Biosimilar development is not generic drug development with more paperwork. It is a $100M-to-$300M scientific undertaking that demands the simultaneous mastery of high-resolution analytical chemistry, mammalian bioprocessing, global regulatory strategy, and adversarial intellectual property litigation. The molecule a biosimilar company is trying to match — the reference biologic — is itself a variable, proprietary entity whose complete blueprint is an inaccessible trade secret. That is the foundational problem, and every technical, regulatory, and legal decision in a biosimilar program flows from it.
The central regulatory doctrine — the ‘totality of the evidence’ standard — has historically been visualized as a pyramid: a massive analytical base supporting smaller tiers of non-clinical and clinical confirmation at the apex. That pyramid is in the process of conceptual inversion. Analytical tools have become so sensitive, and clinical trials so demonstrably blunt at detecting the structural differences that matter, that the apex study is increasingly seen as a $100M+ financial barrier rather than a scientific necessity. The European Medicines Agency (EMA) has embraced this shift with its 2023 reflection paper on tailored clinical approaches. The FDA is following. This is the single most commercially significant regulatory trend in the biosimilar space.
Concurrently, innovator companies have constructed what researchers describe as the most aggressive patent portfolios in the history of the pharmaceutical industry. On average, nine to twelve times more patents are asserted against US biosimilar applicants than against their Canadian or UK counterparts. AbbVie’s Humira (adalimumab) carried a portfolio exceeding 250 patents at peak, covering formulations, devices, manufacturing processes, and methods of use — a playbook that has since been replicated across the industry. Freedom-to-operate analysis is no longer an optional legal exercise; it is a prerequisite for program initiation.
This document is a complete technical and strategic reference for the teams that build, fund, and litigate biosimilar programs.
Section 1: The Biologic and Biosimilar Landscape
1.1 Defining the Terrain: Biologics, Reference Products, and Biosimilars
A biologic is a therapeutic product derived from or manufactured using living organisms — bacteria, yeast, mammalian cells, or more complex systems. The active molecule is typically a large protein: a monoclonal antibody at roughly 150,000 Daltons, a glycoprotein at varying molecular weights, or a fusion protein combining functional domains from multiple sources. Compare this to aspirin at 180 Daltons and the scale of the engineering challenge becomes concrete. The structural complexity of a biologic — its amino acid sequence, its three-dimensional fold, its attached glycan chains, its disulfide bond map — is a direct product of the living system used to make it and the conditions under which that system operates.
The reference product (RP) is the approved innovator biologic against which a biosimilar is measured. FDA and EMA define it as the single biological product licensed on the basis of a full independent preclinical and clinical package. Every biosimilar program is anchored to this specific comparator. This sounds straightforward until you recognize that the RP is not a fixed chemical entity: it is itself a heterogeneous population of molecular variants, produced by a process that has likely been modified multiple times over the product’s commercial life, each modification introducing subtle changes to the molecule’s quality attribute profile.
A biosimilar is a licensed biological product demonstrated to be ‘highly similar’ to the reference product with ‘no clinically meaningful differences’ in safety, purity, and potency. The ‘highly similar’ standard explicitly permits minor structural differences in components judged to be clinically inactive — a concession to the biological reality that no two manufacturing processes will produce identical molecular populations. What biosimilar developers often underestimate is how narrow the acceptable range of ‘minor’ actually is in practice, and how much of the regulatory submission is devoted to proving that specific observed differences fall within it.
Key Takeaway (Section 1.1): The ‘highly similar’ standard is a precision regulatory instrument, not a permissive one. The entire biosimilar development apparatus exists to generate enough evidence to convince regulators that observed differences are clinically irrelevant — a demonstration that requires substantially more analytical and clinical work than most non-specialist estimates assume.
1.2 Why Biosimilars Are Not Generic Drugs: The Complexity Gap
The pharmaceutical industry’s public communications often frame biosimilars as ‘the generic equivalent’ of biologics. This framing is convenient but misleading in ways that matter for investment, legal, and regulatory strategy.
