The Drug Price Competition and Patent Term Restoration Act of 1984, universally called Hatch-Waxman, created the legal architecture that now governs roughly $100 billion in annual generic drug approvals in the United States. Every time a pharmacist fills a prescription with a generic, the transaction traces back to a regulatory and litigation framework that Congress assembled four decades ago and that courts and agencies have been refining ever since.
Understanding Hatch-Waxman means understanding how money moves in pharmaceuticals. Brand companies use it to extend commercial exclusivity. Generic manufacturers use it to enter markets years before patents technically expire. Litigation under the Act has produced some of the largest patent settlements in American legal history. And the FDA’s Orange Book, the statutory mechanism sitting at the center of the framework, is a document that portfolio managers, patent attorneys, and business development teams treat as a primary source of competitive intelligence.
This guide covers the ANDA pathway from filing mechanics through patent certification strategy, 30-month stays, 180-day exclusivity races, inter partes review, and the commercial math that drives every generic launch decision. Where relevant, it notes real cases, real drugs, and real timelines that illustrate how the rules operate in practice.
What Is the Hatch-Waxman Act and Why Does It Still Matter?
Hatch-Waxman solved a problem that had existed since generic drugs became a commercial category: demonstrating that a copy of an already-approved drug is safe and effective should not require repeating the clinical trials that proved the original. The Act formalized an abbreviated approval pathway, the ANDA, that lets generic applicants rely on the brand’s clinical data by proving bioequivalence rather than efficacy from scratch.
In exchange, the Act gave brand companies a set of tools to protect their investment period. Patent term restoration compensates for time lost during FDA review. Data exclusivity blocks FDA from approving generics during defined windows even when patents are absent. And the Paragraph IV litigation mechanism, which we address in detail below, gave courts a structured way to adjudicate patent validity and infringement before a generic product hits the market.
The 1984 compromise has never been revised by statute in any fundamental way, despite repeated legislative attempts. Its longevity partly reflects the fact that both sides of the industry have found it workable, and partly reflects how deeply its concepts, Orange Book listings, paragraph certifications, 30-month stays, 180-day exclusivity, have been embedded in business models, contracts, and case law. Courts interpreting the Act have produced thousands of opinions. The Federal Circuit’s Hatch-Waxman jurisprudence alone constitutes a substantial body of patent and administrative law.
How the 1984 Act Changed Generic Drug Economics
Before Hatch-Waxman, generic market penetration was limited. Generic manufacturers faced significant regulatory uncertainty and were reluctant to invest in products where the approval pathway was unclear. The Waxman component of the Act, sponsored by Rep. Henry Waxman, created the ANDA pathway. The Hatch component, from Sen. Orrin Hatch, provided the brand-side protections.
The result was explosive growth in generic approvals. By the mid-2000s, generics represented roughly 50% of prescriptions. By 2023, according to the Association for Accessible Medicines, generics account for approximately 90% of all prescriptions filled in the United States while representing roughly 20% of total drug spend. That gap between prescription volume and dollar share reflects the pricing mechanics the Act enables: brand drugs command premium prices during exclusivity, then face rapid price erosion at generic entry.
‘Generic drugs saved the U.S. health care system $373 billion in 2021 alone, according to the Association for Accessible Medicines 2022 Generic Drug & Biosimilar Access & Savings Report.’ [Association for Accessible Medicines, 2022]
The Orange Book: What It Lists and Why It Controls ANDA Strategy
The FDA publishes the Orange Book, formally titled ‘Approved Drug Products with Therapeutic Equivalence Evaluations,’ as the official registry of drug products approved under NDA or ANDA pathways. For Hatch-Waxman purposes, the critical content is the patent and exclusivity information the NDA holder submits.
NDA holders must submit patent information to FDA within 30 days of approval, or at the time of NDA submission for patents that already exist. The regulations require listing of patents that claim the drug substance (active ingredient), the drug product (formulation or composition), or a method of using the drug. Process patents and metabolite patents generally do not qualify for Orange Book listing.
Once listed, a patent in the Orange Book controls ANDA timing. A generic applicant cannot obtain final approval before the expiry of any unexpired Orange Book patent unless it successfully challenges that patent via Paragraph IV certification. The listing date, the patent expiration date, and the Paragraph IV challenge history are the three data points that define the competitive landscape for any given molecule.
Tools like DrugPatentWatch aggregate Orange Book data alongside patent prosecution histories, litigation records, and exclusivity status, giving generic companies and brand competitors a consolidated view of the patent estate around any approved drug. Analysts use that data to model loss-of-exclusivity timelines, which in turn drive revenue forecasts and portfolio valuations.
Orange Book Patent Listing Disputes: What Happens When a Patent Shouldn’t Be There
The FDA historically took a ministerial approach to Orange Book listings, accepting submissions from NDA holders without examining whether the listed patents actually met the statutory criteria. This created an obvious problem: brand companies had an incentive to list any patent that might plausibly qualify, because each listing extends ANDA processing timelines and creates litigation leverage.
The 2003 Medicare Modernization Act introduced a process allowing ANDA applicants to challenge improper listings, but the mechanism was limited. Courts were reluctant to second-guess listing decisions as a matter of administrative law.
The FDA Reauthorization Act of 2017 and subsequent regulatory actions tightened the listing rules. In 2021, the FTC and FDA jointly identified improper Orange Book listings as a competitive concern and the agencies signaled more aggressive enforcement. In 2023, the FTC sent letters to several pharmaceutical companies asserting that certain patents, particularly those directed to devices used with drug products, did not meet the statutory listing criteria.
Ampio Pharmaceuticals, Sorrento Therapeutics, and several other companies received FTC challenges to specific Orange Book listings between 2022 and 2024. The enforcement environment around Orange Book listing has shifted, and brand companies now face greater legal risk from aggressive listing strategies than they did a decade ago.
The ANDA Filing Process: Step by Step from Submission to Approval
An Abbreviated New Drug Application is a regulatory submission to the FDA Center for Drug Evaluation and Research, specifically to the Office of Generic Drugs. Unlike an NDA, which requires original clinical data demonstrating safety and efficacy, an ANDA relies on the already-approved reference listed drug, or RLD, for those demonstrations. The ANDA applicant must show that its product contains the same active ingredient, same route of administration, same dosage form, and same strength as the RLD, and that it is bioequivalent.
What Goes Into an ANDA: Chemistry, Manufacturing, and Bioequivalence Requirements
The technical content of an ANDA divides roughly into three areas. The chemistry, manufacturing, and controls section covers the manufacturing process, facility information, raw material specifications, finished product specifications, and stability data. CDER reviewers scrutinize this section heavily because manufacturing failures represent a primary source of generic drug quality problems.
The bioequivalence section contains the pharmacokinetic data demonstrating that the generic product delivers the active ingredient to the systemic circulation at a rate and extent comparable to the RLD. The FDA’s standard requires that the 90% confidence interval for the ratio of AUC and Cmax between test and reference products fall within the 80–125% acceptance criteria. For most drugs, an in vivo crossover study in healthy volunteers satisfies this requirement, though the FDA has approved in vitro methods for locally-acting products and certain topicals.
The labeling section requires that the generic’s prescribing information be the same as the RLD’s, with certain permitted differences for patent-protected indications.
ANDA Review Timeline: How Long Does FDA Approval Really Take?
The Generic Drug User Fee Amendments, GDUFA, fundamentally changed ANDA review timelines. Before GDUFA I took effect in fiscal year 2013, the FDA’s ANDA backlog exceeded 2,900 applications and median approval times stretched beyond four years. GDUFA I committed the agency to performance goals in exchange for user fees from the generic industry.
Under GDUFA III, which covers fiscal years 2023–2027, the FDA targets review of 90% of original ANDAs within 10 months from receipt date for applications classified as standard. Applications that receive complete response letters restart the review clock upon resubmission.
In practice, the median time from ANDA submission to first action has fallen to roughly 12–15 months for standard applications, though complex products, those with complicated bioequivalence requirements or novel formulations, can take longer. Applications that receive refuse-to-file notifications must refile and effectively restart.
The priority review designation for ANDAs, established under GDUFA, provides a shorter target review time, historically around six months, for products where there are fewer than three approved ANDAs and where there is no blocking exclusivity. This pathway matters most for drugs where rapid generic entry serves a public health interest.
ANDA Amendments and Complete Response Letters: What Delays Approvals
A complete response letter is the FDA’s formal notification that an ANDA cannot be approved as submitted. CRLs address deficiencies in bioequivalence data, manufacturing data, labeling, or patent certification status. Responding to a CRL requires a Class 1 or Class 2 resubmission, with different review time targets depending on classification.
