uses for existing drugs.58 For biologics, this is often part of a “platform empire” strategy, where the indications for a molecule are continuously expanded over time.64
Combination Therapies: Patenting the use of the drug in combination with other therapies.60
Process Patents: Securing patents on slight modifications or improvements to manufacturing processes.19 For biologics, given the “product is the process” paradigm, process patents are particularly significant in litigation.11
Device Patents: Obtaining patents on associated drug delivery mechanisms, such as auto-injectors or specialized inhalers.65
AbbVie’s Humira stands as a prominent example of an evergreening strategy, with over 100 patents filed in the US. This extensive patent portfolio significantly delayed biosimilar entry for years after its primary exclusivity expired.36 Similarly, Roche’s oncology blockbuster Herceptin (trastuzumab) boasts an extensive patent portfolio of over 1,500 patents, many of which cover new indications and combination therapies.64
The strategic shift from product to portfolio protection is a defining characteristic of originator companies’ responses to impending biosimilar competition. These companies are moving beyond merely protecting the core molecule to building vast “patent portfolios” or “platform empires” that encompass every conceivable aspect of the drug’s use, manufacturing, and delivery.9 This is a deliberate and aggressive strategy designed to create “impenetrable barriers” 64 and delay biosimilar entry, even when the individual patents may cover only minor improvements.62 This proactive and aggressive IP strategy directly undermines the policy intent of biosimilar pathways to foster competition post-exclusivity. It effectively shifts the battleground from scientific comparability to protracted legal disputes, increasing costs for biosimilar developers and delaying patient access to more affordable treatments, thereby impacting the overall sustainability of healthcare systems.9
3.3 Biosimilar Patent Litigation: The “Patent Dance,” Litigation Outcomes, and Market Entry Delays
The Biologics Price Competition and Innovation Act (BPCIA) introduced a unique framework for resolving patent disputes between biosimilar applicants and reference product sponsors, colloquially known as the “patent dance”.19 This framework provides a structured process for the exchange of confidential information and the identification of patents that could potentially be litigated. The entire “patent dance” process, if both parties fully engage and utilize maximum allotted time, can extend up to 250 days, or approximately eight months.35
The “patent dance” is not mandatory; a biosimilar applicant can choose to opt out. However, opting out has strategic implications, as it shifts control over when and which patents to litigate to the originator company and can affect the biosimilar applicant’s ability to file declaratory judgment actions.35 A key difference from small-molecule generic litigation under the Hatch-Waxman Act is the absence of an “Orange Book” for biologics, no automatic 30-month stay of FDA approval, and a significant focus on process patents in litigation.19 Biosimilar applicants are also required to provide a 180-day notice of commercial marketing to the reference product sponsor before launching their product, a notice that can be given either before or after FDA approval.35
Patent disputes represent a significant challenge that frequently delays biosimilar market entry, even after the statutory exclusivity periods for the reference product have expired.1 A study analyzing 32 biosimilars that entered the market between 2010 and 2022 revealed distinct outcomes and their impact on launch timelines:
Wins for Biosimilar Firms: 16% of biosimilars won their litigation against the brand biologic, with an average duration of 2.3 years from patent expiry to biosimilar launch.66
Losses for Biosimilar Firms: 9% of biosimilars lost their litigation, resulting in a significantly longer average duration of 16.5 years from patent expiry to biosimilar launch.66
Settlements: The majority of biosimilar patent disputes, specifically 75%, resulted in settlements. For these cases, the average duration from patent expiry to launch was 2.5 years.66 These settlements often involve agreements that delay the actual launch date of the biosimilar.36
While manufacturing patents constitute approximately half of all patents asserted in biosimilar litigation, research suggests they have had less impact on market launch delays compared to composition, active pharmaceutical ingredient, and treatment patents.20
The current patent litigation framework, particularly the “patent dance” and the prevalence of patent thickets, transforms legal challenges into a powerful tool for originator companies to maintain market dominance. This effectively extends the originator’s monopoly beyond statutory exclusivity periods, as evidenced by the extreme market entry delays associated with losing a patent dispute (16.5 years).66 Even the most common outcome, settlement, frequently involves delayed launches.36 This means that the realization of cost savings and patient access benefits from biosimilars is often postponed, making the legal landscape a critical, and often unpredictable, determinant of a biologic’s effective lifecycle. Reforms that would allow earlier patent litigation, such as at the start of Phase 3 clinical trials 66, could potentially expedite biosimilar market availability.
Table 2: Anticipated Major Biologic Patent Expirations (2025-2029) and Potential Market Impact
Year of Patent Expiry
Drug Name (Reference Product)
Originator Company
Key Indication(s)
Approx. 2024 Global Sales (USD Billions)
Potential Market Impact
2025
Perjeta (pertuzumab)
Genentech (Roche)
HER2-positive breast cancer
$4.3
High potential for biosimilar entry, significant price competition expected.
2025
Benlysta (belimumab)
GlaxoSmithKline
Systemic lupus erythematosus
$2.156
Potential for biosimilar entry, focus on next-gen T-cell engagers.
2025
Blincyto (blinatumomab)
Amgen
Acute lymphoblastic leukemia
$1
Shift in BiTE lifecycle strategy, room for novel formats or next-gen T-cell engagers.
2026
Kadcyla (ado-trastuzumab emtansine)
Genentech (Roche)
HER2-positive breast cancer
$2.31
Potential for biosimilar entry, competitive reshuffling.
