Every day a blockbuster drug holds patent protection is worth, on average, $8.2 million in revenue. That figure makes the patent expiration date the single most consequential number in pharmaceutical finance, more important than a trial readout, more watched than an FDA PDUFA date, and infinitely more litigated than any other line in a company’s IP portfolio. Patent expiration is not just a legal event; it is the moment a company’s most durable cash flow converts from a monopoly into a commodity.
This guide exists because calculating that date is not arithmetic. It is a discipline spanning patent prosecution strategy at the USPTO, regulatory maneuvering at the FDA, courtroom outcomes in the Federal Circuit, and contractual settlements negotiated in the shadow of antitrust law. Getting it wrong costs companies billions. Getting it right, and knowing how to push it forward, is one of the most defensible sources of alpha available to both pharmaceutical executives and the institutional investors who fund them.
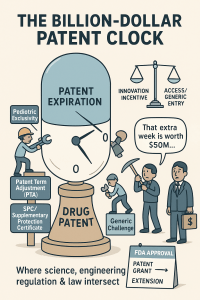
Section 1: The 20-Year Baseline and Why It Lies to You
Under 35 U.S.C. § 154(a)(2), a U.S. utility patent carries a term of 20 years from its earliest effective filing date. The TRIPS Agreement (Article 33) codifies this same minimum for all 164 WTO member nations, creating a nominally harmonized global standard. That uniformity is, in practice, largely illusory for pharmaceutical assets.
The 20-year clock starts on the filing date of the earliest application in a patent’s priority chain, which for a new drug is almost always a provisional or PCT application filed during discovery or early preclinical research. At that moment, the compound has never touched a human. It will not touch one for years. Before it can generate a dollar of revenue, it must survive Phase I, II, and III trials averaging 6 to 10 years combined, an NDA or BLA review that the FDA’s own data shows takes 10 months for standard review and 6 months for Priority Review on standard applications, and the administrative latency between approval and commercial launch.
The result is a structural erosion of effective patent life that the industry calls the ‘patent gap.’ By the time a new molecular entity reaches the market, its primary composition-of-matter patent typically has 7 to 12 years of life remaining. For rare disease drugs that cleared accelerated pathways, the remaining term can be shorter still, because the trial periods that drain the patent clock were front-loaded and compressed.
This erosion is not incidental. It is the core economic challenge that the entire legislative apparatus of Hatch-Waxman, the PTA statute, and the PTE statute was built to partially address. ‘Partially’ is the operative word. No legal mechanism fully restores the lost time.
The Pre-1994 Regime and Why It Changed
Prior to the Uruguay Round Agreements Act of 1994, U.S. patents ran 17 years from the date of grant, not 20 years from filing. Under that system, delays during USPTO examination were economically irrelevant to the patentee. Every month the application sat pending was a month pushed to the back of a 17-year term that had not yet started. Patent applicants and their counsel had little incentive to expedite prosecution.
The shift to ’20 years from filing’ changed the incentive structure entirely. Now examination delay directly eats into the patent term. A pharmaceutical company waiting 36 months for a first Office Action loses 36 months from the back end of its patent. This is precisely why Congress created Patent Term Adjustment in 35 U.S.C. § 154(b), a compensatory mechanism for USPTO-caused delay that we will dissect in full.
Key Takeaways: Section 1
The nominal 20-year term is a planning fiction for drug patents. The number that matters to an IP team, a portfolio manager, or a licensing negotiator is the effective market exclusivity period, which requires calculating the patent’s actual remaining life at NDA/BLA approval, layering on any PTA and PTE, and then offsetting with regulatory exclusivities. That calculation, not the patent filing date, is the foundation of every pharmaceutical valuation model worth building.
Section 2: Hatch-Waxman’s Architecture: The Grand Bargain, Dissected
The Drug Price Competition and Patent Term Restoration Act of 1984, universally called the Hatch-Waxman Act, restructured the economics of pharmaceutical competition more thoroughly than any piece of legislation before or since. Its core mechanism was an explicit trade. Innovator companies received a legal pathway to restore patent term lost during FDA review. Generic manufacturers received a streamlined approval pathway that eliminated the need to repeat costly clinical trials.
Neither side got everything it wanted. Both sides got enough to create a functioning market. That compromise has generated both $1.2 trillion in generic drug savings (FTC estimates through 2019) and a pharmaceutical R&D ecosystem that continues to produce novel therapeutics. Whether the balance is correctly calibrated is a political question this guide does not address. What matters here is how the machinery works.
The ANDA Pathway and Bioequivalence as the Generic Engine
The Abbreviated New Drug Application (ANDA), authorized by 21 U.S.C. § 355(j), allows a generic manufacturer to obtain FDA approval by demonstrating bioequivalence to an already-approved reference listed drug (RLD), rather than repeating the full clinical program. Bioequivalence is typically established by showing that the generic product’s pharmacokinetic parameters (Cmax and AUC) fall within an 80-125% confidence interval of the reference product’s when administered at the same molar dose.
This pathway eliminated a genuine market failure. Before 1984, generic firms had to conduct their own Phase I through III programs even for identical molecular copies of approved drugs, a cost and time barrier so high that generic penetration of off-patent markets was negligible. Hatch-Waxman converted generic entry from a capital-intensive R&D problem into a manufacturing and regulatory execution problem, reducing barriers to entry by orders of magnitude.
The 35 U.S.C. § 271(e)(1) ‘safe harbor’ provision accompanied the ANDA pathway. It protects generic manufacturers from infringement liability for activities ‘reasonably related’ to obtaining regulatory approval, including manufacturing clinical supplies and conducting bioequivalence studies before patent expiration. Without this provision, a generic company preparing to launch on day one of patent expiration would be infringing the patent during its preparation phase. The safe harbor makes ‘at-risk’ preparation legally viable.
Patent Term Extension: The Innovator’s Compensation
35 U.S.C. § 156 provides the PTE mechanism as the innovator’s half of the bargain. Its purpose is straightforward: a patent whose commercial life was truncated by mandatory FDA review receives a partial restoration of that lost time. The statute is specific about what types of regulatory review qualify, what formula governs the calculation, and what caps apply. We address each of those elements in full in Sections 7 through 9.
Key Takeaways: Section 2
Hatch-Waxman is not a consumer protection statute and it is not an innovation protection statute. It is both simultaneously, which is why every policy debate about drug pricing that touches generic entry timelines eventually collides with Hatch-Waxman mechanics. IP teams must understand the Act’s dual purpose because it shapes both the rights they hold and the constraints on how they can assert them.
Section 3: The Orange Book as a Competitive Weapon
The FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, known universally as the Orange Book, is the operational center of all Hatch-Waxman patent strategy. Under 21 U.S.C. § 355(b)(1), an NDA applicant must submit to the FDA information on every patent that claims the drug substance, the drug product, or an approved method of use, and for which a patent infringement claim could reasonably be asserted against an unauthorized manufacturer. The FDA lists those patents in the Orange Book without substantive review of their validity or scope.
This listing process is the first offensive move in pharmaceutical patent strategy. A patent listed in the Orange Book triggers mandatory interaction from any generic applicant. A patent not listed is invisible to the Hatch-Waxman machinery, which means it cannot generate the 30-month litigation stay that provides the brand company with its most valuable procedural protection. Innovator IP teams approach Orange Book listing decisions with the same rigor they apply to claim drafting.
What Qualifies for Orange Book Listing
The FDA’s regulations at 21 C.F.R. § 314.53 specify three categories of listable patents: drug substance patents (covering the active moiety or active ingredient), drug product patents (covering formulations, compositions, or delivery systems), and method of use patents (covering a specific FDA-approved indication). Process patents, metabolite patents, and patents covering intermediates used in synthesis do not qualify for listing.
The method of use listing is particularly strategic. A company can list a method of use patent that covers only a subset of the drug’s approved indications. A generic applicant seeking approval only for an indication not covered by that patent can then file a ‘carve-out’ label (formally, a Section viii statement) rather than a Paragraph IV certification, avoiding the litigation trigger entirely. This asymmetry creates a complex game between innovators expanding their method of use coverage and generic applicants crafting narrowly scoped ANDA labels.