Manufacturing deficiencies are the leading cause of CRLs and the most time-consuming to resolve. When a manufacturing facility receives a Warning Letter or fails a pre-approval inspection, the FDA will not approve any pending ANDAs manufactured at that site. Indian and Chinese manufacturing facilities, which account for a large proportion of generic drug production, have historically had higher rates of inspection deficiencies than domestic sites.
Patent Certifications in ANDA Filings: Paragraphs I Through IV Explained
Every ANDA must include a certification with respect to each patent listed in the Orange Book for the RLD. The four certification options are defined by the statute and are known by their paragraph numbers in the regulatory framework.
A Paragraph I certification states that no patent information has been submitted to FDA for the listed drug, which typically means no relevant Orange Book patents exist. A Paragraph II certification states that the patent has expired. A Paragraph III certification states that the patent will expire on a specific date and that the ANDA applicant does not seek approval until after that date. These three certifications are administratively straightforward and do not trigger litigation.
A Paragraph IV certification is the mechanism that drives pharmaceutical patent litigation. It states that the listed patent is invalid or will not be infringed by the generic product. Filing a Paragraph IV certification is, by statute, an artificial act of infringement, which gives the patent holder an immediate right to sue.
What Is a Paragraph IV Certification and What Happens After You File One?
Short answer: Filing a Paragraph IV certification initiates a formal notice process and creates the predicate for patent litigation that can delay ANDA approval by up to 30 months.
After filing, the ANDA applicant must send a detailed notice letter to both the NDA holder and each patent owner listed for the relevant patents. The notice letter must describe the factual and legal basis for the invalidity or non-infringement position. Courts have found that defective notice letters can toll the statutory deadlines that govern the patent holder’s response, so generic companies invest significantly in drafting thorough notice letters.
The patent holder then has 45 days to decide whether to file an infringement suit. If no suit is filed within 45 days, FDA can approve the ANDA when it is otherwise ready, and the patent holder’s leverage under Hatch-Waxman largely evaporates.
If the patent holder sues within 45 days, an automatic 30-month stay of FDA approval takes effect. During the stay period, FDA can complete its review but cannot grant final approval. The stay ends at the earlier of: 30 months from the date of the notice letter, a court decision that the patent is invalid or not infringed, or a court decision in the patent holder’s favor (in which case FDA cannot approve until the patent expires).
The 30-Month Stay: How Brand Companies Use It and How Generics Fight Back
The 30-month stay is the single most commercially significant provision in Hatch-Waxman from a brand-side perspective. A company holding an NDA can delay generic entry for up to 2.5 years simply by suing within 45 days of receiving the Paragraph IV notice, regardless of the merits of its patent claims.
Before the 2003 Medicare Modernization Act amendments, brand companies could list multiple patents and get a new 30-month stay for each successive listing and Paragraph IV certification. A practice emerged in the 1990s of listing patents after an ANDA was filed, a strategy sometimes called the ‘listing a patent after notice’ tactic, to generate successive stays. The 2003 amendments limited each NDA to a single 30-month stay per ANDA.
Generic companies fight the 30-month stay in several ways. They can challenge Orange Book patent listings as improper, seeking to have FDA delist a patent and thereby eliminate the stay predicate. They can file motions in the district court litigation to shorten or eliminate the stay based on failure to comply with discovery obligations. And they can file inter partes review petitions at the Patent Trial and Appeal Board, which, while not directly affecting the district court stay, creates an alternative invalidity challenge that may produce a faster adverse result for the brand patent.
Can a 30-Month Stay Be Extended or Shortened by Court Order?
Yes, in both directions. Courts can shorten the stay if the patent holder has not diligently prosecuted the litigation. Courts can extend the stay in limited circumstances where equity favors additional time. The practical reality is that most Hatch-Waxman cases settle before any court ruling on the merits, which means the stay’s commercial effect runs for a negotiated period rather than the full statutory 30 months.
180-Day Exclusivity: The Prize That Drives Generic Filing Strategy
The 180-day exclusivity period is the incentive Congress built into Hatch-Waxman to encourage generic companies to challenge brand patents. The first ANDA applicant to file a substantially complete application containing a Paragraph IV certification earns a 180-day period during which FDA will not approve any other ANDA for the same drug.
During those 180 days, the first filer faces no generic competition. The commercial value of 180-day exclusivity depends on the drug’s sales volume, but for a blockbuster product it can represent hundreds of millions of dollars in revenue at prices well above the eventual competitive generic price.
How Is First-Filer Status Determined When Multiple ANDAs Arrive the Same Day?
Before the 2003 amendments, the first-filer rule was strictly chronological: the first ANDA with a Paragraph IV certification, by filing timestamp, earned the exclusivity. This created scenarios where generic companies would hand-deliver applications at midnight on the day they planned to file, racing each other to FDA’s document room.
The 2003 amendments changed the rule so that all ANDAs submitted on the same day are considered simultaneous first filers. If multiple companies file Paragraph IV certifications on the same day, all of them share the 180-day exclusivity period and can launch simultaneously when the exclusivity window opens.
This change eliminated the midnight races but created strategic incentives for coordinating filing dates, either directly (which raises antitrust concerns) or indirectly by monitoring public information about competitors’ development timelines.
When Does 180-Day Exclusivity Forfeiture Apply?
The 2003 amendments introduced forfeiture provisions that stripped first filers of 180-day exclusivity if they failed to act with reasonable commercial diligence. A first filer forfeits exclusivity if it fails to market the drug within 75 days after a final court judgment of invalidity or non-infringement, or if it fails to obtain tentative approval within 30 months of filing.
The failure-to-obtain-approval forfeiture provision has been controversial because it can strip exclusivity from a first filer that has done everything right but faces an FDA review delay. Courts have generally been reluctant to read forfeiture provisions broadly, given the commercial importance of exclusivity.
Forfeiture also occurs if the first filer enters a settlement agreement with the brand company that is found to be an unlawful reverse payment. This provision was added in the wake of the FTC’s campaign against so-called ‘pay-for-delay’ settlements.
Shared Exclusivity: Strategic Implications When Multiple First Filers Exist
When multiple companies share 180-day exclusivity, the commercial dynamics shift significantly. Multiple generic entrants compete on price from day one of the exclusivity window, which accelerates the price erosion that typically begins at generic entry. The revenue pie is divided among more players, and each individual company’s return on its litigation investment is lower.
Companies respond to the shared exclusivity problem in several ways. Some seek to enter authorized generic agreements with the brand company, which are not subject to the 180-day exclusivity because they are technically a version of the brand product. Others focus on speed to market, trying to have inventory and distribution in place for the first day of the exclusivity period. And some choose not to file simultaneously with expected competitors, instead pursuing a different patent certificate or a later filing date to avoid the sharing dynamic.
Reverse Payment Settlements: The FTC’s Campaign Against Pay-for-Delay
A reverse payment settlement, also called a pay-for-delay agreement, is a settlement of Hatch-Waxman litigation in which the brand company pays value to the generic challenger in exchange for the generic company agreeing to delay market entry. The term ‘reverse’ refers to the fact that money flows from the patent holder to the alleged infringer, which is the opposite of a normal patent settlement.
The FTC campaigned against these settlements for over a decade, arguing that they were anticompetitive because they converted a patent challenge, which might result in early generic entry if the patent was found invalid or not infringed, into a guaranteed delay of competition. The FTC filed dozens of enforcement actions and consistently asked courts to declare reverse payment settlements presumptively unlawful.
FTC v. Actavis: What the Supreme Court Actually Decided in 2013
In FTC v. Actavis (2013), the Supreme Court resolved a circuit split over how courts should analyze reverse payment settlements under antitrust law. The case involved AndroGel (testosterone gel), where AbbVie’s predecessor Solvay Pharmaceuticals paid Actavis and other generic challengers substantial sums in exchange for delayed entry.
The Court, in a 5-3 decision authored by Justice Breyer, held that reverse payment settlements could violate the Sherman Act and must be analyzed under the ‘rule of reason.’ The Court rejected both extreme positions: it did not adopt the FTC’s proposed presumption of illegality, but it also rejected the Eleventh Circuit’s view that settlements within the scope of the patent were automatically immune from antitrust scrutiny.
Actavis changed the settlement calculus for both brand and generic companies. Large cash payments from brand to generic became much riskier from an antitrust perspective. The industry responded by shifting toward non-cash value transfers, including authorized generic agreements, royalty-free licenses for other products, and supply agreements, which are harder to quantify and therefore harder for the FTC to challenge as anticompetitive.