2026
Taltz (ixekizumab)
Eli Lilly
Plaque psoriasis, psoriatic arthritis
$3.2
Biosimilar entry expected, differentiation will hinge on formulation tech, delivery device IP, and pricing.
2027
Myalept (metreleptin)
Amryt Pharma
Generalized lipodystrophy
Biosimilar entry expected, especially from India- and China-based players.
2027
Sylvant (siltuximab)
EUSA Pharma
Castleman disease (Multicentric)
$2.34
Biosimilar entry expected, especially from India- and China-based players.
2027
Trulicity (dulaglutide)
Eli Lilly
Type 2 Diabetes
$7.13
Intense biosimilar activity, differentiation on formulation, delivery device IP, and pricing.
High potential for biosimilar entry, significant price competition expected.
Note: Sales figures are approximate 2024 global sales as per source.68
4. Manufacturing and Supply Chain Dynamics
The manufacturing and supply chain aspects of biologics are inherently complex, posing significant challenges for both originator products and biosimilars, while simultaneously shaping competitive strategies.
4.1 Complexities of Originator Biologic Manufacturing (Cell Line Development, Fermentation, Purification, Quality Control)
Biologic manufacturing is an exceptionally intricate process, encompassing multiple sophisticated steps from the initial genetic engineering of a cell to produce the desired biologic, through various purification stages, and culminating in extensive quality testing.12 Key stages typically include cell line development, fermentation (cell culture), purification (often involving chromatography), and final formulation.6 The profound complexity of these processes means that even minor alterations in manufacturing conditions can significantly impact the final product’s safety, purity, and potency. This underpins the fundamental principle that for biologics, “the product is the process”.4 Ensuring the safety and efficacy of these complex molecules necessitates the implementation of robust quality control measures, regular inspections, and strict adherence to Good Manufacturing Practices (GMPs) throughout every stage of production.4
The inherent complexity and sensitivity of biologic manufacturing serve as a substantial barrier to entry for new companies.28 The need for unique approaches in cell expression and protein purification 6, coupled with the rigorous control required to ensure product consistency, means that developing and operating a biologic manufacturing facility demands significant scientific expertise, specialized infrastructure, and substantial capital investment. This complexity also positions the manufacturing process itself as a significant source of intellectual property for originator companies. This leads to the filing of extensive process patents 19, which further complicates the landscape for biosimilar developers. These developers must independently devise their own manufacturing processes to achieve “high similarity” without infringing on the innovator’s proprietary methods, thereby increasing the complexity and cost of biosimilar development and potentially delaying market entry.
The manufacturing of biosimilars presents a unique set of challenges beyond merely achieving high similarity. The development process typically begins with an exhaustive characterization of the reference product to guide the creation of a cell line capable of producing a highly comparable biosimilar.16 The concept of comparability, initially a regulatory tool for managing manufacturing changes in existing biologics while ensuring consistent critical quality attributes (including immunogenicity), is fundamentally the same scientific principle applied to biosimilarity.18
Post-Translational Modifications (PTMs), such as glycosylation, are a critical challenge. These modifications profoundly influence a biologic’s stability, efficacy, and immunogenicity.70 Achieving consistent PTM profiles in biosimilars is exceptionally complex due to inherent differences in expression systems, cell culture conditions, and various bioprocessing variables.70
Innovative Formulation Strategies are also a focus for biosimilar manufacturers, particularly the development of high-concentration, buffer-free formulations aimed at improving patient adherence and ease of use, especially for subcutaneous administration.70 However, these innovations introduce their own challenges, including managing increased viscosity, preventing protein aggregation, and mitigating potential immunogenicity risks.70
Scale-Up Challenges are pivotal. The transition from small-scale development batches to commercial-scale production is a critical hurdle, requiring meticulous management to ensure consistent critical quality attributes regardless of batch size.69 This involves navigating complex equipment requirements, potential facility constraints, and the ongoing challenge of recruiting and retaining a specialized bioprocessing workforce.71
Achieving Cost-Efficiency in biosimilar manufacturing is paramount for market competitiveness. This necessitates overcoming the inherent manufacturing complexity and scaling challenges.69 Strategies to enhance efficiency and reduce costs include the adoption of single-use technologies, continuous processing, and automated systems.14
The manufacturing process for biosimilars presents a significant “catch-22.” While biosimilars are intended to be cost-effective alternatives, their development and manufacturing are inherently complex and expensive, demanding substantial investment in specialized infrastructure and expertise.17 The regulatory requirement to demonstrate “high similarity” 1 without direct access to the originator’s proprietary manufacturing process 71 forces biosimilar developers to undertake independent, high-cost R&D in process development. This situation creates a dynamic where the very nature of biosimilars (complex biologics) drives high manufacturing costs, which then necessitates aggressive cost-efficiency strategies and large-scale production to achieve the desired price reduction and market viability. This challenge ultimately limits the number of companies capable of entering the biosimilar market, potentially hindering overall competition and patient access in the long run.
4.3 Critical Supply Chain Vulnerabilities: Cold Chain Requirements, Raw Material Sourcing, and Geopolitical Risks
The supply chain for biologic drugs is characterized by extreme complexity and inherent vulnerabilities, largely due to their delicate nature and stringent handling requirements.