Generic drugs are small molecules synthesized through chemical reactions. Their active pharmaceutical ingredient (API) is fully characterized, absolutely reproducible from batch to batch, and identical in two products made by different manufacturers from the same synthesis route. Demonstrating bioequivalence — that the generic reaches the same blood concentration at the same rate — is sufficient for FDA approval. The development cost runs in the $1M-$5M range. The approval timeline is typically two to four years.
Biosimilars are entirely different. The molecular weight difference alone (180 Da vs. 150,000 Da for a monoclonal antibody) understates the complexity gap. Aspirin has one chiral center and no post-translational modifications. A typical therapeutic antibody has four polypeptide chains, approximately 1,300 amino acids, multiple glycosylation sites each carrying heterogeneous oligosaccharide chains, dozens of disulfide bonds, and a three-dimensional structure maintained by non-covalent interactions that are exquisitely sensitive to temperature, pH, ionic strength, and shear stress. The manufacturing process — the specific CHO cell clone, the culture media composition, the bioreactor geometry, the purification train — is not incidental to the product. It is the product. Change the process and you change the molecule.
This ‘process is the product’ reality creates the defining challenge of biosimilar reverse engineering. The innovator’s manufacturing process is a trade secret protected by a combination of patent law, regulatory exclusivity, and practical opacity. A biosimilar developer cannot access it. They must develop an independent process that produces a different molecular population — because different cells, different media, and different equipment will always introduce differences — and then prove that the differences between their molecular population and the innovator’s molecular population are not clinically meaningful. This is an inherently probabilistic and evidence-intensive exercise, not a deterministic one.
The immunogenicity dimension compounds this. Small-molecule generics pose essentially no immunogenicity risk. Therapeutic proteins, by contrast, can elicit anti-drug antibody (ADA) responses that range from benign (accelerated drug clearance with no clinical consequence) to severe (neutralizing antibodies that cross-react with the patient’s endogenous protein, as occurred with recombinant erythropoietin causing pure red cell aplasia). The structural differences introduced by a distinct manufacturing process can, in theory, expose neo-epitopes that the innovator product does not present, elevating immunogenicity risk. Mandatory comparative immunogenicity assessment in clinical studies is the regulatory response to this risk.
Key Takeaway (Section 1.2): Development cost ($100M-$300M per program), timeline (seven to twelve years from concept to approval), and manufacturing capital expenditure ($200M-$500M for a GMP biologics facility) are the primary reasons biosimilar markets support far fewer competitors than generic drug markets. These economics determine which molecules get challenged and by whom.
1.3 The Economic and Therapeutic Imperative
The biosimilar market exists because innovator biologics are among the most expensive medicines ever sold. Humira’s US list price exceeded $80,000 per patient per year at its peak. Herceptin (trastuzumab) and Avastin (bevacizumab) carry annual costs in the $50,000-$100,000 range. The cumulative spend on just the top ten biologic targets represents hundreds of billions of dollars annually across major markets.
The patent cliff created by the expiration of primary composition-of-matter patents on these blockbusters represents, on paper, one of the largest market access opportunities in pharmaceutical history. Humira lost its primary US patent protection in 2023. Enbrel (etanercept), Remicade (infliximab), Avastin, Herceptin, Rituxan (rituximab), and Stelara (ustekinumab) have all seen or will see primary patent expiration across major markets between 2015 and 2028. IQVIA estimated the cumulative ‘biologic patent cliff’ opportunity at over $200 billion in annual global sales through 2028.
The market dynamics following biosimilar entry, however, look nothing like the generic drug experience. A small-molecule generic entering the US market after patent expiration typically captures 80-90% market share within twelve months, driven by automatic pharmacy substitution and multi-source price competition that can reduce prices by 80% or more. Biosimilar markets are structurally different. Price erosion in the US has averaged 20-30% off the reference product’s list price in the near term, substantially less than European markets where centralized procurement and mandatory substitution policies produce 40-70% price reductions. The reasons are structural: higher barriers to entry (fewer competitors), the absence of automatic substitution for non-interchangeable biosimilars, the rebate wall maintained by PBMs, and the requirement for active physician and patient education that generics do not face.