What Counts as a ‘Large and Unjustified’ Payment After Actavis?
The Supreme Court’s test focuses on whether the reverse payment is ‘large and unjustified,’ with the size of the payment relative to anticipated litigation costs serving as a useful proxy for anticompetitive intent. The Court suggested that a payment substantially exceeding saved litigation costs suggests the brand is paying for delay rather than settling a genuinely disputed claim.
Post-Actavis, courts have wrestled with how to quantify non-cash benefits. The Third Circuit, in cases involving the In re: Lipitor Antitrust Litigation, accepted that authorized generic licenses can constitute reverse payments subject to antitrust scrutiny. Courts in other circuits have reached varying conclusions about co-promotion agreements, supply contracts, and royalty waivers.
FTC enforcement has continued aggressively post-Actavis. The agency filed suit against AbbVie and Besins Healthcare over AndroGel patent listings, successfully arguing that the underlying patents were listed improperly and that the resulting settlements were anticompetitive. The D.C. Circuit ruled in the FTC’s favor in 2020 on certain claims, establishing important precedent on both Orange Book listing abuse and reverse payment theory.
Patent Term Restoration Under Hatch-Waxman: How Brand Companies Recover Lost Time
The Hatch component of Hatch-Waxman provides two mechanisms for brand companies to recover exclusivity time lost during regulatory review. Patent term extension, under 35 U.S.C. § 156, restores up to five years of patent life to compensate for time spent in FDA clinical review. Data exclusivity, the NCE exclusivity discussed below, provides a regulatory exclusivity period independent of patent status.
How Patent Term Extension Calculation Works Under 35 U.S.C. § 156
The calculation of patent term extension involves several variables. The restoration period equals half the time spent in clinical investigation (Phase I through filing of NDA) plus the full regulatory review period (from NDA filing to approval). The restoration is capped at five years, and the resulting extended patent term cannot exceed 14 years from the date of approval.
Only one patent per product can receive term extension, and the applicant must choose which patent to extend. This creates a strategic decision with major commercial implications: the best candidate is typically the patent with the latest expiration date, to maximize the period of exclusivity, but other factors including the breadth of the patent’s claims and its vulnerability to challenge may argue for a different choice.
Applications for patent term extension must be filed within 60 days of FDA approval. Late filing forfeits the right entirely, a deadline that brand companies occasionally miss through administrative error. The USPTO reviews the extension calculation and can dispute the applicant’s computation of clinical investigation time.
NCE and NME Exclusivity: Five-Year Bar on ANDA Filings
New chemical entity exclusivity provides a five-year period during which FDA will not accept an ANDA for a drug containing the same active moiety as the approved product. For products with NCE exclusivity, ANDA applicants cannot even file their applications for four years, and Paragraph IV certifications can only be submitted in the fifth year of the exclusivity period.
The NCE designation turns on whether the active moiety, meaning the molecular entity responsible for the drug’s activity, has been previously approved by FDA. Salts, esters, and other modifications of a previously approved moiety do not qualify for NCE exclusivity. Companies developing prodrugs and reformulations routinely test the boundaries of this definition with FDA.
Three-year exclusivity applies to applications for new indications, new formulations, or new dosage forms where the NDA contains new clinical investigations essential to approval. Unlike NCE exclusivity, three-year exclusivity does not bar ANDA filing, but it does bar approval for the specific condition of use or dosage form covered by those new investigations.
Pediatric Exclusivity: Six Months That Can Be Worth Hundreds of Millions
The Best Pharmaceuticals for Children Act, incorporated into the FDA Reauthorization Act, grants brand companies an additional six months of exclusivity, attached to all existing patents and exclusivity periods, in exchange for conducting FDA-requested pediatric studies. This six-month addition applies regardless of whether the pediatric studies show efficacy in children.
For a drug with $2 billion in annual sales, six months of additional exclusivity has a net present value in the range of $700 million to $1 billion after accounting for the generic entry pricing curve. Pediatric exclusivity requests have accordingly become standard practice for approved drugs, with companies routinely accepting FDA’s Written Requests for pediatric studies on high-revenue products even when the pediatric market itself is small.
Inter Partes Review and the PTAB: How Generic Companies Challenge Patents Outside Court
The America Invents Act of 2011 created inter partes review, a trial proceeding before the Patent Trial and Appeal Board in which any party can challenge the validity of an issued patent on the basis of prior art. IPR became an immediately significant tool in pharmaceutical patent litigation because it provided a faster, cheaper, and often more favorable forum for invalidity challenges than district court litigation.
Before IPR, generic companies challenging brand patents faced a single primary forum: district court litigation as part of the Hatch-Waxman 30-month stay proceedings. District court invalidity challenges required full discovery, claim construction, expert testimony, and trials that could take three to five years and cost tens of millions of dollars. The Federal Circuit’s high standard for overcoming a patent’s presumption of validity made district court challenges difficult.
IPR vs. Hatch-Waxman District Court Litigation: Which Forum Do Generic Companies Prefer?
IPR proceedings must be initiated within one year of service of a complaint in district court litigation, which in the Hatch-Waxman context means within one year of the brand company’s infringement suit. The PTAB institutes review if it finds a reasonable likelihood that the petitioner would prevail on at least one challenged claim. The trial then proceeds on a schedule targeting a final written decision within 12 months of institution.
The PTAB applies a ‘preponderance of the evidence’ standard for invalidity, which is lower than the ‘clear and convincing’ standard required in district court under Microsoft Corp. v. i4i Ltd. Partnership (2011). PTAB proceedings also tend to be narrower, focusing on prior art combinations identified in the petition, which can be advantageous for well-researched invalidity positions.
Generic companies often file IPR petitions simultaneously with Paragraph IV certifications, running the two proceedings in parallel. If the PTAB issues a final written decision finding claims unpatentable before the 30-month stay expires in district court, the stay effectively ends early. The coordination of IPR and district court strategy is now a standard element of generic patent challenge planning.
The Estoppel Problem: Why IPR Can Backfire in District Court
IPR petitioners face a significant risk if they proceed through a final written decision: statutory estoppel under 35 U.S.C. § 315(e) bars the petitioner from asserting in any later district court proceeding that the challenged claims are invalid based on any ground the petitioner raised or reasonably could have raised in the IPR. This estoppel is broad and has been applied by courts to encompass prior art references that were publicly available and reasonably searchable, even if not specifically cited in the IPR petition.
For generic companies, IPR estoppel means that filing a thorough IPR petition forecloses many invalidity arguments in subsequent district court litigation. Companies must therefore decide, at the outset, whether they want to preserve district court invalidity arguments by filing a narrow IPR or to maximize IPR success probability with a broad petition that creates estoppel risk. There is no clean answer to this tradeoff, and practice has varied across generic companies and their counsel.
PTAB Institution Rates in Pharmaceutical Cases: What the Data Shows
The PTAB’s institution rate for pharmaceutical IPR petitions has historically been lower than for technology sector patents, reflecting the fact that pharmaceutical patents often involve complex chemistry and biochemistry where prior art is less abundant and the arguments for obviousness are more difficult to establish on a paper record. Studies of PTAB institution rates from 2013 through 2022 show institution rates for pharmaceutical petitions in the 50–60% range, compared to 65–70% for technology petitions.
Among instituted pharmaceutical IPRs, final written decisions result in claim cancellation at rates roughly comparable to other technology sectors, approximately 60–65% of instituted cases produce at least partial cancellation of challenged claims. The challenge is getting to institution in the first place, which requires assembling a credible prior art combination and claim mapping in the petition itself.
Loss-of-Exclusivity Forecasting: How Analysts Model Generic Entry Risk
Loss of exclusivity, LOE, is the event horizon around which pharmaceutical revenue models are organized. For a brand drug facing patent expiration or a successful Paragraph IV challenge, LOE triggers a pricing cliff that typically produces a 70–90% price reduction within 12–24 months of first generic entry, as competition intensifies. Modeling the timing and extent of generic entry accurately is one of the most commercially consequential tasks in pharmaceutical business development.
What Drives Generic Entry Timing? The Variables That Matter Most
Patent expiration date is the obvious starting point, but it rarely tells the complete story. The relevant date is the expiration of the last patent listed in the Orange Book that covers the approved formulation and uses, not just the compound patent. Formulation patents, method-of-use patents, and packaging patents can each extend effective exclusivity well beyond the compound patent’s expiration.