The Purple Book: Its Biologic Counterpart
The Orange Book applies only to small molecule drugs approved under the Federal Food, Drug, and Cosmetic Act via the NDA pathway. Biologics approved under the Public Health Service Act via BLA operate under a separate regime governed by the Biologics Price Competition and Innovation Act (BPCIA) of 2009. The FDA maintains the Purple Book as the analogous listing of approved biological products and their reference status, though the BPCIA patent resolution mechanism (the ‘patent dance’) operates through a direct exchange between the originator and biosimilar applicant, not through a centralized Orange Book-style listing and certification system. We address the BPCIA patent dance in Section 16.
Key Takeaways: Section 3
Every decision about which patents to list in the Orange Book, when to list them, and at what scope, is a strategic choice with direct financial consequences. Listing too broadly invites litigation challenges that might invalidate the listed patent. Listing too narrowly leaves protection gaps a generic can exploit. The Orange Book listing strategy is inseparable from the lifecycle management strategy.
Section 4: Paragraph IV Filings: Mechanics, Timelines, and Strategic Intent
When a generic company files an ANDA, it must certify its position with respect to each Orange Book-listed patent for the reference drug. The four possible certifications are: (I) no patent information has been filed; (II) the patent has expired; (III) the patent will expire on a stated date, with the generic agreeing not to enter until then; and (IV) the listed patent is invalid, unenforceable, or will not be infringed by the generic’s proposed product.
The Paragraph IV (PIV) certification is, by design, a litigation trigger. Congress declared it a ‘constructive act of infringement’ under 35 U.S.C. § 271(e)(2), allowing the patent holder to sue before any infringing product reaches the market. This artificial infringement doctrine is the mechanism that makes pre-market patent resolution possible under Hatch-Waxman.
The 30-Month Stay: Mechanics and Limits
Upon receipt of a PIV Notice Letter (which the generic must send to the NDA holder and patent owner within 20 days of the FDA accepting the ANDA for filing), the innovator has 45 days to file suit. If suit is filed within that window, the FDA is automatically prohibited from granting final ANDA approval for 30 months from the date the NDA holder received the Notice Letter, or until the patent dispute is resolved in the generic’s favor, whichever comes first.
The 30-month stay is automatic and requires no court action. It provides the innovator with approximately 2.5 years to litigate the patent’s validity and scope before facing commercial generic competition. The 2003 Medicare Prescription Drug Improvement and Modernization Act (MMA) limited each listed patent to a single 30-month stay, eliminating the multi-stay strategy that some brands had used to serially relist patents and trigger successive stays against the same generic applicant.
180-Day First-Filer Exclusivity: Value and Forfeiture
The first ANDA applicant to file a substantially complete ANDA with a PIV certification against a given patent earns 180 days of marketing exclusivity. During this window, the FDA cannot approve any subsequent ANDA for the same drug. This exclusivity is enormously valuable: a first-filer operating as the sole generic alternative to a brand drug typically captures substantial market share at a price point meaningfully above what will prevail once multiple generics enter.
IMS Health (now IQVIA) data has consistently shown that first-filer generics capture 75 to 85% of prescription volume within the first 90 days and price at 15 to 30% below brand. Once a second generic enters, pricing quickly moves to 70 to 80% discounts from brand WAC. The financial difference between being the first and second filer in a major market can reach hundreds of millions of dollars over the 180-day period.
The 180-day exclusivity is subject to forfeiture under multiple statutory conditions, including failure to market within a specified period after receiving FDA approval or court judgment, and entering into an agreement that the FTC determines is anticompetitive. The forfeiture provisions were designed to prevent ‘parking’ of 180-day exclusivity, a practice where first-filers delayed market entry indefinitely to maintain leverage in settlement negotiations with brand companies.
Investment Strategy Note: Tracking PIV Filings
Institutional investors in generic pharmaceutical companies should track PIV filings as leading indicators of near-term revenue events. A company that files a PIV certification against a major drug, achieves first-filer status, and successfully litigates or settles its way to a favorable entry date has a defined, high-probability revenue event. Platforms such as DrugPatentWatch make these filings traceable in real time, allowing analysts to identify first-filer positions in their early stages.
Section 5: Patent Term Adjustment (PTA): Clawing Back USPTO Delay
Patent Term Adjustment exists to solve a specific problem: under the ’20 years from filing’ regime, every day the USPTO takes to examine and issue a patent is a day subtracted from the back end of the patent’s commercial life. 35 U.S.C. § 154(b) provides that a patent shall be adjusted for delays beyond specified timeframes that are attributable to the USPTO, minus any delay attributable to the applicant. The final PTA appears as a number of days added to the 20-year term on the face of the issued patent.
For pharmaceutical patents with long prosecution histories, PTA can be substantial. Extensions of 400 to 900 days are common for complex NDA-stage patents that were prosecuted through multiple Office Actions and responses over several years. Each day of PTA translates directly into an additional day of market exclusivity at the back end of the patent’s life, where the drug is generating peak revenue in a competitive market just prior to generic entry.
The PTA Formula: (A + B + C) Minus Overlap Minus Applicant Delay
The calculation proceeds under 35 U.S.C. § 154(b)(1):
A Delays accrue when the USPTO misses specific response deadlines: failure to issue a first Office Action within 14 months of filing; failure to respond to an applicant’s submission within 4 months; failure to act within 4 months after a PTAB or court decision where at least one claim was found allowable; and failure to issue the patent within 4 months after payment of the issue fee. Each missed deadline generates a day-for-day A delay.
B Delays accrue at a rate of one day per day for each day the application remains pending beyond three years from the actual filing date, subject to exclusions for time consumed by a Request for Continued Examination (RCE), an interference or derivation proceeding, a secrecy order, and appellate review. The B delay provision ensures that even if A delays are modest, an exceptionally long pendency still generates compensatory PTA.
C Delays accrue for time consumed by interferences, derivation proceedings, secrecy orders, and successful PTAB or court appeals that reverse adverse patentability determinations. These are relatively uncommon but can generate substantial PTA for the patents involved.
The statute provides that to the extent these categories of delay ‘overlap,’ the adjustment reflects only the actual net delay. This overlap provision was the subject of the most consequential PTA litigation in the statute’s history.
Applicant Delay: The Self-Inflicted Reduction
Under 37 C.F.R. § 1.704, any period of delay attributable to the applicant is subtracted from accumulated PTA on a day-for-day basis. The most common sources of applicant delay are: taking longer than three months to respond to an Office Action (beyond the three-month safe harbor, any extension of time counts as applicant delay); submitting a preliminary amendment that requires a supplemental Office Action; filing a reply that contains an omission; and submitting a supplemental reply not requested by the examiner.
The three-month response practice is particularly important to manage. Every month beyond three that a response takes adds directly to the applicant delay reduction. For a prosecution that spans 48 months with multiple Office Actions, the cumulative impact of repeatedly taking four- and five-month response periods can wipe out hundreds of days of earned PTA. Law firms handling pharmaceutical prosecution for sophisticated clients manage this at the docket level.
Key Takeaways: Section 5
PTA is not a passive benefit. It must be actively earned through diligent prosecution and carefully preserved by avoiding applicant delay. Companies that allow prosecution to drift, take liberal extensions, or file unnecessary preliminary amendments forfeit PTA that could represent millions of dollars in extended exclusivity. Every extension-of-time request during prosecution should be evaluated against its expected impact on the PTA balance.
Section 6: Wyeth v. Kappos and Novartis v. Lee: How Litigation Rewrote the PTA Rulebook
The USPTO spent years applying a methodology for calculating PTA that was favorable to the agency’s administrative efficiency and adverse to patentees. Two Federal Circuit decisions dismantled that methodology and forced a recalibration that added significant patent life to thousands of pharmaceutical patents.