This market structure creates a specific investment thesis. The biosimilar opportunity is not about commodity volume with thin margins, as in small-molecule generics. It is about being one of two to four credible manufacturers capturing a premium margin in a market that will still be priced at a meaningful discount to the reference product. Scale, first-mover advantage, interchangeability status, and commercial infrastructure are the determinants of success.
Key Takeaway (Section 1.3): Institutional investors modeling biosimilar programs should apply market share and pricing assumptions calibrated to a ‘specialty pharmaceutical’ competitive dynamic, not a generic drug dynamic. Assume two to five approved competitors at steady state, price reductions of 25-50% from WAC, and adoption curves measured in years rather than months, particularly in the US.
Section 2: The Regulatory Compass: Totality of the Evidence
2.1 The Foundational Principle: Demonstrating Biosimilarity, Not De Novo Efficacy
The regulatory architecture for biosimilar approval rests on a single, powerful premise: the safety and efficacy of the reference biological active substance have already been established. The biosimilar applicant does not need to re-prove that mechanism of action works. They need to prove their version of the molecule is close enough to the reference product that it will perform identically in the clinic.
This has a profound practical consequence. The biosimilar regulatory dossier is not organized around clinical outcomes. It is organized around molecular similarity. The clinical data, where required, serves to confirm the absence of clinically meaningful differences — a negative proof — rather than to establish therapeutic benefit. This inversion of the evidentiary logic relative to a new drug application is what makes biosimilar programs scientifically distinct and what makes strong analytical data so central to regulatory strategy.
The ‘totality of the evidence’ standard operationalizes this premise. FDA, EMA, and WHO all apply it. No single study type is determinative. The package as a whole must persuade the agency that the biosimilar will perform like the reference product across its approved indications. The nature and extent of studies required are case-specific, driven by the complexity of the reference molecule and the strength of the analytical similarity demonstration. A highly complex glycoprotein with multiple mechanisms of action requires a more extensive comparability package than a structurally simple, single-mechanism cytokine.
Key Takeaway (Section 2.1): ‘Totality of the evidence’ means the quality of the analytical data package determines how much clinical data regulators will demand. Programs with a compelling structural and functional similarity case can negotiate reduced clinical requirements. This makes early investment in analytical capability a direct lever on total program cost.
2.2 The Development Pyramid: Structure, Sequence, and Strategic Weighting
The ‘totality of the evidence’ approach is operationally structured as a development pyramid with four tiers, each building on the one below it.
The base — the largest and most resource-intensive tier — is comprehensive analytical characterization. This is where the biosimilar developer characterizes the reference product across every measurable structural and functional attribute, then demonstrates that their candidate falls within or matches that attribute range. The analytical package for a monoclonal antibody typically includes primary sequence confirmation, higher-order structure assessment via at least five orthogonal techniques, full post-translational modification profiling including site-specific glycan analysis, charge variant quantitation, size variant quantitation using both SEC and AUC, and a complete functional panel covering target binding kinetics, Fc receptor binding, and cell-based potency for each known mechanism of action. This work commonly generates thousands of data points across hundreds of samples from multiple reference product lots.
The second tier covers non-clinical (animal) studies. Regulatory guidance increasingly acknowledges that animal studies have limited utility in the biosimilar context — they are too insensitive to detect the small differences that matter and too prone to species-specific confounds. FDA and EMA guidance now explicitly indicates that repeat-dose toxicology studies in animals are generally not needed when the analytical and functional similarity case is strong. The EMA’s 2023 reflection paper went further, advising that PK/PD bridging in relevant animal models may also be waived for well-characterized molecules. This is not universally applied, but it represents the direction of travel.
The third tier is comparative clinical pharmacology: head-to-head PK and, where measurable, PD studies in humans. These studies confirm that the body handles the biosimilar identically to the reference product, translating the structural and functional similarity evidence into the clinical pharmacology domain. PK studies in healthy volunteers are the standard approach for systemic biologics; patient populations are used when the molecule’s biology or safety profile makes healthy volunteer studies inappropriate.