Cold Chain Requirements are paramount for biologics. These medications are exceptionally sensitive to temperature fluctuations and demand rigorous cold chain management throughout their entire lifecycle, from the point of manufacturing to final patient delivery.72 Even minor deviations from the specified temperature range can compromise a biologic’s efficacy and safety, potentially leading to costly recalls, significant product waste, and serious risks to patient well-being.73
Global Distribution Challenges further compound these complexities. The worldwide distribution of biologics involves numerous in-transit transfers and hand-offs across diverse geographies and varying temperature zones.72 This exposes products to a range of environmental variations, including extreme ambient temperatures and humidity. Challenges include limitations in transport vehicles, exposure to freezing cargo hold temperatures during air transport, unprotected tarmac exposure during offloading, and delivery delays caused by weather, traffic, or mechanical issues.72
Raw Material Sourcing introduces another layer of vulnerability. The biotech supply chain is intricate, relying on a consistent flow of specialized raw materials and consumables.74 The presence of unstructured data within these supply chains can lead to unreliable market forecasting, fragmented communication among stakeholders, and poor traceability, all of which magnify existing vulnerabilities.75
Geopolitical Risks have emerged as a significant threat to supply chain security. Global events, such as the COVID-19 pandemic, and ongoing geopolitical uncertainties, including tariffs and evolving trade policies, can severely disrupt supply chains, leading to critical availability issues and prolonged delivery delays.74
The supply chain for biologics functions not merely as a logistical necessity but as a strategic differentiator and a risk multiplier. The extreme cold chain requirements 72 and the complexities of global distribution 72 elevate the supply chain to a critical, high-cost component of biologic drug lifecycle management. Geopolitical risks 75 introduce an additional layer of unpredictability, impacting raw material sourcing and overall supply chain resilience. Failures in maintaining supply chain integrity can result in substantial product loss, critical shortages, and direct patient safety risks, ultimately undermining market trust and financial viability. This elevates supply chain management from a purely operational function to a core strategic imperative in the highly competitive biologic landscape, influencing investment decisions in areas such as domestic manufacturing capabilities and diversified sourcing strategies.76
4.4 Enhancing Supply Chain Security and Resilience: The Role of Data Management and Diversification
To mitigate the inherent vulnerabilities within the biologic supply chain, biopharmaceutical companies are actively implementing a range of strategies focused on enhancing security and resilience.
Diversifying the Supplier Base is a key approach, involving the expansion of qualified vendors across multiple geographies. This is particularly crucial for critical raw materials, specialized excipients, and components sourced from geopolitically sensitive regions, ensuring business continuity and regulatory compliance.76 Concurrently,
investing in domestic manufacturing capabilities aims to reduce reliance on overseas supply chains, thereby mitigating geopolitical risks and ensuring uninterrupted service to clients.76 Companies are also adopting a proactive stance by
sourcing materials earlier in the development phase, extending through to commercial manufacturing, to ensure a robust and continuous supply and de-risk their supply chains.76 Furthermore, continuous monitoring of tariff developments and assessing their impact on suppliers and products based on country of origin is becoming standard practice, leveraging supply chain engineering to navigate international trade laws and alleviate risk.76
The role of data management platforms has become indispensable in this endeavor. These platforms are transforming fragmented and unstructured data into a powerful resource that provides personalized insights, enables real-time data collection for enhanced traceability, simplifies communication across stakeholders, and offers advanced search capabilities.75 By providing comprehensive supply chain analysis, these platforms help companies identify weaknesses, find alternatives for at-risk raw materials or suppliers, and proactively optimize inventory based on reliable market forecasts.75 They also streamline regulatory submissions and ensure compliance.75
The reliance on data as the backbone of modern biologic supply chains is a significant development. The inherent vulnerabilities of biotech supply chains, stemming from temperature sensitivity, global distribution complexities, and geopolitical risks, are significantly magnified by the presence of unstructured data.75 The implementation of robust data management platforms is presented as a critical solution to these vulnerabilities.75 This signifies a fundamental shift towards data-driven supply chain management as a non-negotiable component for ensuring product integrity, operational efficiency, and regulatory compliance in the biologic and biosimilar space. It implies that strategic investment in digital infrastructure and advanced data analytics is as crucial as investment in physical manufacturing facilities for maintaining a competitive edge and safeguarding patient safety.
5. Market Access, Pricing, and Competition
The market dynamics surrounding biologics have been dramatically altered by the introduction of biosimilars, leading to significant shifts in pricing, market share, and access strategies.
5.1 Global Biosimilar Market Growth Projections and Cost Savings (Value, CAGR, US Savings Statistics)
The global biosimilars market is projected for robust and sustained growth in the coming decade. Forecasts indicate an expansion from approximately USD 35.04 billion in 2025 to USD 72.29 billion by 2035, representing a Compound Annual Growth Rate (CAGR) of 7.5%.3 Some analyses project even higher growth, estimating the market to reach USD 175.79 billion by 2034 with a CAGR of 17.6%.8 This growth is primarily fueled by the impending patent expirations of numerous blockbuster biologics, the escalating global demand for more affordable therapeutic options, and a supportive environment of increasing regulatory approvals.3
In the US, biosimilars have already generated substantial cost savings for the healthcare system. In 2022, biosimilars accounted for $9.4 billion in savings, contributing to a cumulative total of $23.6 billion since the first biosimilar entry in 2015.30 Other reports indicate even higher savings, with $12.4 billion in 2023 alone, bringing cumulative savings to $36 billion since 2015.29 These savings are distributed across various payer types, including $130 billion for Medicare and $194 billion for commercial plans in 2022.30
Despite the significant savings already generated and the robust market growth projections, there remains a notable disparity between the full savings potential of biosimilars and their currently realized impact. While biosimilars have demonstrably delivered billions in savings 29, their overall market penetration, as exemplified by the 23% market share for adalimumab biosimilars after nearly two years 32, and the general lag in US uptake compared to Europe 2, suggests that the full economic benefits are not yet being achieved. This indicates that despite the clear financial advantages, systemic barriers—such as PBM incentives, persistent knowledge gaps among stakeholders, and ongoing litigation—are preventing faster and broader biosimilar adoption. The projected market growth 3 is contingent upon effectively addressing these underlying issues to unlock the full potential for cost containment and expanded patient access.