Data exclusivity operates independently of patents. A drug with an expired compound patent but active NCE exclusivity is still protected from ANDA approval until the NCE period runs out. Pediatric exclusivity attached to any of the relevant exclusivity periods further extends the effective protection date.
The Paragraph IV challenge pipeline matters enormously. If multiple generic companies have filed Paragraph IVs and are in active litigation, the probability of pre-expiration entry rises substantially. Litigation outcomes are uncertain, but the historical data shows that generic challengers succeed in establishing invalidity or non-infringement in a meaningful percentage of cases.
Tools like DrugPatentWatch compile patent term extension records, exclusivity expiration dates, ANDA filing histories, and Paragraph IV litigation status for every approved drug, providing the raw data inputs that analysts use to build LOE models. The platform’s patent expiration analysis is used by buy-side analysts, investment banks, and business development teams to generate quantitative estimates of the window during which a brand drug faces generic competition risk.
Historical Generic Entry Patterns: Price Erosion After LOE by Market Structure
The pricing dynamics of generic entry depend heavily on the number of approved ANDA filers at the time of first generic launch. With a single generic entrant during 180-day exclusivity, the generic typically launches at 80–85% of brand price, and the brand itself maintains significant market share, particularly among patients with insurance coverage that tiers branded and generic products similarly.
When the exclusivity period ends and additional generics enter, price erosion accelerates rapidly. With three to five generic competitors, generic prices typically fall to 40–60% of brand. With ten or more competitors, which is common for older molecules with large markets, generic prices fall to 10–20% of brand, and the brand’s market share collapses to single digits except for patients with strong brand loyalty or narrow formulary tier advantages.
The atorvastatin (Lipitor) LOE is the canonical large-scale example. Pfizer’s patent expired in November 2011, Ranbaxy launched as the first-filer generic, and within 18 months of full generic competition the market price for atorvastatin had fallen to less than 5% of Lipitor’s branded price. Pfizer’s atorvastatin revenue fell from approximately $10 billion annually to less than $2 billion.
LOE Cliff Risk Assessment: What Makes Some Drugs More Vulnerable Than Others
Generic substitution rates vary by therapeutic category. Primary care drugs for chronic conditions, statins, ACE inhibitors, proton pump inhibitors, face high substitution rates because prescribers are comfortable with generics and payers aggressively push generic substitution. Specialty drugs, particularly those in oncology or neurology, may face lower initial substitution rates because of prescriber inertia and payer coverage patterns, but this changes as biosimilar-like competitive dynamics develop.
Formulation complexity also affects substitution. Modified-release products, transdermal patches, and inhaled formulations face higher manufacturing barriers that limit the number of generic entrants. Products like Advair (fluticasone/salmeterol inhalation powder) and Suboxone (buprenorphine/naloxone sublingual film) generated extended periods of effective exclusivity not because of strong patent positions but because of the technical difficulty of demonstrating bioequivalence for complex formulations.
Complex ANDAs: 505(b)(2) Applications and the Hybrid Approval Pathway
Not every application for a drug that relies on prior clinical data proceeds through the full ANDA pathway. Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act allows applications that rely at least partially on data in the published literature or on FDA’s prior findings of safety and effectiveness for a previously approved drug. The 505(b)(2) pathway is the right choice when the proposed product differs from the RLD in a way that requires some new clinical data, but where full NDA-level data generation would be unnecessarily burdensome.
When to Use 505(b)(2) Instead of ANDA: Reformulations, Combinations, and New Routes
The classic 505(b)(2) candidate is a reformulation of an approved drug, for example, an extended-release version of an immediate-release approved product, or a new salt or ester of an approved active moiety. These products need some clinical work to demonstrate safety and efficacy of the new formulation or chemical form, but they can rely on the already-established pharmacology of the parent compound.
Fixed-dose combination products, new routes of administration, and products with new indications for approved drugs also commonly use the 505(b)(2) pathway. The pathway has been used by many specialty pharma companies to differentiate their products from commoditized generics while avoiding the cost of a full NDA development program.
For intellectual property purposes, 505(b)(2) applications are subject to the same Orange Book listing requirements and Paragraph IV certification obligations as ANDAs. A 505(b)(2) applicant must certify with respect to all Orange Book patents for the reference product, and a Paragraph IV certification by a 505(b)(2) applicant triggers the same 30-month stay mechanism as for an ANDA.
505(b)(2) Patent Certification Strategy vs. Full ANDA: Key Differences
The 505(b)(2) pathway and the ANDA pathway differ in one commercially important respect: the 180-day exclusivity provision does not apply to 505(b)(2) applications. A 505(b)(2) applicant that files a Paragraph IV certification for the first time is not eligible for the 180-day exclusivity that a first-filing ANDA applicant would earn. This reduces the incentive for 505(b)(2) patent challenges relative to ANDA challenges.
The absence of 180-day exclusivity for 505(b)(2) applicants means that brand-side strategies involving 505(b)(2) follow-on products as authorized generics or reformulations have different competitive dynamics. Brand companies sometimes pursue 505(b)(2) applications for slightly modified versions of their own products, a strategy called ‘product hopping,’ to shift prescription volume to a formulation without generic competition before the original product’s patents expire.
Product Hopping: How Brand Companies Use Reformulations to Extend Exclusivity
Product hopping refers to a strategy in which a brand pharmaceutical company introduces a reformulated or modified version of an existing drug and then withdraws or disadvantages the original product, forcing prescribers and patients to use the new formulation, which has its own patent protection and exclusivity.
The tactic works because generic substitution in pharmacies is typically limited to drugs that are rated therapeutically equivalent in the Orange Book. A reformulated product with a different dosage form, different strength, or different active ingredient form will have its own Orange Book listing, and existing ANDAs for the original formulation cannot be automatically substituted for the reformulation.
Namenda and Doryx: Landmark Product Hopping Litigation Outcomes
The Namenda (memantine) product hop is the most extensively litigated example of the strategy. Forest Laboratories, later acquired by Actavis/Allergan, held patents on Namenda IR, the immediate-release formulation used for Alzheimer’s disease. As Namenda IR’s patents approached expiration in 2015, Forest introduced Namenda XR, an extended-release formulation with later patent expiration, and announced plans to discontinue Namenda IR.
The State of New York sued Forest, arguing that the forced switch to Namenda XR constituted illegal monopolization under Section 2 of the Sherman Act because it would eliminate the possibility of generic substitution at the pharmacy level. The Second Circuit Court of Appeals ultimately ruled that the planned discontinuation of Namenda IR could constitute anticompetitive conduct, and Forest reached a settlement that allowed Namenda IR to remain on the market long enough for generic substitution to occur.
The Doryx (doxycycline hyclate delayed-release tablets) litigation involved a different variant of the strategy, where Warner Chilcott repeatedly changed the tablet strength and formulation of its products, forcing ANDAs to be refiled or amended and effectively resetting the competitive timeline. The Third Circuit’s analysis of the Doryx product hops in Mylan Pharmaceuticals v. Warner Chilcott provided detailed guidance on when product reformulations cross from legitimate competition into anticompetitive conduct.
Antitrust Risk of Product Hopping: How Courts Distinguish Legitimate Reformulation from Illegal Exclusion
Courts have not adopted a categorical rule against product reformulations. A company is generally free to introduce new products and to compete aggressively on price, features, and promotion. The antitrust violation arises when the company uses conduct beyond competition on the merits, such as withdrawing a product solely to eliminate generic substitution or engaging in exclusive dealing that forecloses the original formulation from distribution, to prevent consumers from accessing lower-cost generics.
The distinction between procompetitive product improvement and anticompetitive product hopping is fact-intensive and turns on evidence of intent, timing relative to generic entry, and the nature of any alleged market foreclosure. Companies considering product transitions near LOE dates should evaluate this risk explicitly, with antitrust counsel, as part of the transition strategy.
Authorized Generics: The Brand Weapon That Dilutes 180-Day Exclusivity
An authorized generic is a version of a brand drug that is marketed by the brand company, or by a third party under license from the brand company, under the brand’s NDA rather than an ANDA. Because it is not an ANDA product, it is not subject to the 180-day exclusivity restriction. An authorized generic can launch on the first day of the first filer’s exclusivity window, directly competing with the first filer generic during the period when the first filer expected to have the market to itself.
How Authorized Generics Affect the Commercial Value of 180-Day Exclusivity
The impact of an authorized generic on first-filer economics is substantial. Studies by the FTC and academic economists have consistently found that first-filer revenue during 180-day exclusivity is significantly lower when an authorized generic is present. A first filer facing an authorized generic competitor may earn 50–70% less revenue during exclusivity than a first filer without an authorized generic.