Wyeth v. Kappos (2010): Rejecting the ‘Greater Of’ Approach
Before 2010, the USPTO calculated PTA under an interpretation that treated the A and B delay periods as largely mutually exclusive. The agency’s position was that because the B clock measures total pendency beyond three years and the A clock measures specific communication failures, any A delays that occurred within the three-year period were ‘overlapping’ with the B period and should not be added separately. In practice, the USPTO awarded patentees the greater of their total A delays or their total B delays, not the sum.
The Federal Circuit rejected this methodology in Wyeth v. Kappos, 571 F.3d 1367 (Fed. Cir. 2010). The court held that the statutory text is clear: B delays, by definition, cannot begin to accrue until the three-year anniversary of filing. Therefore, A delays that occur in the first three years of prosecution cannot ‘overlap’ with B delays that have not yet started. The correct method is to add all A delays that accrued in the first three years to all B delays that accrued after year three (plus any A delays that occurred after year three), then subtract genuine overlap where both types of delay were accruing simultaneously.
The financial impact was immediate. The USPTO had to rewrite its PTA calculation software and issue revised PTA figures for potentially tens of thousands of patents. For pharmaceutical companies, the recalculated PTA added months of additional exclusivity to patents they had already assumed were properly calculated. This decision created a one-time windfall for innovator companies and a corresponding delay for generic entry timelines that had been modeled on the pre-Wyeth figures.
Novartis v. Lee (2014): The RCE Clock-Pause Rule
The 2014 decision in Novartis AG v. Lee, 740 F.3d 593 (Fed. Cir. 2014), addressed how the filing of a Request for Continued Examination (RCE) interacts with B delay accrual. The USPTO’s position was that an RCE permanently tolls the B delay clock for all time after the RCE’s filing, including the period between allowance of the application and actual issuance of the patent.
The Federal Circuit split the question. The court agreed that time during which an application is under active continued examination after an RCE is appropriately excluded from B delay accrual, the USPTO’s position on that point. But the court held that examination ends, presumptively, when a notice of allowance is mailed. The period between the mailing of the notice of allowance and the patent’s issuance date is not ‘time consumed by continued examination.’ Therefore, the B delay clock restarts on the date the notice of allowance is mailed and runs until the patent issues.
For applications prosecuted through one or more RCEs, this ruling added back weeks to months of B delay PTA. The practical significance is highest for pharmaceutical patents that spend extended periods in allowance-to-issuance limbo while final fees are processed and printing queues are cleared.
The Ongoing Imperative for Independent PTA Audits
The lesson from Wyeth and Novartis is not just historical. The USPTO’s automated PTA calculation system operates on programmed rules that can diverge from the latest statutory interpretation when the law is unclear or evolving. A company that passively accepts the PTA printed on its issued patent is relying on an algorithm that may not reflect current case law.
For patents covering high-revenue pharmaceutical products, an independent PTA audit by specialized patent counsel is standard practice. If the independent calculation differs from the USPTO’s figure, the patentee can petition the USPTO to reconsider under 37 C.F.R. § 1.705(b), generally within two months of the patent’s issuance. A successful petition can add months to the patent’s term. For a drug generating $3 billion annually, a single additional month of exclusivity is worth approximately $250 million. The cost of the audit is trivial by comparison.
Key Takeaways: Section 6
Wyeth and Novartis established that the USPTO’s PTA calculations are not authoritative. They are a starting point. Any pharmaceutical company that treats the USPTO’s printed PTA figure as final without independent verification is leaving money on the table, potentially hundreds of millions of dollars for major products.
Section 7: Patent Term Extension (PTE): Reclaiming the FDA Years
Patent Term Extension under 35 U.S.C. § 156 is the most direct response to the fundamental economic problem of pharmaceutical patents: the FDA requires years of clinical development and regulatory review before a drug can be sold, and those years consume the patent term at full speed while generating no revenue. PTE partially compensates for this by restoring a calculated portion of the ‘regulatory review period’ (RRP) to the patent’s term, allowing the patent to expire later than its PTA-adjusted date would otherwise indicate.
The word ‘partially’ is load-bearing. PTE does not fully restore all lost time. The statute’s formula gives only half-credit for clinical trial time and full credit for FDA review time, and then imposes two caps that, in many cases, reduce the award below even that partial restoration. For drugs that move quickly through development, the 14-year post-approval cap is often the binding constraint.
Eligibility Requirements
Before the PTE formula applies, the patent must clear four eligibility gates under 35 U.S.C. § 156(a):
First, the patent must claim the approved drug product, a method of using it, or a method of manufacturing it. Second, the patent must not have previously been extended under this section (only one PTE per patent). Third, the product’s first permitted commercial marketing or use must be the approval under review. This means a patent covering a reformulation of a previously approved active ingredient generally does not qualify for PTE based on the reformulation’s approval, because the active ingredient has already been approved. Fourth, the patent must have some remaining term at the time of FDA approval.
One PTE Per Product
A critical constraint applies even before the formula: only one patent per approved product may receive PTE for any given regulatory approval. If multiple patents cover the approved drug, the patent holder must elect which single patent to extend. This election decision is among the most consequential a pharmaceutical IP team makes. The strategic analysis involves comparing the remaining terms, claim breadth, litigation vulnerability, and competitive entry dynamics of each candidate patent.
A company may file multiple PTE applications within 60 days of FDA approval to preserve its options, then make the final election when the USPTO requests it. The 60-day filing window from the approval date is a hard deadline. Missing it forfeits the right to PTE for that approval event.
Section 8: The PTE Formula, Step-by-Step, with a Full Worked Case
The PTE calculation proceeds in five stages: defining the regulatory review period, calculating initial creditable time, applying deductions, applying the statutory caps, and determining the final extension.
Defining the Regulatory Review Period (RRP)
The RRP has two components:
The Testing Phase runs from the effective date of the IND application to the date the NDA or BLA is initially submitted. The IND becomes effective 30 days after submission absent a clinical hold, so the testing phase clock starts approximately one month after the IND is filed. This phase captures the duration of the clinical development program.
The Approval Phase runs from the NDA or BLA submission date to the date of FDA marketing approval. This phase captures the FDA’s own review time.
Calculating Initial Creditable Time
The formula credits one-half day per day of the Testing Phase and one full day per day of the Approval Phase:
Initial Credit = (Testing Phase Days x 0.5) + (Approval Phase Days x 1.0)
Applying the Pre-Grant Deduction
The statute does not allow credit for any portion of the RRP that occurred before the patent was issued. For the Testing Phase (which is credited at half-time), the pre-grant portion is first identified, then that number of days is multiplied by 0.5, and the result is subtracted from the initial credit. For any Approval Phase time that occurred before the patent grant, those days are subtracted on a 1:1 basis.
Applying the Due Diligence Deduction
If the applicant failed to act with due diligence during the RRP, the period of non-diligence is subtracted day-for-day. The FDA makes this determination. Companies with documented, contemporaneous records of their FDA interactions are far better positioned to defend their diligence than companies relying on reconstructed timelines.
Full Worked Case: Hypothetical Drug ‘Carvelox’
Given Data:
IND Effective Date: January 15, 2015
Patent Grant Date: September 1, 2017
NDA Submission Date: March 10, 2020
NDA Approval Date: February 8, 2022
Due Diligence Delays: 0 days
PTA-Adjusted Patent Expiration: November 14, 2037
Step 1: Duration of RRP Phases
Testing Phase = NDA Submission (March 10, 2020) minus IND Effective (January 15, 2015) = 1,881 days
Approval Phase = NDA Approval (February 8, 2022) minus NDA Submission (March 10, 2020) = 700 days
Step 2: Initial Creditable Time
Testing Credit = 1,881 x 0.5 = 940.5 days (round down to 940 per USPTO practice) Approval Credit = 700 x 1.0 = 700 days Total Initial Credit = 1,640 days
Step 3: Deductions
Pre-Grant Testing Period = Patent Grant (September 1, 2017) minus IND Effective (January 15, 2015) = 959 days Pre-Grant Testing Deduction = 959 x 0.5 = 479.5 days (round down to 479 days)
Adjusted Credit = 1,640 – 479 – 0 = 1,161 days
Step 4: Apply Statutory Caps
Cap 1 (5-Year Maximum): The 5-year cap is 1,826 days. Our 1,161-day credit is below this cap; Cap 1 is not binding.