Upon market entry, biosimilars typically introduce lower list prices (Wholesale Acquisition Cost, or WAC) compared to their reference products.32 Initial launch discounts can be modest, often less than 10% of the Average Sales Price (ASP), followed by average annual price drops ranging from 10% to 15%.8 The introduction of biosimilar competition fundamentally alters market dynamics, compelling originator companies to reduce their prices or offer competitive discounts and rebates to retain market share.9
This competition leads to significant reductions in Average Sales Prices (ASPs) for both brand biologics and biosimilars. Many brand biologic ASPs have decreased by over 45% following biosimilar launch.32 For instance, trastuzumab biosimilars have averaged a 52% discount, rituximab 66%, and bevacizumab 49%.32 Some biosimilar manufacturers, particularly for adalimumab, have adopted dual or multiple pricing models, offering both high and low WAC options to cater to different market segments.32 Private label strategies also play a role in shaping these pricing dynamics.32
The price erosion in the biologics market, driven by biosimilar entry, is dynamic and often unpredictable. While biosimilars consistently lead to price reductions 32, the
rate and magnitude of this erosion are highly variable. Originator responses differ significantly, ranging from steep ASP cuts to minimal adjustments.33 This volatility, combined with the rising costs of biosimilar production, can exert immense pressure on biosimilar manufacturers, potentially forcing them to exit markets if profitability becomes unsustainable.10 This unpredictability in pricing dynamics creates significant uncertainty for all stakeholders, from manufacturers needing a predictable return on investment to healthcare providers operating under “buy and bill” models who require assurance of cost recovery.10 This suggests that while competition is fierce, the current market mechanisms may not always foster sustainable conditions for biosimilar developers, potentially limiting future investment and product diversity.
Biosimilar launches have demonstrated a significant impact on market share, achieving an average of 53% market share and a 53% reduction in average drug costs after five years of competition across various therapeutic areas.32
Oncology has emerged as the therapeutic area with the fastest growing biosimilar uptake, averaging an impressive 81% market share after five years.32 Specific examples include trastuzumab (86% market share), bevacizumab (90%), rituximab (76%), and pegfilgrastim (85%).32 Oncologists generally express willingness to switch to biosimilars, especially in combination therapies, provided there is FDA approval, comparable toxicity profiles, and favorable payer coverage.80
In contrast, Immunology has shown slower biosimilar market growth, averaging only 26% market share after five years.32 For adalimumab, a prominent immunology biologic, the market share for its ten biosimilars reached 23% after nearly two years, primarily split between Hyrimoz and Hadlima.32 Uptake in immunology is heavily influenced by payer coverage policies and the emergence of newer branded therapies that offer improved efficacy profiles, such as Skyrizi competing with Stelara biosimilars.80
Originator manufacturers employ diverse strategies in response to biosimilar competition:
Sole Preferred Coverage Strategy: Some originators, like those for infliximab (Remicade) and pegfilgrastim (Neulasta), implement steep ASP reductions (50% or more) to maintain preferred status and market share. Despite some market share decline, these products often remain dominant within their respective drug families.81
Non-Sole Preferred Coverage Strategy: Other originators, such as those for trastuzumab (Herceptin) and filgrastim (Neupogen), pursue moderate ASP reductions. This leads to steeper declines in payer preference and market share, with biosimilars rapidly gaining dominance. For example, Kanjinti (trastuzumab biosimilar) and Zarxio (filgrastim biosimilar) quickly overtook their originator counterparts.81
The dynamics of market share and competitive responses highlight that the biosimilar market is not a simple “race to the bottom” on price alone. While biosimilars consistently drive significant price reductions 32, the ultimate market share outcomes are heavily dependent on the originator’s pricing strategy 33 and the formulary decisions of payers.37 Some originators aggressively cut prices to defend market share, while others may concede market share, particularly if they have clinically differentiated next-generation therapies in their pipeline.80 This complex interplay means that the long-term sustainability of the biosimilar market requires manufacturers to consider not only competitive pricing but also patient support programs and supply chain reliability as crucial differentiators.80 This nuanced competitive environment impacts R&D investment for both originator and biosimilar products.