This commercial reality has changed how generic companies value Paragraph IV litigation. When a brand company signals that it will launch an authorized generic upon generic entry, the expected value of winning the Paragraph IV challenge is reduced, which in turn affects the settlement negotiations. Generic companies negotiating settlements may seek explicit commitments from brand companies not to launch authorized generics, which in the post-Actavis environment creates additional antitrust risk if combined with other value transfers.
The FTC has studied authorized generics extensively and concluded in its 2011 report that they reduce consumer savings during the 180-day exclusivity period. But because authorized generics are FDA-compliant product launches, not anticompetitive conduct per se, regulatory action to restrict them has been difficult to justify legally.
Authorized Generic Agreements and Reverse Payment Concerns Post-Actavis
After FTC v. Actavis limited direct cash payments from brand to generic companies, authorized generic licenses emerged as a common non-cash value transfer in settlements. Under a typical post-Actavis settlement, the brand company grants the generic challenger the right to market an authorized generic during the 180-day exclusivity period as part of the consideration for the generic agreeing to delay its own entry.
Courts and the FTC have recognized that authorized generic licenses constitute value transfers that can substitute for the type of direct cash payments condemned in Actavis. The Third Circuit’s analysis in In re: Lipitor Antitrust Litigation specifically addressed how to value an authorized generic license for purposes of the Actavis reverse payment analysis, concluding that the license’s value must be quantified and weighed against the competitive delay agreed to in the settlement.
Biosimilar vs. Small-Molecule Generic: How the BPCIA Differs From Hatch-Waxman
The Biologics Price Competition and Innovation Act, BPCIA, enacted as part of the Affordable Care Act in 2010, established an abbreviated pathway for biosimilar approval modeled loosely on Hatch-Waxman but significantly different in key respects. Understanding how BPCIA differs from Hatch-Waxman is essential because the two frameworks operate in parallel and govern different segments of the drug market.
The Patent Dance: BPCIA’s Information Exchange vs. Hatch-Waxman’s Paragraph IV
BPCIA’s patent resolution mechanism, colloquially called the ‘patent dance,’ requires the biosimilar applicant to provide the reference product sponsor with a copy of its application and manufacturing information. The parties then exchange lists of patents they believe are infringed or subject to challenge, and engage in a negotiation process to determine which patents will be litigated before launch.
Unlike Hatch-Waxman’s 30-month stay, the patent dance is not mandatory. The Supreme Court held in Sandoz v. Amgen (2017) that biosimilar applicants can opt out of the dance, and that the only consequence is that the reference product sponsor may seek a preliminary injunction under patent law. The Supreme Court also held that the 180-day advance notice of commercial marketing is mandatory, establishing a minimum period between biosimilar approval and first commercial sale.
The commercial exclusivity for reference biologics under BPCIA is 12 years, substantially longer than the five-year NCE exclusivity for small molecules. This reflects both the greater development costs for biologics and the political dynamics of the ACA’s passage. Interchangeability designation, which would allow pharmacist-level substitution for biosimilars equivalent to the AB ratings that govern small-molecule generic substitution, requires additional clinical data beyond the base biosimilarity showing.
Patent Thickets in Biologics: Why AbbVie’s Humira Estate Was Different
AbbVie’s patent protection strategy for Humira (adalimumab) became the most studied example of a biologic patent thicket. AbbVie built an estate of more than 130 U.S. patents covering various aspects of adalimumab, its formulations, manufacturing processes, and methods of use. While the original compound patent expired in 2016, the later formulation and method patents did not expire until the 2030s.
Rather than litigate the entire patent estate, AbbVie settled with biosimilar developers through a series of agreements that allowed U.S. biosimilar entry in January 2023 and European entry earlier. The settlements, which involved licenses to AbbVie’s patent portfolio in exchange for royalties and other terms, allowed AbbVie to extract revenue from biosimilar entrants even after generic-like competition began.
The Humira situation raised questions about whether the BPCIA’s patent dance and exclusivity provisions were functioning as Congress intended. Multiple investigations and academic studies have concluded that large biologic patent estates can delay biosimilar entry well beyond what the statutory exclusivity framework contemplates, effectively extending monopoly periods through strategic patent prosecution rather than through the exclusivity periods Congress specified.
Interchangeable Biosimilars: How FDA Determines Substitutability and What It Means Commercially
FDA grants interchangeability designation to biosimilars that demonstrate, through a switching study, that alternating between the reference product and the biosimilar produces no greater risk than using the reference product alone. Interchangeable biosimilars can be substituted at the pharmacy level without prescriber intervention, similar to how small-molecule generics are substituted for brand drugs.
The commercial significance of interchangeability is substantial. Without it, biosimilar substitution requires prescriber action, which creates a substantial adoption barrier because most physicians are not focused on biosimilar-to-reference substitution for existing patients. With interchangeability, payers can implement formulary substitution policies that dramatically accelerate biosimilar market share.
The first FDA-approved interchangeable biosimilar was Viatris’s Semglee (insulin glargine) in July 2021. Several insulin biosimilars and one adalimumab biosimilar have received interchangeability designation since then. The commercial impact of these designations will take several years to fully materialize as payer formularies update and pharmacy systems adapt.
FDA Exclusivity Types: A Reference Guide to All Regulatory Exclusivity Periods
The pharmaceutical regulatory system provides multiple distinct exclusivity protections that can operate independently or in combination. Each has its own eligibility criteria, duration, and effect on ANDA acceptance and approval. Understanding all of them is necessary for accurate LOE modeling.
Complete Exclusivity Table: Types, Durations, and Statutory Triggers
Exclusivity Type
Duration
Trigger
Effect
NCE (New Chemical Entity)
5 years
Approval of NDA for new active moiety
Bars ANDA filing for 4 years; Paragraph IV CAN be filed in year 5
New Clinical Investigation (3-year)
3 years
NDA or supplement relying on new clinical investigations
Bars ANDA approval for new indication/dosage form/route
Pediatric Exclusivity
6 months (added to existing exclusivity)
Completion of FDA-requested pediatric studies
Attaches to all patent and exclusivity protections; delays all generic approvals
Orphan Drug Exclusivity
7 years
FDA designation and approval for rare disease indication
Bars approval of same drug for same indication; does not block other indications
180-Day Generic Exclusivity
180 days
First ANDA with Paragraph IV certification
FDA will not approve other ANDAs during exclusivity window
Biologic Reference Product Exclusivity
12 years
Approval of biologic NDA under section 351(a)
Bars biosimilar approval for 12 years; biosimilar applications cannot be submitted for 4 years
QIDP Exclusivity
5 additional years (added to NCE, NCI)
Qualified Infectious Disease Product designation; approval of antibiotic or antifungal for serious infection
Extends underlying exclusivity period by 5 years
Orphan Drug Exclusivity vs. NCE Exclusivity: When Both Apply to the Same Drug
A drug that qualifies for both orphan drug exclusivity and NCE exclusivity gets both protections, but the effects are complementary rather than additive in a simple sense. Orphan exclusivity blocks approval of the same drug for the same indication; it does not block approval of the same drug for different indications. NCE exclusivity blocks ANDA filings for four years and ANDA approvals for five years, regardless of indication.
For a drug with both designations, the relevant question is which exclusivity expires last and whether an ANDA could be approved for a different indication during the overlap period. These questions become significant for drugs where the orphan indication is a narrow subset of potential uses and where off-label use for non-orphan conditions is commercially significant.
QIDP and GAIN Act Exclusivity: The Antibiotic-Specific Framework
The Generating Antibiotic Incentives Now Act, GAIN Act, passed in 2012 as part of the FDA Safety and Innovation Act, established the Qualified Infectious Disease Product designation. QIDP drugs treating serious or life-threatening infections caused by specified resistant bacteria receive an additional five years of exclusivity attached to any other applicable exclusivity. For an NCE antibiotic, this means ten years of total exclusivity rather than five.
The QIDP designation was intended to address the pipeline problem in antibiotics, where commercial incentives were insufficient to drive investment in new drugs for resistant pathogens. Whether the GAIN Act has meaningfully stimulated antibiotic development remains disputed; studies have found limited correlation between QIDP designation rates and actual increases in antibiotic NDA submissions for the most commercially challenging resistant organism indications.
Paragraph IV Litigation Outcomes: Win/Loss Rates and What They Mean for Generic Strategy
The empirical record of Paragraph IV litigation outcomes shapes how generic companies evaluate their legal positions before filing. Understanding historical outcome rates, by patent type, claim type, and therapeutic area, is a prerequisite for rational investment in Paragraph IV challenges.