Cap 2 (14-Year Post-Approval): The remaining patent term at approval is calculated as: PTA-Adjusted Expiry (November 14, 2037) minus Approval Date (February 8, 2022) = approximately 5,758 days (15.77 years). The maximum extension permitted by the 14-year cap is: (14 x 365.25) = 5,114 days minus 5,758 days = negative. When the remaining patent term at approval already exceeds 14 years, the 14-year cap allows zero extension.
Step 5: Final Determination
The governing constraint is the 14-year post-approval cap. Because Carvelox’s patent had approximately 15.77 years of remaining term at the time of approval, the 14-year cap permits no PTE whatsoever, despite the drug spending over 7 years in clinical and regulatory development.
This outcome is counterintuitive but consistent with the statute and illustrates a critical planning insight: a drug that is protected by a patent filed relatively late in its development cycle, and therefore retains a long remaining term at approval, derives zero benefit from PTE regardless of how long it spent in clinical trials. The PTE mechanism is structurally most valuable for drugs protected by patents filed very early in development, where the remaining patent term at approval has been substantially depleted.
Section 9: The Statutory Caps: Why the 14-Year Rule Hits Harder Than the 5-Year Cap
The 5-year maximum extension cap under 35 U.S.C. § 156(c)(3) receives most of the attention in PTE discussions, but in practice the 14-year post-approval cap under 35 U.S.C. § 156(c)(3)(B) is more frequently the binding constraint. Understanding when each cap applies requires modeling the full term profile of each candidate patent before making the PTE election.
The 14-year cap reflects a policy judgment that the total marketable life of a drug under a single extended patent should not exceed 14 years from FDA approval. Congress was concerned that PTE, without this limit, could allow drugs with long remaining patent terms to receive additional extensions that were economically unjustified by the rationale of compensating for regulatory delay. The cap effectively penalizes late-filed patents, which retain more of their 20-year term by the time of approval.
The practical implication for lifecycle management is significant. Companies that file continuation patents on improved formulations late in a drug’s development cycle, after the original composition-of-matter patent has already been substantially eroded, may find those continuation patents eligible for larger PTE awards (or at least not blocked by the 14-year cap) even if the continuation was not the patent the company would have chosen to extend for other strategic reasons.
Strategic Patent Filing Timing and PTE Optimization
PTE optimization is best addressed proactively, not retroactively. The IP team’s filing strategy for a new drug candidate should explicitly model the expected approval date and remaining patent term at that date for each patent in the portfolio. Where the primary composition-of-matter patent is filed early in development, it will likely hit the 14-year cap. Secondary patents on formulations or delivery systems, filed later in development, may receive larger PTE awards.
This creates a strategic tension: composition-of-matter patents filed early provide the broadest protection during development (when they are not yet generating revenue) but may receive limited PTE. Formulation patents filed later may receive more PTE but offer narrower claims. Sophisticated IP teams design their filing sequences to optimize across both dimensions simultaneously.
Section 10: FDA Regulatory Exclusivities: The Parallel Universe
Patents and FDA regulatory exclusivities both restrict generic and biosimilar entry, but they arise from different laws, are administered by different agencies, and protect different interests. A patent protects an invention and is enforced through the courts. An exclusivity protects a regulatory approval and is enforced by the FDA’s own approval policies.
The two systems can run concurrently, sequentially, or in tension. A drug may lose patent protection but remain shielded by exclusivity, or may have exclusivity expire while substantial patent term remains. Accurately modeling the first date of possible generic or biosimilar entry requires tracking both systems simultaneously.
New Chemical Entity (NCE) Exclusivity
Five-year NCE exclusivity under 21 U.S.C. § 355(c)(3)(E)(ii) attaches to the first NDA approval for a drug containing an active moiety that has never before been approved by the FDA. ‘Active moiety’ means the molecule responsible for the drug’s pharmacological effect, excluding any ester, salt, or other noncovalent derivative. During the five-year exclusivity period, the FDA cannot accept an ANDA for filing. After four years, an ANDA containing a Paragraph IV certification can be filed, allowing the litigation clock to start before the exclusivity expires.
NCE exclusivity is one of the most commercially significant protections in the FDA’s toolkit because it operates on top of any patent protection and applies regardless of whether the drug is patented at all. A drug with weak patent coverage but strong NCE exclusivity still has five years of market protection from the FDA’s side.
Orphan Drug Exclusivity (ODE)
Seven-year ODE under 21 U.S.C. § 360cc attaches to FDA approval of a drug for a disease or condition affecting fewer than 200,000 persons in the United States, or for which there is no reasonable expectation that the cost of developing and making the drug will be recovered from U.S. sales. During the seven-year period, the FDA cannot approve another application for the same drug for the same orphan indication. The protection is indication-specific: a second company could obtain approval of the same drug for a different orphan indication.
ODE has been the subject of significant debate over its interaction with biologics and whether it blocks biosimilar approval. FDA has taken the position that ODE for biologics blocks the approval of a ‘same’ biological product, and that biosimilars can differ sufficiently from the reference biologic to avoid the ODE block even for the same indication.
Pediatric Exclusivity: The Six-Month Multiplier
Pediatric exclusivity under 21 U.S.C. § 505A is the most operationally unusual exclusivity in the FDA’s portfolio. It does not create a stand-alone exclusivity period. Instead, it attaches to and extends every existing patent and exclusivity for the active moiety by six months. A drug with five patents and two exclusivity periods that earns pediatric exclusivity gets six months added to each of those seven protections simultaneously.
For a major branded drug protected by 10 or more patents, pediatric exclusivity can shift the entire loss-of-exclusivity timeline by half a year across every listed patent. The FTC has estimated that pediatric exclusivity delays on major drugs cost payers and consumers hundreds of millions of dollars per drug per six-month extension. Companies respond that the pediatric study data required to earn the exclusivity provides genuine clinical value by establishing dosing and safety information for pediatric populations.
The FDA initiates the process by issuing a Written Request (WR) specifying the studies it wants conducted. The sponsor has the right to accept or decline. If the sponsor accepts and conducts the studies to the FDA’s satisfaction, the six-month exclusivity attaches regardless of whether the studies show benefit in pediatric populations.
Three-year exclusivity under 21 U.S.C. § 355(c)(3)(E)(iii) attaches to approval of an NDA supplement or new NDA that required submission of new clinical investigations (other than bioavailability studies) essential to approval. It protects only the new information, not the underlying drug. A generic can reference the original approval data freely; it is blocked only from relying on the new clinical data for the new indication, new dosage form, or new formulation.
This exclusivity is the standard tool for protecting lifecycle management investments in reformulation, new indications, and combination products. A company that spends $200 million conducting Phase III trials for a new indication of an existing drug earns three years of protection for that indication, during which no generic can obtain approval for the same new indication by relying on the sponsor’s new clinical data.
Biologics Exclusivity Under BPCIA
The BPCIA provides two layers of exclusivity for reference biologic products: four years of data exclusivity, during which no biosimilar application can be accepted by the FDA, and 12 years of market exclusivity, during which no biosimilar can be approved even if the application was accepted. The 12-year period has been the subject of ongoing debate. Several legislative proposals have sought to reduce it to 7 years, citing the enormous revenue opportunity for biosimilar manufacturers and the pricing burden on payers, but none has yet passed Congress.
Key Takeaways: Section 10
The exclusivity stack varies significantly by drug type. A standard small molecule NDA earns NCE exclusivity if the active moiety is new, three-year exclusivity for subsequent supplements, and potentially pediatric exclusivity if studies are completed. A biologic earns 12 years of BPCIA protection. An orphan drug earns seven years for the designated indication. Each of these layers interacts differently with patent protection and must be modeled separately.