Table 3: Impact of Biosimilar Entry on US Market Share and Average Sales Price (ASP) for Selected Biologics (through 2022 Q3)
Reference Product (Example)
First Biosimilar Entry (Year/Quarter)
Brand Biologic ASP Change (%)
Biosimilar ASP Change (%)
Biosimilar Market Share (%) (as of 2022 Q2)
Therapeutic Area
Product A (e.g., Infliximab)
2015 Q3
-1%
-60% to -73%
82%
Immunology
Product B (e.g., Adalimumab)
2017 Q1
-57%
-41% to -62%
42%
Immunology
Product C
2018 Q3
-36%
-38%
32%
Various
Product D
2018 Q4
-66%
-56% to -65%
42%
Various
Product E
2019 Q4
-14%
-52% to -63%
82%
Various
Product F
2019 Q4
-21%
-41% to -69%
80%
Various
Product G
2020 Q2
-13%
-44% to -58%
64%
Various
Note: Data from.33 Therapeutic areas for Products C-G are not specified in the source, but generally include oncology and immunological conditions.
5.4 Formulary Decisions: The Influence of Pharmacy Benefit Managers (PBMs) and Payers
Pharmacy Benefit Managers (PBMs) and other payers wield significant influence over biosimilar uptake through their formulary decisions.37 PBMs have increasingly shown a willingness to place biosimilar medications on preferred formulary tiers shortly after their market launch, a shift from their earlier hesitancy to cover these products.37
However, a key challenge to widespread biosimilar adoption arises when PBMs maintain the reference biologic product on the same preferred tier as its biosimilar counterparts. This practice can significantly slow biosimilar uptake.37 PBMs approach formulary decisions strategically, meticulously analyzing factors such as the wholesale acquisition costs (WACs) and concentrations of biosimilar products, and tailoring their choices to align with the specific lines of business they serve.37 For example, major PBMs like CVS Health Corp.’s Caremark, The Cigna Group’s Express Scripts, and UnitedHealth Group’s Optum Rx all added Humira (adalimumab) biosimilars to their national preferred formularies upon launch, but simultaneously kept the original Humira product on the same tier.37
Concerns have been raised that PBMs may have inherent incentives to favor medicines with high list prices that offer substantial rebates, potentially hindering the uptake of biosimilars despite their lower net costs.10 This dynamic positions PBMs as de facto gatekeepers of biosimilar access. While biosimilars offer significant cost savings 29, their actual market penetration is heavily influenced by these PBM formulary decisions.37 The practice of placing both biosimilars and originators on the same preferred tier, or prioritizing high-rebate products 10, can undermine the very cost-saving potential that biosimilars promise. This suggests that PBM practices can create “market distortions” 33 that impede expedient biosimilar uptake, limiting patient access to lower-cost options and potentially discouraging future biosimilar development.10 This highlights the critical need for policy interventions to align PBM incentives with broader healthcare cost-saving and patient access goals.
5.5 Barriers and Facilitators to Biosimilar Uptake (Lack of Knowledge, Interchangeability Concerns, PBM Incentives)
The uptake of biosimilars in various markets, particularly in the US, has been slower than anticipated despite their proven safety and efficacy. This is attributable to a complex interplay of barriers and facilitators.
Key Barriers:
Lack of Knowledge and Comfortability: A significant impediment is the widespread lack of knowledge and comfort among both healthcare providers (HCPs) and patients regarding biosimilar structural differences, their safety, and efficacy.7 This knowledge gap can lead to a “nocebo response,” where patients perceive biosimilars as less effective due to their lower cost.10
Interchangeability Concerns: Confusion surrounding the interchangeability designation and perceived safety of switching between a reference biologic and a biosimilar persists, despite scientific evidence supporting the safety of such switches.22
PBM Incentives: Limited market access often results from PBM practices that may prioritize high-rebate products or maintain originators on preferred tiers, thereby hindering cost-saving competition.10
Perverse Economic Incentives for Providers: In “buy and bill” models, particularly within Medicare Part B, providers may face a precarious economic situation. Continuous declines in biosimilar Average Sales Prices (ASPs) can lead to insufficient reimbursement to cover acquisition costs, creating an incentive for providers to choose more expensive reference products.10
Legal and Regulatory Barriers: Ongoing patent disputes and certain state laws that restrict automatic substitution can further delay biosimilar market entry and uptake.1
Key Facilitators:
Awareness and Education: Comprehensive and unbiased educational initiatives are crucial for healthcare professionals and patients to build trust and understanding regarding biosimilar safety, efficacy, and interchangeability.1 Professional societies play a vital role in disseminating this information.10
Cost-Effectiveness and Improved Access: The inherent cost savings offered by biosimilars and their potential to expand patient access to essential treatments are strong drivers for adoption.3
Regulatory Support: Clear regulatory frameworks and supportive government policies are essential facilitators.3 Recent policy changes, such as increased Medicare Part B reimbursement for biosimilars under the Inflation Reduction Act (IRA) of 2022, are positive steps towards incentivizing their use.10
Professional Influence of Pharmacists: Pharmacists are key educators and often advocate for and support biosimilar substitution practices.7
Real-World Evidence: The growing body of real-world evidence supporting the safety and efficacy of biosimilars helps build confidence among prescribers and patients.80
The adoption of biosimilars is not determined by a single factor but by a complex interplay of scientific understanding, regulatory clarity, economic incentives, and stakeholder perceptions.7 A lack of alignment or the presence of perverse incentives in one area, such as PBM practices, can negate progress achieved in others, like FDA approvals. Therefore, achieving widespread biosimilar adoption requires a holistic, coordinated effort across the entire healthcare ecosystem. Policy interventions must address not only regulatory pathways but also economic incentives for providers and payers, alongside comprehensive education campaigns for all stakeholders. Failure to address these interconnected barriers will continue to limit the full benefits of biosimilars, despite their proven safety and cost-effectiveness.