Historical Paragraph IV Win Rates for Generic Challengers: What the Data Shows
Multiple academic and regulatory studies have examined Paragraph IV litigation outcomes. An FTC study published in 2011 found that generic challengers were successful, achieving a court ruling of invalidity or non-infringement, in approximately 48% of cases that were fully litigated to a decision, based on cases filed between 1992 and 2000. Studies of more recent litigation have found somewhat lower success rates for generic challengers as brand companies have strengthened their patent portfolios and developed more sophisticated litigation strategies.
The 75% of Paragraph IV cases that settle before a final court decision make statistical analysis of ‘win rates’ inherently limited. Settlement terms are confidential, but the timing and structure of settlements provide indirect evidence about which party held a stronger legal position at the time of settlement. Settlements closer to the end of the 30-month stay, with generic entry dates near patent expiration, generally indicate stronger brand-side positions. Settlements with generic entry dates several years before patent expiration generally indicate stronger generic-side positions.
Patent Types Most Vulnerable to Paragraph IV Challenge: Compound vs. Formulation vs. Method
Compound patents, those claiming the active ingredient itself, are generally the most defensible because the composition they protect is the core innovation. Successfully arguing that a compound patent is invalid typically requires identifying prior art that discloses the specific compound or makes its synthesis obvious, which is often difficult for novel small molecules.
Formulation patents are more vulnerable. Courts have frequently found formulation patents obvious when the components of the formulation were individually known and the rationale for combining them was apparent to a skilled formulator. The Federal Circuit’s obviousness analysis in pharmaceutical formulation cases has become notably demanding of evidence that the claimed formulation produces unexpected results beyond what would be expected from the prior art components.
Method-of-use patents face a specific vulnerability in the Hatch-Waxman context: the carve-out provision. An ANDA applicant can file a Paragraph III certification for a method-of-use patent, stating that it does not seek approval for the patented use, and then include skinny labeling that omits the patented indication. Courts have debated the extent to which skinny-labeled generics can be liable for induced infringement when doctors prescribe the generic for the patented indication based on the full label’s disclosure in the brand product’s prescribing information.
Skinny Labeling and Induced Infringement After GSK v. Teva
GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc., decided by the Federal Circuit in a divided opinion in 2021, addressed the induced infringement liability of skinny-labeled generic carvedilol. GSK held a method-of-use patent for treating heart failure with carvedilol. Teva’s ANDA omitted the heart failure indication from its label (skinny label) but included the approved indications for hypertension and left ventricular dysfunction post-MI.
The Federal Circuit’s majority upheld the jury verdict finding Teva liable for induced infringement, reasoning that Teva’s marketing and communications contributed to physicians prescribing carvedilol for heart failure even with the skinny label. The dissent argued that allowing induced infringement based on promotion of a non-patented use undermined the statutory carve-out mechanism Congress had created.
GSK v. Teva created significant uncertainty about skinny labeling as a litigation avoidance strategy. Generic companies facing method-of-use patents began investing more heavily in clear communications to prescribers about which indications were covered by the skinny label, and some reconsidered whether skinny labeling was a viable strategy for high-revenue indications.
The Commercial Math: How Generic Companies Value Paragraph IV Challenges
Generic pharmaceutical companies allocate development resources and litigation budgets based on expected value calculations that weigh the commercial opportunity against the costs and risks of patent challenges. Understanding this math clarifies why some drugs attract dozens of Paragraph IV filers while others see none.
Generic Drug Development Cost vs. Potential 180-Day Exclusivity Revenue
The all-in cost of an ANDA, from bioequivalence studies through manufacturing scale-up, regulatory filing, and patent litigation, varies substantially by product complexity. For a simple oral solid-dosage form product without significant bioequivalence challenges, development costs of $2–5 million are achievable. For a complex product requiring clinical endpoint bioequivalence studies, modified-release formulation development, or inhalation dose delivery system development, costs can reach $20–50 million or more.
On the revenue side, 180-day exclusivity for a drug with $1 billion in annual brand sales, assuming generic pricing at 80% of brand and market share of 50–60% during exclusivity, generates gross revenue of roughly $200–240 million during the six-month window. After accounting for cost of goods, distribution costs, and the need to share exclusivity with co-first-filers, net contribution from exclusivity can range from $20 million to well over $100 million depending on the market structure.
These calculations explain the concentration of Paragraph IV activity around high-revenue branded drugs. According to FDA data and analyses available through resources like DrugPatentWatch, the top 50 drugs by revenue collectively attract the majority of Paragraph IV certifications in any given year. Smaller-market drugs, where exclusivity revenues would not cover litigation costs, generate little generic challenge activity.
What Happens to Generic Pricing After 180-Day Exclusivity Ends?
The end of 180-day exclusivity typically triggers a cascade of additional ANDA approvals and a rapid decline in generic prices. FDA approval of additional ANDAs is not automatic on day 181; those applications must have received final approval or tentative approval and must be ready to launch. In practice, 5–15 additional generic filers often have tentative approvals waiting and launch within weeks of the exclusivity window’s expiration.
Price erosion in the post-exclusivity period follows a pattern studied extensively by FDA, the FTC, and academic economists. Generic prices stabilize at a competitive equilibrium determined by manufacturing costs, distribution economics, and the number of active competitors. For high-volume oral solid-dosage form generics, that equilibrium often produces prices 80–90% below brand levels. For sterile injectable generics or complex formulation generics, the equilibrium is often higher because the supply chain is thinner.
Key Litigation Venues: Which Courts Handle Most Hatch-Waxman Cases
Hatch-Waxman patent litigation concentrates in a small number of federal district courts. The District of Delaware and the District of New Jersey handle the majority of Paragraph IV cases, reflecting the incorporation of most major pharmaceutical companies in Delaware and the historical concentration of the pharmaceutical industry in New Jersey. The Eastern District of Virginia’s ‘rocket docket’ was historically favored for its fast trial scheduling but has been less central to pharmaceutical litigation in recent years.
District of Delaware Hatch-Waxman Practice: Why So Many Cases End Up There
Delaware’s concentration of pharmaceutical Hatch-Waxman cases reflects several factors. Most major pharmaceutical companies are incorporated in Delaware, giving plaintiffs the option to sue there under personal jurisdiction. Delaware’s judges have substantial experience with pharmaceutical patent litigation and have developed specialized procedures for handling complex Hatch-Waxman cases efficiently. The court’s sophisticated local rules regarding claim construction, expert discovery, and trial procedure have created predictability that both plaintiffs and defendants value.
The fact-specific and technically complex nature of pharmaceutical patent litigation has made judicial experience and consistency particularly valuable. Practitioners note that Delaware judges who have handled dozens of Hatch-Waxman cases develop expertise in distinguishing strong from weak invalidity and non-infringement arguments, which generally produces more predictable outcomes than cases before generalist judges encountering pharmaceutical patent law for the first time.
Federal Circuit Appeals in Hatch-Waxman: How Appellate Decisions Shape the Framework
The Court of Appeals for the Federal Circuit has exclusive jurisdiction over appeals in patent cases, including Hatch-Waxman cases, and its jurisprudence directly shapes the legal rules that brand and generic companies operate under. Federal Circuit decisions on obviousness, written description, enablement, and claim construction apply to pharmaceutical patents as directly as to any other technology.
Several Federal Circuit precedents have had outsized commercial significance for the pharmaceutical industry. The court’s decision in Pfizer Inc. v. Apotex Inc. (2007), holding that Pfizer’s amlodipine besylate salt patent was obvious over the prior art compound, cost Pfizer hundreds of millions of dollars in projected exclusivity and established demanding standards for salt selection patents. Decisions in the Warner-Lambert v. Apotex series established important precedent on induced infringement for method-of-use patents. And the court’s consistent application of Amgen Inc. v. Hoechst Marion Roussel (2006) on written description requirements has shaped how biologic companies draft their patents.
Claim Construction and Its Outsized Role in Hatch-Waxman Outcomes
Claim construction, the court’s determination of the meaning and scope of patent claims, frequently determines the outcome of Hatch-Waxman cases. A broad claim construction typically favors the patent holder by bringing the accused generic product within the patent’s scope. A narrow construction may allow the generic to design around the patent or establish non-infringement.