The apex is the confirmatory comparative clinical efficacy and safety study. This is the most expensive single element of the development program, commonly costing $50M-$150M for a single-arm or crossover study in a patient population sensitive to treatment differences. Its scientific utility is increasingly questioned. A regulatory-powered equivalence study in, for example, rheumatoid arthritis patients comparing a Humira biosimilar to the reference product is powered to detect a 15-20% difference in ACR20 response at week 24. The structural differences between a well-characterized biosimilar and its reference product are on the order of single-digit percentage variations in specific glycan species — differences that would need to translate into a substantial clinical effect to be detected by this blunt instrument. The probability of observing such an effect, if the analytical similarity is genuine, is very low. This is the scientific basis for the push to waive or eliminate these studies.
Key Takeaway (Section 2.2): Teams evaluating biosimilar program budgets should model two scenarios: the full-pyramid approach with a confirmatory clinical study ($200M-$300M total) and the analytically streamlined approach without it ($80M-$150M). The gap represents the clinical study cost plus timeline extension. Regulatory strategy — specifically, how convincingly the analytical package argues for waiver of the apex study — is a direct financial variable.
2.3 Critical Quality Attributes: The Blueprint for Equivalence
Before any head-to-head comparison can be executed, the biosimilar developer must define exactly what they are measuring. This process begins with constructing a Quality Target Product Profile (QTPP) for the biosimilar, derived from exhaustive characterization of the reference product.
The QTPP identifies all Critical Quality Attributes (CQAs): the physical, chemical, biological, and microbiological properties that must be controlled within defined limits to ensure the product’s identity, purity, potency, and safety. For a monoclonal antibody, the CQA list is extensive. It includes the primary amino acid sequence and the correct disulfide bond pairing (identity), the glycan profile at each N-glycosylation site (potency and safety — glycans influence Fc effector function and immunogenicity), the charge variant distribution (stability and clinical PK), the aggregate level measured by both SEC and AUC (safety), the fragment content (potency), the host cell protein (HCP) concentration (safety and immunogenicity), the residual host cell DNA level, and the binding affinity and potency across all characterized mechanisms of action.
Each CQA must be assigned a risk ranking. High-risk CQAs are those where a deviation from the reference product range could plausibly affect clinical performance. Glycosylation, aggregate content, and functional potency are universally high-risk. Charge variant distribution is typically moderate-risk, with context-dependent exceptions. Minor sequence-adjacent PTMs like oxidation at non-active-site methionines are typically low-risk. This risk ranking drives the tiered analytical strategy: high-risk CQAs are assessed with multiple orthogonal methods and require the most stringent similarity demonstration.
Key Takeaway (Section 2.3): The CQA risk-ranking process is where analytical science and regulatory strategy first converge. Developers who invest early in a comprehensive, publication-quality characterization of the reference product across a statistically adequate number of lots build the strongest foundation for arguing analytical similarity to regulators and for defending against innovator patent claims that assert process-specific quality differences.
Section 3: The Art of Deconstruction: Analytical Characterization
3.1 The Reference Product Characterization Challenge: Sourcing a Moving Target
The first analytical challenge has nothing to do with the biosimilar candidate. It is about the reference product itself.
Regulators expect developers to characterize a minimum of three to ten distinct lots of the RP, ideally spanning the product’s commercial shelf life, to capture the full range of acceptable batch-to-batch variability. This lot number requirement has practical and strategic dimensions that are easy to underestimate. A major innovator’s drug substance (DS) batch for a high-volume product may be 2,000 to 10,000 liters of bioreactor culture, yielding enough drug substance to fill dozens or hundreds of drug product (DP) lots. When a developer purchases ten lots from the commercial market, they may be purchasing from a single DS batch — capturing fill-and-finish variability but not upstream process variability. The quality attribute range they establish from such a sample is artificially narrow. This matters because the developer’s manufacturing process will need to consistently produce a molecule within whatever target range they define. Define it too narrowly and they set themselves an impossible manufacturing control problem.