6. Impact on Research & Development Investment
The emergence of biosimilars has significantly influenced R&D investment decisions across the biopharmaceutical industry, impacting both novel biologic development and the pipeline for next-generation therapies.
6.1 Influence on R&D Investment for Novel Biologics
The impending prospect of biosimilar competition, once a reference product’s exclusivity period expires, directly influences the R&D strategies of originator companies.1 This competitive pressure acts as a powerful incentive for these companies to intensify their R&D efforts. Instead of merely defending existing products, originators are driven to innovate by developing next-generation therapies or exploring new indications for their existing biologics. This strategic pivot aims to maintain market differentiation and extend product lifecycles beyond the traditional “patent cliff”.60
The development of novel biologics is a protracted and capital-intensive endeavor, typically spanning 10 to 15 years and costing billions of dollars.60 To recoup these substantial investments, robust patent protection and extensive exclusivity periods are essential.9 The presence of biosimilars, therefore, acts as a catalyst for “next-gen” innovation. By challenging the market share of established biologics, biosimilars compel originator companies to invest in R&D for therapies that offer enhanced efficacy, novel mechanisms of action, or more convenient administration methods.60 This dynamic ultimately benefits patients through continuous therapeutic advancements and the availability of improved treatment options.
6.2 The “Development Gap”: Unaddressed Opportunities in the Biosimilar Pipeline
Despite the substantial number of biologics slated to lose patent protection in the coming years—estimated at 69 in Europe and 118 in the US by 2030 2—a significant “development gap” persists in the biosimilar pipeline. Alarmingly, only 29% of molecules nearing expiry in Europe currently have a biosimilar in development.50
Several factors contribute to this gap:
Commercial Viability: Many of the upcoming off-patent biologics are categorized as “low-revenue drugs,” generating less than €500 million annually. This makes them less attractive for biosimilar developers, given the substantial investment required for biosimilar development (ranging from $100 million to $300 million per product).17 Unlike small-molecule generics, biosimilars are less likely to foster a highly competitive market for these lower-revenue products.
Pipeline Concentration: The vast majority (92%) of biosimilar R&D efforts are concentrated in just two therapeutic areas: oncology and immunology. This leaves many other therapeutic areas largely underserved, meaning that even if a biologic in a less-focused area loses patent protection, it is unlikely to have a biosimilar alternative in the pipeline.50
Scientific and Regulatory Hurdles: The inherent complexities associated with developing biosimilars for orphan indications and highly complex biologics, such as antibody-drug conjugates (ADCs) and cell and gene therapies, present unique scientific and regulatory hurdles that deter investment.50
The existence of this “development gap” represents a market failure in capturing the full potential of biosimilars. Despite the clear public health benefits and significant cost-saving potential of biosimilars 29, the high cost of biosimilar development, coupled with the unpredictable market entry due to litigation and the relatively smaller market size of some biologics, creates a disincentive for investment. This means that numerous opportunities for cost reduction and expanded patient access are being missed. To address this, policy interventions may be necessary to de-risk biosimilar development for “low-revenue” biologics or those in underserved therapeutic areas, perhaps through targeted incentives or further streamlined regulatory pathways, to ensure that the full societal benefits of biosimilars are realized.
6.3 R&D Investment in Next-Generation Therapies (Gene and Cell Therapies) and Biosimilar Potential
Pharmaceutical innovation is increasingly shifting towards advanced biotechnology-derived medicines, with a growing focus on gene and cell therapies.49 By the end of the current decade, some of these highly complex cell and gene therapies are anticipated to face follow-on competition for the first time, marking a new chapter in the history of biosimilars.49
However, the development of biosimilars for these cutting-edge Cell and Gene Therapies (CGTs) faces significant and unique challenges across regulatory, manufacturing, intellectual property, and market size dimensions 91:
Regulatory Hurdles: The FDA needs to provide greater clarity in its guidance for CGT biosimilar development, as manufacturing, testing, and demonstrating safety and efficacy for these therapies are exceptionally complex.91 The applicability of the current biosimilar paradigm to CGTs remains uncertain, posing significant hurdles for standardization and evidence collection.
Manufacturing Complexities: Manufacturing CGT biosimilars involves exceptionally high costs and intricate processes, with limited standardization impeding development. Substantial investment in CGT infrastructure is crucial to foster competition and innovation in this nascent biosimilar space.91
Intellectual Property (IP) Landscape: The IP landscape for CGTs is even more intricate than for traditional therapeutic protein biologics. Beyond patents, manufacturers heavily rely on trade secrets to protect their proprietary processes, offering indefinite protection as long as confidentiality is maintained. This is particularly common in CGTs due to the extreme complexity of their production.91
Market Size and Pricing/Payment Dynamics: Markets for CGT biosimilars targeting rare diseases may be limited, which inherently reduces incentives for competition. Existing programs like Medicare and Medicaid face challenges in managing the high costs of CGTs, and the Inflation Reduction Act could indirectly affect CGT biosimilars through future price negotiations.91
These significant hurdles in regulation, manufacturing, intellectual property, and market size directly influence R&D investment decisions for biosimilar gene therapies. The high costs and complexities associated with these challenges can deter companies from investing in the development of CGT biosimilars.91 Without strategic approaches to overcome these barriers, R&D investment may remain limited, hindering the potential for biosimilar competition to lower prices and improve patient access to these innovative therapies, especially as CGTs begin to target more common diseases with larger patient populations.91
The emergence of highly complex gene and cell therapies presents both a new frontier for biosimilar development and a significant investment dilemma. While the potential for cost reduction and expanded access is high, the unique scientific, manufacturing, and IP challenges 91 make biosimilar development for these modalities far more complex and risky than for traditional protein biologics. This means that a “biosimilar void” 91 could extend to next-generation therapies, limiting their affordability and access if the current biosimilar development paradigm isn’t adapted. R&D investment will likely flow primarily into novel CGTs, with biosimilar development lagging significantly unless specific policy incentives or technological breakthroughs drastically reduce the risk and cost of CGT biosimilarity.