Because claim construction is a question of law reviewed de novo by the Federal Circuit, district court claim construction rulings are subject to reversal on appeal. This creates uncertainty that persists after district court judgments, as the prevailing party must consider the possibility that an appeal will produce a different claim construction and a different outcome on infringement or validity. Hatch-Waxman settlements often occur after district court claim construction rulings precisely because those rulings give the parties a clearer picture of the Federal Circuit’s likely view.
Supply Chain and Manufacturing Exclusivity: The Non-Patent Barriers That Matter
Patents and regulatory exclusivity are not the only barriers to generic entry. Manufacturing economics, supply chain constraints, and API sourcing create practical barriers that can delay or limit generic competition even when legal exclusivity has expired.
API Sourcing and Generic Entry Risk: How Raw Material Supply Constrains Launch
Active pharmaceutical ingredient manufacturing for generic drugs is concentrated in India and China, with Indian manufacturers accounting for a majority of global generic API production. This concentration creates supply chain risk: FDA enforcement actions against specific Indian or Chinese manufacturing facilities can prevent approval or supply of the API needed for generic production.
Several high-profile drug shortages in the U.S. market have been attributed at least partly to API supply chain concentration. Valsartan, the angiotensin receptor blocker, became the subject of a major global recall in 2018 when carcinogenic nitrosamine impurities were discovered in API produced by a Chinese manufacturer, Zhejiang Huahai Pharmaceuticals. The subsequent regulatory requirements for nitrosamine risk assessments across thousands of drug products created additional supply chain complexity.
Complex Product Barriers: Why Some Generic Markets Stay Thin for Years After LOE
Some drug categories present manufacturing barriers that limit generic competition even long after patent expiration. Inhaled drug products, sterile injectables, modified-release oral formulations, and transdermal patches all require specialized manufacturing capabilities and quality systems that not all generic manufacturers possess.
The inhaled drug category illustrates these barriers particularly well. Demonstrating bioequivalence for a metered-dose inhaler or dry powder inhaler requires showing that the particle size distribution, deposition pattern, and pharmacodynamic effect of the generic product match those of the reference. FDA’s bioequivalence requirements for complex inhaled products require multiple studies, including pharmacokinetic studies, pharmacodynamic studies, and in vitro device characterization, that can take years to complete and cost tens of millions of dollars. The result is that some inhaled drugs with expired patents have few or no approved generics.
Monitoring the Competitive Landscape: Intelligence Tools and Data Sources
Tracking Orange Book patent listings, ANDA filings, Paragraph IV notifications, and litigation developments requires systematic data aggregation that goes beyond what FDA’s public databases provide. The pharmaceutical patent intelligence ecosystem has developed several specialized tools to fill this need.
How to Use Orange Book Data for Competitive Intelligence
FDA’s electronic Orange Book provides patent and exclusivity data for every approved drug, but it requires interpretation to generate actionable competitive intelligence. Raw expiration dates must be adjusted for patent term extensions, pediatric exclusivity additions, and the timing of any pending patent term adjustments at the USPTO. Orange Book data must also be cross-referenced with litigation records to determine whether any listed patents are subject to outstanding invalidity challenges.
DrugPatentWatch integrates Orange Book data with USPTO patent records, FDA exclusivity records, PACER litigation data, and PTAB proceeding information to create consolidated patent estate views for specific drugs. The platform allows users to model different LOE scenarios based on varying assumptions about litigation outcomes, helping brand and generic companies develop strategic plans that account for the range of possible competitive timelines.
IQVIA and IQVIA subsidiary data sources provide market sales data that, when combined with patent intelligence from sources like DrugPatentWatch, allow analysts to build revenue-at-risk models: quantitative estimates of the dollar value of sales exposed to generic competition in each year of a forecast period. These models are standard inputs to buy-side pharmaceutical equity research, investment banking valuations for M&A, and portfolio planning by large pharmaceutical companies.
Reading Paragraph IV Notification Letters: What Public Filings Reveal About Generic Strategy
When a generic company files a Paragraph IV certification, it must send a detailed notice letter to the NDA holder and patent owner. These notice letters are not publicly filed, but their existence, and sometimes their contents, becomes apparent from the patent infringement suits that brand companies file in response. The complaints and related court documents in Hatch-Waxman litigation are public records that reveal the specific patents challenged, the legal theories the generic is advancing, and the commercial opportunity that justified the challenge.
Competitive intelligence teams at brand companies routinely monitor PACER, the federal court’s case management system, for new Hatch-Waxman complaints against their products. The 45-day response window creates urgency in this monitoring function, because failure to file suit within 45 days of receiving the notice letter forfeits the 30-month stay.
What This Means for Pharmaceutical Investors and Business Development Teams
Hatch-Waxman’s mechanics have direct, quantifiable implications for pharmaceutical equity valuations, M&A due diligence, and portfolio strategy. Every branded pharmaceutical asset’s value is contingent on the patent and exclusivity timeline, and every generic pharmaceutical company’s pipeline value depends on its success in navigating that timeline.
LOE Impact on Branded Pharma Valuations: How to Model Revenue at Risk
Standard discounted cash flow valuation of branded pharmaceutical assets incorporates a patent expiration assumption that drives the model’s terminal value. Analysts typically model a step-down in revenue at the assumed LOE date, with the magnitude of the step-down based on historical generic erosion curves for similar products in similar markets.
The error term in these models is substantial. If a Paragraph IV challenge succeeds and generic entry occurs three years before the compound patent expiry, and if the model had not captured that risk, the resulting revenue shortfall can be severe. The opposite error, failing to credit the effective protection provided by formulation and method-of-use patents after compound patent expiry, leads to undervaluation.
Investors who build proprietary LOE models using orange book and litigation data, rather than relying solely on management guidance or sell-side consensus, capture an informational advantage. The analysis is not simply patent expiration date lookup; it requires understanding which patents are vulnerable to challenge, which litigation positions are strong, and how the settlement dynamics in any pending Paragraph IV case are likely to resolve.
Generic Pharma Pipeline Valuation: How to Value a Paragraph IV First-Filer Position
Valuing a first-filer position in 180-day exclusivity requires modeling the following variables: brand drug sales at time of expected generic entry; generic pricing relative to brand at launch; market share during exclusivity; whether authorized generics will be present; the number of co-first-filers, if any; and the probability that the first-filer’s patent challenge will succeed or settle on favorable terms.
The probability-weighting is critical. A first-filer position with strong non-infringement arguments and a clearly invalidity-vulnerable patent may have a 70–80% probability of resulting in pre-patent-expiry entry. A position based primarily on a narrow invalidity theory against a well-prosecuted formulation patent may have a 30–40% probability. Net present value calculations that apply appropriate probability weights to these outcomes generate rational investment thresholds for generic pipeline development.
Key Takeaways
Hatch-Waxman’s ANDA pathway, patent certifications, 30-month stay, 180-day exclusivity, and patent term restoration form an integrated system that simultaneously enables generic entry and provides brand companies tools to protect exclusivity periods.
A Paragraph IV certification is an artificial act of infringement that gives the patent holder the right, but not the obligation, to sue within 45 days and trigger a 30-month stay of ANDA approval.
The 2003 Medicare Modernization Act amendments limited brand companies to a single 30-month stay per ANDA and added first-filer forfeiture provisions to the 180-day exclusivity framework.
FTC v. Actavis (2013) subjected reverse payment settlements to antitrust rule-of-reason analysis, reshaping settlement negotiations toward non-cash value transfers including authorized generic licenses.
Inter partes review at the PTAB provides generic companies a faster, cheaper forum for patent validity challenges, but the estoppel provisions can limit subsequent district court invalidity arguments.
Orange Book listing abuse and product hopping strategies have faced increasing regulatory and antitrust scrutiny since 2019, with the FTC taking a more aggressive enforcement posture toward both practices.
The BPCIA’s biosimilar pathway differs from Hatch-Waxman in its patent dance mechanism, 12-year biologic reference product exclusivity, and lack of an equivalent to 180-day generic exclusivity.
LOE forecasting requires integrating patent expiration data, exclusivity status, Paragraph IV litigation history, and market structure analysis to produce probabilistically weighted timelines that can support investment decisions.
Platforms like DrugPatentWatch aggregate the data inputs necessary for this analysis, providing commercial-grade patent intelligence that goes beyond FDA’s public Orange Book database.
Manufacturing barriers, API supply chain constraints, and complex product bioequivalence requirements create practical barriers to generic entry that can delay competition even when legal exclusivity has expired.