Section 11: Exclusivity Stacking: How IP Teams Build a Moat
The most sophisticated pharmaceutical companies do not rely on a single patent or a single exclusivity period. They build what the academic literature calls a ‘multi-layered protection system’ where multiple patents and multiple exclusivities overlap in time, each filling the gaps left by the others. The goal is to ensure that no single court decision, regulatory development, or generic filing can simultaneously eliminate all protection.
A realistic protection stack for a major branded small molecule drug might include the following elements, all operating simultaneously during the drug’s commercial peak:
The primary composition-of-matter patent, covering the active ingredient itself, typically filed during discovery. This is the most valuable patent and the first target for generic PIV challenges. Its expiration, adjusted for PTA and PTE, defines the outer boundary of the protection period.
Formulation patents covering specific pharmaceutical compositions, delivery systems, or particle size specifications required to achieve the drug’s clinical profile. These are typically filed during Phase II or Phase III development, when the formulation is locked. They expire later than the composition-of-matter patent and are often harder to design around because the formulation is FDA-approved and changes require their own approval.
Method of treatment patents covering the specific therapeutic indications or patient populations for which the drug is approved. These are valuable for drugs with multiple indications because a generic seeking approval for a non-patented indication cannot carve out the method of use claim if the claim is drafted to cover the drug’s primary use.
NCE exclusivity, which runs for five years from approval and blocks ANDA filing entirely for four years. For a drug approved in year 12 of its composition-of-matter patent, the NCE exclusivity provides full protection for an additional four years beyond the patent’s capacity to block ANDA filings.
Pediatric exclusivity, which shifts every element of the above stack by six months.
The interplay creates a protection timeline that looks less like a single cliff and more like a series of steps, each step representing the expiration of one element of the stack. Managing the slope of those steps is the central task of pharmaceutical lifecycle management.
Section 12: IP Valuation: Quantifying Patent Term as a Balance Sheet Asset
For portfolio managers, M&A analysts, and licensing executives, the patent portfolio is not an intangible afterthought. It is the primary source of future cash flow protection and the most direct determinant of a branded drug’s terminal value. Valuing that portfolio requires translating legal analysis into financial projections with defensible assumptions.
The DCF Framework for Patent-Protected Revenue
The standard approach to valuing pharmaceutical patent protection is a risk-adjusted discounted cash flow (DCF) model where the patent term is the single most sensitive input. The model projects annual revenue through the period of market exclusivity, then models the revenue decline curve after generic entry. The shape of the post-LOE decline curve varies by drug type: small molecules typically lose 80-90% of their brand revenue within 12 months of first generic entry; biologics lose 30-70% over 18 to 36 months due to the slower adoption dynamics of biosimilar interchangeability.
The present value of each additional year of exclusivity is determined by the drug’s peak revenue and the discount rate applied to pharmaceutical cash flows. For a drug generating $4 billion annually, an additional year of exclusivity at a 12% WACC is worth approximately $3.6 billion in NPV terms. The PTA and PTE calculations that determine whether a patent expires in year X or year X+1 are therefore exercises in billion-dollar valuation.
IP Valuation in M&A Due Diligence
When a pharmaceutical company acquires another company or licenses a drug asset, the patent position is typically the most thoroughly scrutinized element of the deal. Due diligence must address at minimum: the accuracy of the PTA calculations printed on each key patent; the eligibility and optimal election strategy for PTE; the scope of Orange Book listings and any pending PIV certifications; the existence and timing of any regulatory exclusivities; and the litigation risk profile of the composition-of-matter and formulation patents.
A miscalculation in any of these areas can materially alter the value of the acquired asset. If the acquirer’s model assumes 10 years of remaining exclusivity but the actual exclusivity is 7 years due to an unchallenged PTA error or a missed PTE election, the NPV of the acquisition changes by billions of dollars. IP due diligence in pharma M&A is not a legal formality; it is the core of financial underwriting.
The Humira IP Estate: A Valuation Case Study
AbbVie’s adalimumab franchise (Humira) is the most cited example of IP portfolio construction in the pharmaceutical industry. The original composition-of-matter patent for adalimumab, U.S. Patent 6,090,382, was scheduled to expire in 2016. AbbVie pursued an aggressive strategy of filing continuation, divisional, and improvement patents covering formulations, dosing regimens, manufacturing processes, and new indications, accumulating over 130 U.S. patents.
The result was effective U.S. market exclusivity through January 2023, nearly seven years beyond the original patent expiration. During that extension period, Humira generated approximately $100 billion in global revenue. The IP value added by the secondary patent portfolio relative to the original composition-of-matter expiration date was on the order of $30 to 40 billion in U.S. revenue alone.
Biosimilar manufacturers including Amgen, Samsung Bioepis, and Sandoz had biosimilar products approved years before they could launch in the U.S., because AbbVie’s settlement agreements with each of them specified licensed entry dates that corresponded to the effective protection period of the secondary patent portfolio. Each settlement was a separate licensing agreement granting a specific U.S. entry date in exchange for the biosimilar manufacturer not continuing to litigate the secondary patents’ validity.
This outcome has generated substantial regulatory and legislative scrutiny. The FTC has characterized AbbVie’s patent accumulation strategy as a ‘patent thicket’ that delayed competition and inflated healthcare costs. AbbVie’s position is that each individual patent covers a genuine, separately patentable innovation. The legal and policy debate remains unresolved. The financial outcome is clear: the secondary patent portfolio was worth tens of billions of dollars.
Investment Strategy: IP Valuation Metrics for Analysts
Analysts covering pharmaceutical equities should track four patent-specific metrics for each major product in a company’s portfolio: the primary composition-of-matter patent’s PTA-adjusted and PTE-adjusted expiration date; the latest-expiring Orange Book-listed patent for each approved indication; the date and outcome of any pending PIV certifications; and the specific dates of all applicable regulatory exclusivities. The earliest date on which a generic can receive final ANDA approval, sometimes called the ‘ANDA approval date,’ is a more precise indicator of competitive exposure than the patent expiration date itself, because it accounts for the interaction of exclusivities and the 30-month stay.
Section 13: Lifecycle Management and Evergreening: Technology Roadmaps and Tactics
Lifecycle management (LCM) is the set of activities a pharmaceutical company undertakes to extend the commercial life of a drug franchise beyond the expiration of its primary composition-of-matter patent. The term covers a spectrum of activity ranging from genuine therapeutic innovation (new indications, pediatric formulations, combination products) to purely strategic IP maneuvers (minor formulation changes patented to extend protection). The industry uses ‘evergreening’ as a neutral description of the latter category, though critics use it pejoratively.
The LCM Technology Roadmap: From Molecule to Franchise
A well-constructed LCM roadmap should begin during Phase II development, not at the approach of patent expiration. The roadmap identifies the timeline of every patent and exclusivity in the portfolio, maps the competitive landscape of potential generic or biosimilar entrants, identifies clinical development opportunities that could generate new patents and exclusivities, and coordinates the IP strategy with the commercial strategy for the franchise.
The specific LCM tactics available to an innovator company include:
Extended-release formulations that reduce dosing frequency from multiple daily doses to once daily or less. These require their own Phase III trials demonstrating bioequivalence or superiority, NDA approval, and generate three-year exclusivity upon approval. The formulation patents covering the delivery system typically expire 5 to 8 years after the original composition-of-matter patent.
Fixed-dose combination products that pair the primary drug with a second agent in a single pill. FDCs require separate clinical programs demonstrating the contribution of each component and generate their own NDA approvals and three-year exclusivities. The combination patents expire independently of either component’s original patents.
New approved indications that expand the drug’s labeled use. Each new indication requires clinical trial data, generates three-year exclusivity, and potentially generates new method-of-use patents. For a drug with a broad therapeutic profile, new indications can add multiple layers of protection over time.
Enantiomer switches, where the original drug is a racemate and the company develops and patents the pharmacologically active enantiomer as a separate product. Escitalopram (Lexapro) was developed as the active enantiomer of citalopram (Celexa) as Celexa approached patent expiration, generating a new composition-of-matter patent and NCE exclusivity for the enantiomer. The tactic is well-established and controversial in equal measure.