6.4 Strategies for Streamlining Biosimilar Development to Accelerate Market Entry
Streamlining the biosimilar development process is crucial for accelerating market entry, reducing costs, and enabling the development of a wider variety of biological drugs.27 This involves a strategic re-evaluation of current regulatory requirements and leveraging accumulated scientific experience.
Key strategies include:
Eliminating Routine Comparative Clinical Efficacy Studies: Extensive clinical efficacy trials, often modeled on Phase 3 trials for new drugs, have consistently proven to be predictable and unnecessary when robust analytical and PK/PD data already demonstrate high similarity.27 Their elimination would significantly reduce development time and costs, allowing manufacturers to pursue biosimilars for a broader range of biologics, including those with smaller market sizes.27
Utilizing a Risk-Based Approach for Immunogenicity Testing: While immunogenicity is a critical concern for biologics, two decades of experience with approved biosimilars have shown that their immunogenicity profiles reliably match those of their reference products.27 Moving to a risk-based approach, focusing testing only when specific uncertainties cannot be resolved analytically, would streamline development without compromising patient safety.27
Eliminating the US “Interchangeable Biologic” Designation: The unique US “interchangeable” designation, which often requires additional switching studies, is not scientifically necessary and acts as an unnecessary barrier and source of confusion for healthcare professionals and patients.27 Its removal would simplify the regulatory landscape and accelerate market entry.
Eliminating Comparative PK Testing of US and EU Reference Products: Originator reference products are typically approved globally based on a single development program. Requiring additional comparative PK testing between US and EU sourced reference products for biosimilar development is scientifically redundant and significantly inflates costs and delays.27 Leveraging existing global approval data would streamline clinical study design.
Leveraging Real-World Data and Evidence: Increased utilization of real-world data and evidence can provide valuable post-approval confirmation of biosimilar safety and effectiveness. This can build trust among healthcare professionals and patients, potentially reducing the perceived need for extensive pre-approval clinical trials and thus accelerating acceptance and uptake.27
Implementing Quality Management Systems (QMS): A balanced QMS, incorporating robust Document Management, Learning Management, Equipment Asset Management, and Change Management Systems, can significantly improve time to market. These systems ensure traceable, auditable data, reduce development costs, and help navigate regulatory barriers more efficiently.93
The regulatory environment is undergoing a process of “catch-up,” recognizing that accumulated experience and scientific advancements now permit streamlining that was not possible when biosimilar pathways were initially established.27 The proposed strategies directly address the high cost and long timelines associated with biosimilar development.17 If these streamlining measures are widely adopted and harmonized globally, they could dramatically lower the cost and time required for biosimilar development, making a wider range of biologics commercially viable for biosimilarization.27 This would accelerate market entry, intensify competition, and significantly expand patient access to a broader array of affordable biologic therapies, fundamentally transforming the biopharmaceutical landscape.
7. Strategic Implications and Recommendations
The transformative impact of biosimilars on biologic drug life cycle management necessitates adaptive and forward-looking strategies for all stakeholders in the biopharmaceutical ecosystem.
7.1 For Originator Biologic Manufacturers: Adapting to the Biosimilar Era
Originator biologic manufacturers must evolve their strategies to thrive in an era of increasing biosimilar competition.
Shift R&D Focus: A primary recommendation is to pivot R&D investments towards novel biologics that offer truly differentiated clinical benefits or to next-generation therapies, such as gene and cell therapies, to create new, protected market segments.80
Proactive Lifecycle Management: Implement advanced lifecycle management strategies that genuinely enhance patient value. This includes exploring new indications, developing improved formulations (e.g., subcutaneous, high-concentration versions), and creating novel combination therapies. The emphasis must be on demonstrable patient benefits beyond mere patent extension.60
Strategic IP Defense: Develop robust, multi-layered patent portfolios to protect innovations, but simultaneously be prepared for and strategically manage complex patent litigation. This may involve exploring early settlement options to mitigate risks and maintain a strategic market presence.9
Competitive Pricing and Value Proposition: Be prepared to adjust pricing strategies, including significant Average Sales Price (ASP) reductions and competitive rebates, to compete effectively with biosimilars. Simultaneously, emphasize and invest in value-added services, such as comprehensive patient support programs, to differentiate the originator product beyond price.10
Supply Chain Resilience: Invest strategically in diversifying raw material sourcing and, where feasible, in domestic manufacturing capabilities. This is crucial for mitigating geopolitical and broader supply chain risks, ensuring uninterrupted supply and maintaining patient trust.75
7.2 For Biosimilar Manufacturers: Navigating Challenges and Maximizing Opportunities
Biosimilar manufacturers must adopt agile and strategic approaches to navigate the complex market and capitalize on opportunities.