Frequently Asked Questions
1. What is the difference between a Paragraph III and Paragraph IV certification?
A Paragraph III certification states that the generic applicant will not seek approval until after a specific listed patent expires, meaning the generic delays entry voluntarily until patent expiration. A Paragraph IV certification challenges the listed patent as invalid or not infringed by the generic product, potentially allowing approval and market entry before the patent expires. Only a Paragraph IV certification triggers the 30-month stay mechanism and potentially earns 180-day exclusivity.
2. How many Paragraph IV certifications does a drug typically attract?
The number varies widely by commercial attractiveness. A drug with $1 billion or more in annual sales will typically attract 5 to 20 or more Paragraph IV filers. Smaller-market drugs may see one or two challengers, or none at all if the litigation economics do not support a challenge. FDA does not publish a centralized list of pending Paragraph IV certifications, but the patent infringement suits filed in response to notice letters are public records.
3. Can a brand company list new patents in the Orange Book after an ANDA is filed?
Yes, brand companies can list new patents that issue after the original NDA approval, provided those patents meet the Orange Book listing criteria. When a new patent is listed after an ANDA is already pending, the ANDA applicant must amend its application to certify to the new patent. However, per the 2003 MMA amendments, a brand company can only receive one 30-month stay per ANDA, so listing new patents after the stay has already been triggered does not generate additional stays.
4. What is tentative approval and how does it affect generic launch timing?
Tentative approval is FDA’s notification that an ANDA is otherwise approvable but cannot receive final approval due to existing patent or exclusivity protections. A tentatively-approved ANDA applicant is fully ready to launch upon expiration of the blocking period and does not need to await any additional FDA review. For 180-day exclusivity purposes, tentative approval within 30 months of filing is required to avoid forfeiture.
5. What happens when a generic wins a Paragraph IV challenge after the 30-month stay expires?
If the 30-month stay expires before the district court issues a final judgment, FDA can grant final approval to the ANDA and the generic can launch ‘at risk,’ meaning it enters the market while the litigation continues. If the brand company then wins the patent suit, the generic may face damages for sales during the at-risk period. At-risk launches are commercially significant decisions that generic companies make based on their assessment of the patent’s vulnerability and the likely damages exposure.
6. How does orphan drug exclusivity interact with Paragraph IV challenges?
Orphan drug exclusivity bars FDA approval of the same drug for the same rare disease indication for seven years. A generic company can file a Paragraph IV certification for a drug with orphan exclusivity, but FDA cannot grant final approval for the protected indication until the seven years expire, regardless of the patent challenge outcome. The generic can seek approval for a different indication if one is approved without orphan protection, but this limits commercial utility for drugs where the orphan indication is the primary use.
7. What is a ‘skinny label’ ANDA and when is it used?
A skinny-label ANDA is an application that omits from the generic labeling a method-of-use indication that is covered by a valid, unexpired Orange Book patent. By omitting the patented indication, the applicant can file a Paragraph III certification (agree to wait for patent expiry) for that patent while proceeding with Paragraph IV certifications for other patents. The strategy allows earlier approval and entry for non-patented uses, while technically avoiding direct infringement of the method-of-use patent. Post-GSK v. Teva, the induced infringement risk of skinny labeling requires careful legal analysis.
8. How does the FDA decide which ANDA gets 180-day exclusivity when multiple companies file the same day?
All ANDAs that are substantially complete and contain a Paragraph IV certification, submitted on the same calendar day, are treated as simultaneous first filers and share the 180-day exclusivity. Each shares the exclusivity window equally, meaning all can launch on the same day when the exclusivity window opens. Shared exclusivity reduces the commercial value of the exclusivity period because of immediate price competition among multiple generic entrants.
9. Can a settlement of Hatch-Waxman litigation require FDA action to be enforceable?
Most Hatch-Waxman settlements are contracts between private parties that do not require FDA to take any action. The settlement specifies when the generic company may launch and may include a license to the brand’s patents. If the settlement includes an authorized generic component, the brand company must have the commercial capability to market an authorized generic product, but FDA approval is not required for this because the brand’s NDA already covers the product. The FTC must be notified of certain pharmaceutical patent settlements under the Medicare Modernization Act, but notification is not approval.
10. How long does it typically take to resolve a Paragraph IV case in the District of Delaware?
Cases that are not settled typically take 18 to 36 months from filing of the complaint to a trial decision, with an additional 12 to 18 months for Federal Circuit appeal. Many cases settle within 12 to 18 months of filing, particularly after claim construction rulings clarify the legal landscape. The total timeline from notice letter to final judicial resolution, in the rare cases that go that route, can therefore span four to five years, often outlasting the 30-month stay that triggered the litigation.
References
Association for Accessible Medicines. (2022). 2022 Generic Drug & Biosimilar Access & Savings Report. Association for Accessible Medicines.
Drug Price Competition and Patent Term Restoration Act of 1984, Pub. L. No. 98-417, 98 Stat. 1585 (1984).
Medicare Prescription Drug, Improvement, and Modernization Act of 2003, Pub. L. No. 108-173, 117 Stat. 2066 (2003).
FTC v. Actavis, Inc., 570 U.S. 136 (2013).
Federal Trade Commission. (2011). Authorized Generic Drugs: Short-Term Effects and Long-Term Impact. U.S. Federal Trade Commission.
Federal Trade Commission. (2011). Emerging Health Care Issues: Follow-On Biologic Drug Competition. U.S. Federal Trade Commission.
Pfizer Inc. v. Apotex Inc., 480 F.3d 1348 (Fed. Cir. 2007).
GlaxoSmithKline LLC v. Teva Pharmaceuticals USA, Inc., 7 F.4th 1320 (Fed. Cir. 2021).
Sandoz Inc. v. Amgen Inc., 582 U.S. 1 (2017).
Biologics Price Competition and Innovation Act of 2009, Pub. L. No. 111-148, 124 Stat. 804 (2010).
Generating Antibiotic Incentives Now Act, Title VIII of the FDA Safety and Innovation Act, Pub. L. No. 112-144, 126 Stat. 1100 (2012).
Microsoft Corp. v. i4i Ltd. Partnership, 564 U.S. 91 (2011).
Mylan Pharmaceuticals Inc. v. Warner Chilcott Public Ltd. Co., 838 F.3d 421 (3d Cir. 2016).
U.S. Food and Drug Administration. (2023). Approved Drug Products with Therapeutic Equivalence Evaluations (Orange Book), 43rd ed. U.S. Department of Health and Human Services.
U.S. Food and Drug Administration. (2023). Generic Drug User Fee Amendments: GDUFA III Commitment Letter. U.S. Department of Health and Human Services.
Hemphill, C. S., & Sampat, B. N. (2012). Evergreening, patent challenges, and effective market life in pharmaceuticals. Journal of Health Economics, 31(2), 327–339.
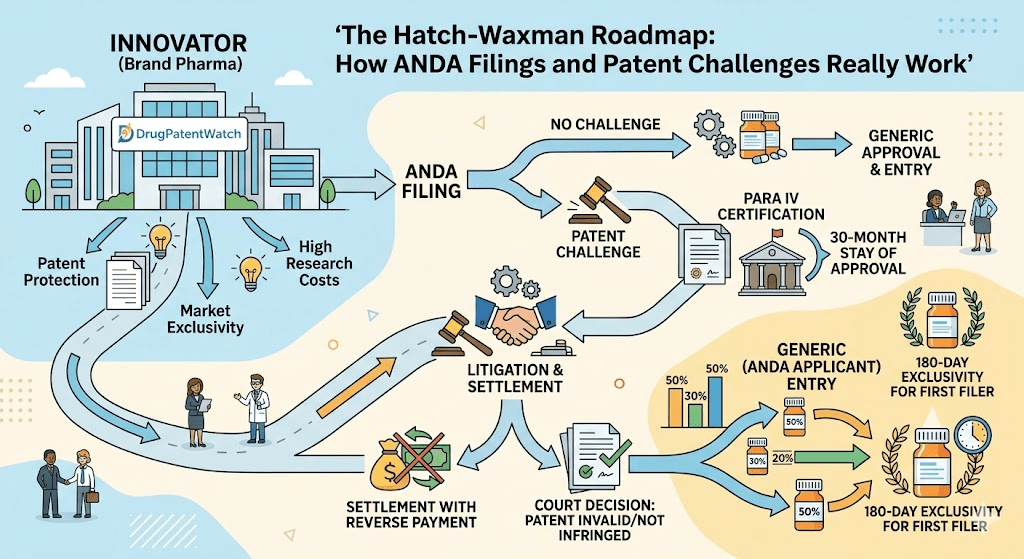



















")