Polymorph patents covering specific crystalline forms of the active ingredient that may offer improved stability, bioavailability, or manufacturability. These patents are among the most frequently challenged in IPR proceedings and PIV litigation because generic manufacturers often discover that the specific polymorph used in their manufacturing process differs from the patented form, creating a design-around opportunity.
Biologic Evergreening: Sequence Variants and Next-Generation Products
For biologics, evergreening mechanics differ from small molecule strategies because the composition-of-matter patents on large molecules are more complex and the regulatory pathway for biosimilars is structurally different from the ANDA pathway. The primary LCM tools for biologics include:
New sequence variants: Developing modified versions of the original biologic with altered receptor binding characteristics, improved half-life (through PEGylation or Fc region engineering), or reduced immunogenicity. These next-generation biologics may qualify as new biological entities with independent BPCIA exclusivity.
Device and delivery system patents: Biologics are frequently delivered via proprietary prefilled syringes, autoinjectors, or pen devices. Patents on these delivery systems are separate from the molecule patents and are typically listed in the Purple Book. A biosimilar manufacturer that develops a biosimilar product must use its own delivery device or obtain a license, and device patents can extend meaningful protection several years beyond the biologic’s molecule patents.
Indication expansion: Adding new indications to an existing biologic product generates new clinical data, new method-of-use patents, and three-year NDA exclusivities for each approved supplement, on a parallel timeline to the 12-year BPCIA exclusivity.
The strategic imperative for biologic manufacturers is to ensure that the next-generation product, whether a high-concentration formulation, a subcutaneous version of an intravenous drug, or a PEGylated derivative, reaches the market before or concurrent with the first biosimilar of the original product. If the next-generation product is demonstrably superior and has its own distinct IP position, the commercial franchise can survive biosimilar entry largely intact.
Section 14: Pay-for-Delay Settlements Post-Actavis: What the FTC Sees Now
The Supreme Court’s 2013 decision in FTC v. Actavis, Inc., 570 U.S. 136 (2013), held that reverse payment settlements in patent litigation are not immune from antitrust scrutiny. Before Actavis, brand and generic companies could settle PIV litigation by having the brand pay the generic a sum of money in exchange for the generic’s agreement to delay market entry. The payments were often in the hundreds of millions of dollars. The FTC estimated these arrangements cost U.S. consumers $3.5 billion annually.
Actavis changed the legal landscape by subjecting these settlements to rule-of-reason antitrust analysis. The Court held that a large, unjustified reverse payment is itself evidence that the brand company feared losing the patent case, and that the settlement was a mechanism to share monopoly profits at consumers’ expense. Since Actavis, the mechanics of settlement compensation have evolved significantly.
Explicit cash payments from brand to generic have declined sharply. The FTC’s monitoring reports show that alternative forms of value transfer have proliferated. These include ‘no-AG’ commitments, where the brand agrees not to launch an authorized generic during the first-filer’s 180-day exclusivity period, making that window far more profitable. A no-AG agreement can be worth hundreds of millions of dollars to the first-filer generic by ensuring it faces no price competition during its exclusivity period. The FTC has been clear that it views no-AG commitments as compensation equivalent to cash.
Quantity restrictions, where the settling generic agrees to sell only a specified maximum volume of product during an agreed-upon entry period before full launch, are another evolved form. The FTC has characterized quantity restrictions as a mechanism to maintain elevated prices by limiting supply, since a genuinely competitive market would not feature contractual volume caps.
Co-promotion deals and supply agreements have also appeared as potential compensation mechanisms. The FTC scrutinizes any business deal entered into by a brand and generic company contemporaneously with a patent settlement for evidence that the business deal’s value exceeds what would be arm’s-length terms, with any excess representing disguised reverse payment.
The practical result is that direct post-Actavis patent settlements still occur and still include entry dates, but the compensation structures are more sophisticated and harder to characterize as per se unlawful. Analysts modeling loss of exclusivity dates for drugs in PIV litigation must evaluate the probability and expected timing of a settlement, not just the litigation outcome, because a negotiated entry date is often the actual market outcome regardless of the patent’s legal validity.
Section 15: Biosimilar Entry Under BPCIA: The 12-Year Exclusivity Wall and the Patent Dance
The Biologics Price Competition and Innovation Act (BPCIA) created a framework for biosimilar approval that differs structurally from the Hatch-Waxman ANDA framework in ways that directly affect competitive entry timing, patent litigation mechanics, and the strategic value of biologic IP portfolios.
The 12-Year Exclusivity Barrier
The BPCIA’s 12-year market exclusivity period for reference biologics is the most consequential difference from the Hatch-Waxman framework. No biosimilar application relying on the reference biologic’s safety and efficacy data can receive FDA approval until 12 years after the reference biologic’s first licensure date. This period is not reducible by Orange Book-style PIV challenges or 30-month stays. It is an absolute bar on biosimilar approval, regardless of the state of patent litigation.
The 12-year period was the result of intense lobbying by the brand biologic industry during the BPCIA’s passage, which argued that the complexity of biologic manufacturing and the higher development costs of biologics justified a longer exclusivity period than the 5-year NCE exclusivity available to small molecules. Critics argued that 7 years would have been sufficient and that the 12-year period was excessive given that biosimilars are not required to conduct the same level of clinical trials as the reference product.
The BPCIA Patent Dance: A Structured Exchange
Unlike Hatch-Waxman, where patent disputes are initiated through Orange Book listings and PIV certifications, the BPCIA provides a detailed, multi-step process for resolving patent disputes between reference biologic manufacturers and biosimilar applicants. This process is colloquially called the ‘patent dance’ and is codified at 42 U.S.C. § 262(l).
The patent dance works as follows: Upon FDA acceptance of a biosimilar application, the applicant must provide the reference biologic manufacturer with a copy of the application and related manufacturing information, allowing the manufacturer to identify patents it believes are infringed. The parties then exchange lists of patents they believe are relevant, negotiate down to a list to be litigated immediately, and exchange infringement and invalidity contentions. The entire process is governed by specific statutory deadlines.
Participation in the patent dance has been contentious. Some biosimilar applicants have declined to initiate the exchange, arguing that participation is not mandatory. The Federal Circuit held in Amgen Inc. v. Sandoz Inc. (2015) that the biosimilar applicant has the option but not the obligation to participate in the patent dance, but that a non-participating applicant forfeits certain procedural protections, including the ability to assert an immediate patent dance resolution. The practical consequence is that most sophisticated biosimilar applicants participate in the dance because non-participation triggers the reference manufacturer’s right to seek immediate injunctive relief.
Biosimilar Interchangeability: The Additional Layer
Beyond the standard biosimilar designation, the BPCIA provides a higher-tier designation called ‘interchangeable biosimilar.’ An interchangeable biosimilar has been shown to produce the same clinical result as the reference product in any given patient and, for a product that involves administration by or at the direction of a healthcare provider, that switching between the reference and the biosimilar does not produce a greater risk than continuing on the reference product. An interchangeable biosimilar can be substituted at the pharmacy level without prescriber intervention in states that permit such substitution.
The first interchangeable biosimilar for a given reference product earns 12 months of exclusivity against approval of other interchangeable biosimilars for the same reference product (with provisions for earlier entry under certain conditions). This exclusivity incentivizes the substantial additional clinical investment required to achieve the interchangeable designation.
For IP analysts and investors, the interchangeability timeline matters: the interchangeable biosimilar will capture a larger market share than a non-interchangeable biosimilar because it can be substituted automatically at the pharmacy, and its 12-month exclusivity against other interchangeable biosimilars provides a meaningful commercial window.
Section 16: U.S. PTE vs. EU SPC: A Global IP Strategy Comparison
Pharmaceutical companies with global products must manage patent term compensation mechanisms across multiple jurisdictions. The U.S. PTE and EU Supplementary Protection Certificate (SPC) are the two most commercially significant systems, but they operate through fundamentally different legal constructs, calculation methodologies, and administrative procedures.