Strategic Product Selection: Prioritize the development of biosimilars for biologics with large market sizes and clear unmet needs, or those with less complex manufacturing and intellectual property landscapes, to ensure commercial viability and a strong return on investment.50
Robust Development and Quality Systems: Invest in state-of-the-art analytical capabilities and implement comprehensive Quality Management Systems (QMS). This includes robust Document Management, Learning Management, Equipment Asset Management, and Change Management Systems to streamline development, ensure rigorous comparability, and accelerate time to market.16
Proactive IP Strategy: Develop a deep understanding of the originator’s patent landscape. Engage strategically and proactively in the “patent dance” process, and be prepared for both litigation and settlement discussions to manage legal risks and optimize market entry timelines.9
Differentiated Market Access: Collaborate closely with payers and Pharmacy Benefit Managers (PBMs) to secure favorable formulary placement and actively address perverse incentives that may hinder biosimilar uptake.10 Consider developing patient support programs as a key differentiator beyond price alone.80
Advocacy for Regulatory Streamlining: Actively engage with regulatory bodies and policymakers to advocate for the elimination of unnecessary and redundant studies (e.g., routine comparative clinical efficacy trials, US-specific interchangeability studies) and to promote greater global regulatory harmonization. Such efforts can significantly reduce development costs and accelerate market access.27
7.3 For Healthcare Systems and Policymakers: Fostering a Sustainable Biosimilar Market
Healthcare systems and policymakers play a pivotal role in shaping a sustainable biosimilar market that maximizes patient benefits and cost savings.
Policy Alignment: Implement consistent and harmonized policies across all levels of government (federal, state, regional) that actively encourage and facilitate biosimilar uptake.10
Reimbursement Reform: Modify reimbursement methodologies, particularly for programs like Medicare Part B, to ensure stable and predictable payments for biosimilars. This is crucial for incentivizing their use by providers and ensuring the long-term viability of biosimilar manufacturers.10 Consider mechanisms that allow providers to share in the cost savings generated by biosimilars.10
Address PBM Incentives: Investigate and implement reforms for PBM practices that may hinder biosimilar adoption. This includes ensuring that cost savings from biosimilars are genuinely passed on to patients and healthcare systems, rather than being absorbed by high rebates that favor originator products.10
Promote Education: Fund and support comprehensive, unbiased educational programs targeting healthcare professionals and patients. These programs are essential for building trust, increasing comfort with biosimilars, and dispelling misconceptions about their safety and efficacy.1
Streamline Regulatory Pathways: Actively advocate for and implement policies that reduce redundant testing requirements, such as eliminating routine comparative efficacy trials and US-specific interchangeability studies. Promote greater global regulatory convergence to reduce development costs and accelerate market entry.27
Address the “Development Gap”: Explore mechanisms, such as targeted incentives or revised exclusivity periods for niche biologics, to encourage biosimilar development for low-revenue or highly complex biologics that currently lack follow-on competition.50
7.4 For Patients and Healthcare Providers: Enhancing Trust and Access
Patients and healthcare providers are at the forefront of biosimilar adoption and play a crucial role in realizing their benefits.
Seek Education: Actively seek out and engage with reliable, evidence-based information on biosimilars from trusted sources, including regulatory agencies (FDA, EMA), professional medical societies, and pharmacists. This is vital for understanding their safety, efficacy, and appropriate role in treatment regimens.1
Open Communication: Healthcare providers should proactively initiate transparent conversations with patients about biosimilars. This includes addressing any concerns, explaining the scientific basis for their use, and clarifying concepts like interchangeability to build confidence and trust.7
Prioritize Patient Outcomes and Affordability: Recognize that FDA/EMA-approved biosimilars offer comparable clinical results to their reference products at a lower cost. Embracing biosimilars can increase access to essential therapies and reduce the financial burden on patients and the healthcare system.1
Advocate for Favorable Policies: Support policies that promote biosimilar uptake, reduce patient out-of-pocket costs, and ensure consistent and equitable access to these valuable medications.10
8. Conclusion: The Future of Biologic Drug Life Cycle Management
Biosimilars are not merely a supplemental category of medications; they represent a fundamental force that is reshaping the entire biologic drug lifecycle. Their emergence drives innovation, intensifies competition, and significantly expands patient access to essential therapies, moving the biopharmaceutical industry beyond a singular focus on novel blockbuster biologics.1
The future biopharmaceutical landscape will be characterized by intensified competition, continuous evolution of intellectual property strategies, and an increasing emphasis on manufacturing efficiency and supply chain resilience. Success in this dynamic environment will hinge on the cultivation of a collaborative ecosystem where regulatory frameworks are streamlined and harmonized globally, market incentives are appropriately aligned, and all stakeholders—from manufacturers and policymakers to healthcare providers and patients—are well-informed and confident in biosimilar adoption. The current period, often referred to as the “golden era of biosimilars” 2, is poised to unlock significant value in healthcare, but this potential can only be fully realized if systemic barriers are proactively identified and addressed.
The ongoing wave of patent expirations for numerous high-value biologics 2 presents an unprecedented opportunity for healthcare systems worldwide to achieve substantial savings and enhance patient well-being. The impact of biosimilars on biologic drug life cycle management is thus a testament to the complex and dynamic interplay of scientific advancement, regulatory policy, legal strategy, and market forces, all striving towards a more accessible, affordable, and sustainable future for advanced medicines.