The innovator’s process change history compounds this. Innovators modify their manufacturing processes over a product’s commercial life — changes in cell culture media composition, bioreactor control strategy, purification chromatography resins — and each such change requires internal comparability qualification but does not necessarily result in a label change visible to external parties. A biosimilar developer characterizing a reference product over a multi-year development timeline may be comparing batches made by subtly different versions of the innovator’s process without knowing it. This is a real and documented risk. Sandoz encountered this scenario during Zarxio development, and the EMA’s regulatory history contains multiple examples where RP manufacturing changes required biosimilar developers to re-run comparability studies.
The practical solution is to purchase RP lots at multiple time points spanning the development program, retain samples from all lots, and perform real-time monitoring of the RP’s quality attribute profile throughout development. This is costly and operationally complex, but it provides the earliest possible detection of process drift and minimizes the risk of a surprise comparability failure late in the program.
Reference product sourcing for US programs introduces a separate complication. FDA generally requires that the final biosimilarity determination be made against a US-licensed reference product. For developers running global programs against EU-sourced reference material, this mandates three-way bridging studies: EU RP vs. US RP to establish product comparability between geographic markets, biosimilar vs. EU RP, and biosimilar vs. US RP. The EMA is more flexible, permitting use of a non-EU RP with appropriate scientific bridging. For companies building a single global development program, this asymmetry adds cost and timeline and requires careful upfront planning.
Key Takeaway (Section 3.1): RP lot sourcing strategy is a first-order program design decision with direct budget and timeline implications. Developers should plan for a minimum of eight to twelve distinct lots sourced across the full development timeline, with documented lot genealogy to identify potential process changes, and should budget for three-way bridging studies if the program targets both US and EU markets.
3.2 Primary Structure Verification: Mass Spectrometry as the Foundation
Confirmation that the biosimilar candidate has an amino acid sequence identical to the reference product is an absolute prerequisite. Any discrepancy in primary sequence disqualifies the molecule from the biosimilar pathway; it is by definition a different protein.
The gold standard for primary structure confirmation is ‘bottom-up’ proteomics by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The workflow is methodical: the intact protein is reduced to break disulfide bonds, alkylated to prevent re-oxidation, then digested with one or more proteases to generate a predictable mixture of peptides. Trypsin is the standard choice, cleaving at lysine and arginine residues to produce peptides in a mass range amenable to MS analysis. The peptide mixture is then separated by UHPLC and introduced into a high-resolution mass spectrometer — typically an Orbitrap instrument capable of sub-parts-per-million mass accuracy.
Each peptide’s molecular mass is measured with high precision, enabling identification by comparison to the theoretical mass calculated from the known sequence. Tandem MS provides sequence confirmation: selected peptides are fragmented by collision-induced dissociation (CID) or electron transfer dissociation (ETD), and the masses of the resulting b- and y-ion series yield a direct amino acid sequence readout. A comprehensive peptide map covers greater than 95% of the protein sequence, with overlapping peptides from different digestion conditions (Lys-C, Asp-N, Glu-C as orthogonal enzymes) closing coverage gaps.
This peptide mapping workflow accomplishes more than sequence confirmation. The same data reveals most post-translational modifications simultaneously. Deamidation of asparagine residues (converting asparagine to aspartate, adding 0.984 Da) is quantifiable at each site. Oxidation of methionine residues (adding 15.995 Da) is detected and located. Glycation of lysine residues from non-enzymatic reaction with reducing sugars in the culture media is identifiable. N-terminal pyroglutamate cyclization is confirmed. The complete disulfide bond pairing pattern is established by running a separate parallel non-reduced digest. In this way, a single LC-MS/MS peptide mapping experiment generates data on primary sequence identity and the full PTM landscape simultaneously.
Intact mass analysis by native mass spectrometry or denaturing ESI-MS provides a complementary top-down perspective. The measured molecular weight of the intact protein — or of its reduced and deglycosylated subunits — provides an independent check on overall molecular identity and reveals major glycoform populations as a mass envelope rather than a sequence-level readout. For heavily glycosylated proteins, the mass envelope is broad and complex, but comparison of the biosimilar and RP intact mass spectra is a rapid first-line similarity check before the full peptide mapping exercise.