The SPC: A Sui Generis Right
A U.S. PTE extends the term of an existing patent. An EU SPC is a legally distinct intellectual property right that comes into force the day the underlying basic patent expires. The SPC is not an amendment to the patent; it is a new right that the patent holder holds separately, governed by EU Regulation 469/2009 (as amended). This distinction matters for licensing, assignment, and litigation: an SPC can be licensed, transferred, and enforced independently of the patent that preceded it.
SPC Duration Formula
The SPC duration is calculated as:
SPC Duration = (Date of First EEA Marketing Authorization) minus (Basic Patent Filing Date) minus 5 years
Subject to a maximum SPC duration of 5 years and a minimum of zero. The policy objective is to ensure the holder receives a total of 15 years of market exclusivity from the date of first EEA marketing authorization, though in practice many products receive less due to the cap.
If the holder completes an agreed Paediatric Investigation Plan (PIP) to the EMA’s satisfaction, the SPC duration is extended by 6 months, raising the maximum to 5.5 years.
Key Structural Differences
The most practically significant difference between PTE and SPC is the scope of protection during the extension period. A U.S. PTE protects the approved product, its approved uses, and its method of manufacture. An EU SPC protects only the active ingredient or combination of active ingredients that was the subject of the marketing authorization. This means an SPC does not protect improvements, new formulations, or dosage forms unless those are specifically authorized and subject to a separate SPC.
Administration is another key difference. A U.S. PTE is a single federal right covering the entire country, applied for at the USPTO with input from the FDA. EU SPCs have historically required separate national applications in each EU member state, each examined by that state’s national patent office. This creates significant administrative burden and the possibility of divergent outcomes across member states. The EU is moving toward a unified SPC system as part of the unitary patent package, which would allow a single SPC application to cover participating member states, but the system is not yet fully operational.
The SPC in Biosimilar Competition
EU SPCs have been central to some of the most significant biosimilar litigation in Europe. Because the SPC protects the active ingredient and runs independently of the basic patent, biosimilar manufacturers must track both the basic patent’s expiration and the SPC’s expiration to identify the correct market entry date. In several major cases, biosimilar manufacturers have challenged SPC validity on the grounds that the SPC was not granted for the product actually authorized, or that the basic patent did not ‘protect’ the product within the meaning of the regulation.
The CJEU’s jurisprudence on the meaning of ‘protected by a basic patent in force’ has been complex and evolving, with decisions in cases including Neurim Pharmaceuticals (C-130/11), Teva Pharmaceutical Industries (C-121/17), and Royalty Pharma (C-650/17) progressively refining the test. IP teams working on European biologic strategy must track this case law with the same attention they give to U.S. Federal Circuit decisions on PTA and PTE.
Comparison Table: U.S. PTE vs. EU SPC
Attribute
U.S. Patent Term Extension (PTE)
EU Supplementary Protection Certificate (SPC)
Legal Basis
35 U.S.C. § 156 (Hatch-Waxman Act)
EU Regulation 469/2009
Administering Body
USPTO (with FDA determination of RRP)
National patent offices (moving to unified SPC)
Nature of Right
Extension of existing patent term
Sui generis independent IP right
Maximum Duration
5 years
5 years (5.5 years with PIP extension)
Total Exclusivity Cap
14 years post-FDA approval
15 years post-first EEA marketing authorization (effective policy target)
Pediatric Incentive
6 months added to all existing patents and exclusivities
6 months added to the SPC
Calculation Basis
0.5x testing phase + 1.0x approval phase, subject to deductions
(MA date) minus (filing date) minus 5 years
Scope During Extension
Approved product, approved uses, method of manufacture
Active ingredient(s) covered by the marketing authorization
Geographic Coverage
Entire U.S. (single right)
Per-country, with unitary SPC in development
Section 17: Modeling the Loss of Exclusivity (LOE) Date: A Multi-Variable Framework
The ‘LOE date’ is the financial model’s most sensitive input because it determines when monopoly-level revenue collapses. Getting it wrong by even six months can change the NPV of a pharmaceutical asset by hundreds of millions of dollars. Yet LOE modeling is not a single calculation. It is a probability-weighted analysis of multiple possible outcomes, each with its own timeline and financial impact.
The Variables in the LOE Model
An accurate LOE model must integrate at minimum the following inputs:
The PTA-adjusted expiration of the primary composition-of-matter patent, calculated independently of the USPTO’s printed figure and verified against current case law.
The PTE award for the product, calculated under the current statutory formula with attention to the 14-year cap and the pre-grant deduction.
The expiration of every Orange Book-listed patent and its relationship to the composition-of-matter patent’s term (does the formulation patent or method-of-use patent expire later, providing additional protection?).
The date and type of all applicable FDA regulatory exclusivities, with specific attention to NCE exclusivity (which blocks ANDA filing, not just approval), ODE (which blocks approval of a competing product for the same indication), and pediatric exclusivity (which extends every other element of the stack by six months).
The status of PIV challenges: which ANDAs have been filed, which are in litigation, and what is the probabilistic outcome of each case based on the strength of the patent’s composition-of-matter and secondary claims.
The probability and expected terms of any settlement agreement, modeled as a probability-weighted average of possible entry dates rather than a binary outcome.
For biologics, the 12-year BPCIA exclusivity date, the patent dance status, and the probability that any of the reference product’s process or formulation patents will survive BPCIA litigation.
Scenario Analysis for the LOE Model
A rigorous LOE model presents at minimum three scenarios: a base case reflecting the most likely outcome given the current patent and litigation landscape; a bear case reflecting early generic or biosimilar entry due to PIV success in litigation or a settlement with an early entry date; and a bull case reflecting successful defense of all relevant patents and full utilization of all exclusivity periods. Each scenario should be assigned a probability, and the expected LOE date is the probability-weighted average of all scenario outcomes.
For a drug with a complex litigation history and multiple pending ANDA filers, the probability distribution of possible entry dates can be wide, spanning five or more years between the earliest and latest realistic outcomes. Collapsing that distribution to a single LOE date, as many sell-side models do, obscures material risk.
Section 18: Investment Strategy: Using Patent Intelligence for Pharma Equity Analysis
Patent data is among the most underutilized sources of alpha in pharmaceutical equity analysis. Public patent filings, Orange Book updates, PTAB proceedings, and PIV notifications provide a real-time stream of information about competitive entry risks, IP strength, and lifecycle management activity that is available to any analyst willing to process it.
Signals Worth Tracking
PIV notification filings: When a generic company files an ANDA with a PIV certification, it must send a Notice Letter to the brand company within 20 days of FDA acceptance. These notices are disclosed in SEC filings and Orange Book updates. A PIV filing against a major product is an explicit signal that a well-resourced generic company believes the protecting patents are vulnerable. The market typically prices in some probability of early generic entry upon PIV disclosure, but the magnitude of repricing varies and is often insufficient in the early stages of litigation.
PTAB inter partes review (IPR) petitions: IPR is a post-grant administrative challenge to patent validity before the USPTO’s Patent Trial and Appeal Board. For pharmaceutical patents, IPR has become a preferred early-stage validity challenge because it is faster and cheaper than district court litigation and applies a lower validity standard (preponderance of evidence rather than clear and convincing). The filing of an IPR petition against a major drug’s composition-of-matter patent signals that the generic or hedge fund petitioner believes the patent is vulnerable to invalidity on prior art grounds. IPR outcomes are publicly available and carry substantial predictive weight for the subsequent district court litigation.
Orange Book listing changes: New patent listings in the Orange Book signal new IP protection being added to a franchise. Deletions can signal that the company is narrowing its protection posture or has let a patent expire. Monitoring these changes in real time provides early indicators of lifecycle management activity.
Continuation patent filings: Continuation and divisional applications filed in the late stages of a drug’s commercial life, specifically covering formulations or delivery systems that differ from the original molecule, are the mechanical precursors to an evergreening strategy. Tracking these filings through the USPTO’s public patent application database gives analysts a leading indicator of LCM investment.
The Patent Cliff Trade
The patent cliff creates well-documented trading opportunities. As a drug’s LOE date approaches, the brand company’s revenue forecast becomes increasingly certain to decline, and the generic entrant’s revenue forecast for the first 180-day exclusivity period becomes increasingly positive. The trade is not simply ‘short brand, long generic.’ The actual market outcome depends on the number of authorized generics in the market, the first-filer’s ability to achieve adequate manufacturing scale, and the pace of formulary substitution by pharmacy benefit managers.
Analysts who have built accurate LOE models with properly probability-weighted scenario analyses are better positioned to time this trade than those relying on consensus LOE dates that may reflect stale patent analysis or unpublished settlement terms.
Section 19: Comprehensive Key Takeaways
On the 20-Year Baseline: The statutory 20-year patent term is an economic fiction for pharmaceutical products. Effective market exclusivity at launch is typically 7 to 12 years after accounting for clinical development time. PTA and PTE exist to partially restore the gap, but neither mechanism provides full compensation.
On Hatch-Waxman Architecture: The Act created a dual system that simultaneously incentivized innovation (through PTE and 30-month stays) and generic competition (through the ANDA pathway and 180-day first-filer exclusivity). Every element of pharmaceutical patent strategy operates within this framework.
On PTA: PTA compensates for USPTO administrative delay using an A-B-C formula. Applicants must manage their prosecution response timing to minimize applicant delay reductions. The PTA printed on the patent by the USPTO is not authoritative; independent audits against current case law are essential for high-value assets.
On Wyeth and Novartis: These two Federal Circuit decisions fundamentally changed PTA calculation methodology in ways that favored patentees. Companies that had patents issued before 2010 without auditing their PTA under the post-Wyeth methodology may have accepted lower PTA than they were entitled to.
On PTE: PTE restores half of clinical trial time and all of FDA review time, subject to the 5-year cap and the 14-year post-approval cap. The 14-year cap is the more frequently binding constraint for drugs with long remaining patent terms at approval. Only one patent per approved product may receive PTE, making the patent election decision critical.
On Regulatory Exclusivities: Patents and FDA exclusivities are parallel, distinct systems. NCE exclusivity blocks ANDA filing for four years and blocks ANDA approval for five years. Pediatric exclusivity adds six months to every existing patent and exclusivity simultaneously. BPCIA’s 12-year biologic exclusivity is an absolute bar on biosimilar approval that operates independently of the patent system.
On Lifecycle Management: LCM and evergreening strategies range from genuine therapeutic innovation (new indications, pediatric formulations) to strategic IP accumulation (polymorphs, minor formulations). The Humira patent thicket is the canonical example of the latter, having extended effective U.S. exclusivity by approximately seven years beyond the original composition-of-matter patent.
On Pay-for-Delay Post-Actavis: Reverse payments are no longer safe from antitrust scrutiny. The compensation mechanisms have evolved from explicit cash to no-AG commitments, quantity restrictions, and side deals. Analysts must evaluate the probability and expected structure of settlement agreements when modeling LOE dates for drugs in active PIV litigation.
On Biosimilars: BPCIA’s patent dance is optional but practically necessary. Biosimilar interchangeability designation carries significant commercial advantages and a 12-month exclusivity against other interchangeable biosimilars. The 12-year BPCIA exclusivity wall dominates LOE modeling for biologics.
On Global Strategy: The EU SPC and U.S. PTE share a policy goal but differ in legal structure, calculation methodology, scope of protection, and geographic application. Each major market requires its own patent term compensation analysis; a consolidated global IP strategy cannot rely on U.S. analysis alone.
On Investment Strategy: Patent data is a real-time signal stream for pharmaceutical equity analysis. PIV filings, PTAB IPR petitions, Orange Book listing changes, and continuation patent filings all provide leading indicators of competitive entry risk and lifecycle management activity. LOE models should present probability-weighted scenario analyses, not single-point estimates.
Frequently Asked Questions
How do PTA and PTE interact when both apply to the same patent?
They are applied sequentially and cumulatively. The USPTO first calculates PTA and adds the resulting days to the 20-year term, establishing the PTA-adjusted expiration date. When the applicant seeks PTE, the calculation uses the PTA-adjusted expiration date as the baseline. The PTE is then added on top of the PTA-adjusted term. The statute at 35 U.S.C. § 156(a)(2) explicitly defines the term subject to extension as the original expiration date of the patent ‘which shall include any patent term adjustment.’ A patent can accumulate both forms of additional term, and for complex pharmaceutical patents with long prosecution histories, receiving both PTA of several hundred days and PTE of several hundred more days is common.
If multiple patents cover the approved drug, how is the PTE election made?
The company must file a PTE application within 60 days of the drug’s FDA approval date for each patent it wishes to preserve as a candidate. The USPTO will then require the company to elect a single patent to extend. The election analysis should compare: the scope and enforceability of each candidate patent’s claims; each patent’s remaining term at the time of the election; the maximum PTE each patent would receive under the formula; and the expected litigation value of each patent against anticipated ANDA filers. The elected patent should be the one whose extended term provides the most durable and broad protection against competitive entry, which is not always the patent with the most remaining term.
Can a patent subject to a terminal disclaimer receive PTE?
Yes. A terminal disclaimer is a patentee’s agreement, filed during prosecution to overcome an obviousness-type double patenting rejection, that a patent will expire no later than a reference patent. A terminal disclaimer can shorten a patent’s effective term. But the Federal Circuit held in Merck & Co. v. Hi-Tech Pharmacal Co., 482 F.3d 1317 (Fed. Cir. 2007), that a terminal disclaimer does not disqualify a patent from PTE. The PTE is added to the disclaimed term. Note that PTA interacts differently with terminal disclaimers: PTA on a disclaimed patent is generally limited to PTA earned by the disclaimed patent’s own prosecution, and cannot extend the patent beyond the reference patent’s PTA-adjusted expiration without specific analysis.
Does the 14-year post-approval cap ever benefit a company?
The 14-year cap is not designed to benefit the patent holder; it is a ceiling. But it can create a useful planning floor in one indirect sense: a company whose drug has more than 14 years of remaining patent term at approval will receive zero PTE, so it has no PTE application to file and no 60-day deadline to manage for that drug’s primary patent. The strategic implication is that those companies should focus their LCM investment on filing and protecting secondary patents that will be approved later in the drug’s lifecycle, when the 14-year cap is less likely to be binding on the secondary patent.
What is the earliest possible date a generic can enter the market for a drug with NCE exclusivity?
The earliest possible date is 5 years from the NCE approval date if no PIV challenge has been filed, or 4 years from the NCE approval date if a PIV challenge is filed within that window, followed by whatever litigation period is required. The 30-month stay on top of the 4-year PIV filing window means that in practice, the theoretical earliest possible generic entry date is approximately 7.5 years (4 years plus 30 months) after the NCE approval. This is approximate because the 30-month stay runs from the date of receipt of the PIV Notice Letter, not from the 4-year mark. The actual earliest entry date requires precise calculation using the specific filing and notice dates for each ANDA.
How does ‘at-risk’ generic launch work after a PIV win?
When a generic company wins a PIV case at the district court level, invalidating or finding non-infringement of the asserted patent, the FDA can grant final ANDA approval. The generic can then launch immediately, though the brand company will almost certainly appeal. If the brand secures an injunction pending appeal, the generic’s launch is stayed. If not, the generic launches ‘at risk,’ meaning it is selling in the market while the appeal is pending. If the Federal Circuit reverses and upholds the patent, the generic would be liable for damages for the period of infringing sales. At-risk launches are commercially rational when the generic company has high confidence in the legal outcome and the revenue from the exclusivity period exceeds the expected litigation exposure.
This guide consolidates the author’s analysis of publicly available statutes, regulations, court decisions, and industry data. It does not constitute legal advice. Patent term calculations for specific products should be performed by qualified patent counsel. Financial projections should be validated by qualified analysts with access to current market and patent data.