Key Takeaway (Section 3.2): Primary sequence confirmation by multi-enzyme peptide mapping with orthogonal intact mass analysis is non-negotiable and should be completed in parallel on the biosimilar candidate and multiple RP lots. Any unexpected PTM difference identified at this stage is diagnostic data for process development — it tells the cell culture or downstream team where to look for process parameter adjustments.
3.3 Higher-Order Structure: The Orthogonal Arsenal
Primary sequence identity does not guarantee functional equivalence. The same amino acid sequence can fold into multiple, structurally distinct conformations, some active and some not. Demonstrating that the biosimilar’s three-dimensional fold — its secondary, tertiary, and quaternary structures — matches the reference product is a critical and technically demanding component of the analytical package.
Regulators and scientists agree that no single technique provides a definitive assessment of higher-order structure (HOS). Each method is sensitive to different structural features and carries its own artifacts and limitations. The orthogonal requirement — using multiple techniques grounded in different physical principles to build a convergent picture of structural similarity — is the scientifically sound response to this limitation.
Far-UV circular dichroism (CD) spectroscopy measures the differential absorption of left- and right-handed circularly polarized light by the protein backbone. The resulting spectrum is diagnostic for secondary structure: alpha-helices produce a characteristic double minimum at 208 nm and 222 nm, beta-sheets produce a single minimum near 218 nm, and disordered regions produce a minimum near 200 nm. Quantitative deconvolution of the spectrum yields the fractional content of each structural element. Overlapping spectra from the biosimilar and multiple RP lots, with statistical analysis of spectral similarity, constitutes strong evidence of secondary structural equivalence.
Near-UV CD extends the analysis to tertiary structure by measuring the optical activity of aromatic amino acid side chains (tryptophan at 295 nm, tyrosine at 275-285 nm, phenylalanine at 250-270 nm) and disulfide bonds. These chromophores are sensitive to their local three-dimensional environment; their CD signals change when the protein’s fold changes. Near-UV CD spectra are therefore a fingerprint of the protein’s tertiary structure in the region around its aromatic residues, and spectral overlay between biosimilar and RP provides tertiary structural similarity evidence.
FTIR spectroscopy provides an independent assessment of secondary structure through the vibrational frequencies of the protein backbone amide bonds. The amide I band (1600-1700 cm-1) is particularly sensitive to secondary structure, with alpha-helices absorbing around 1656 cm-1 and beta-sheets around 1632 cm-1 and 1680 cm-1. FTIR is especially valuable for quantifying beta-sheet content, where it can be more sensitive than far-UV CD. It is also less susceptible to interference from certain buffer components, making it useful when CD measurements are constrained by formulation requirements.
Two-dimensional NMR spectroscopy occupies a unique position in the HOS toolkit. A 2D 1H-15N HSQC experiment produces a spectrum in which each cross-peak corresponds to a chemically distinct nitrogen-hydrogen pair in the protein — approximately one per backbone amide and additional signals from side chain amide groups. The chemical shift of each peak is exquisitely sensitive to the local electronic environment, meaning the precise three-dimensional position of each atom in the protein. The 2D-NMR spectrum is therefore the highest-resolution structural fingerprint available for solution-state HOS comparison. The FDA’s own laboratory employs 2D-NMR for HOS assessment of biosimilars, and the technique has been validated for this purpose in multiple published regulatory science studies including FDA work on filgrastim biosimilars. If two proteins produce overlapping 2D-NMR fingerprints, the probability that they differ meaningfully in three-dimensional structure is very low.
Hydrogen-deuterium exchange mass spectrometry (HDx-MS) reports on protein dynamics and solvent accessibility at peptide-level resolution. The exchange rate of backbone amide hydrogens for deuterium is governed by hydrogen bonding and solvent exposure — both functions of local and global protein structure. A comparative HDx-MS experiment maps the exchange kinetics across the entire protein sequence for both biosimilar and RP, revealing any regions where the biosimilar’s structural dynamics differ. HDx-MS is particularly valuable for detecting subtle conformational differences in large proteins where the other spectroscopic methods might lack the resolution to localize a difference.
Differential scanning calorimetry (DSC) measures the thermal energy required to unfold the protein as temperature increases. The resulting thermogram, with its characteristic melting temperature (Tm) and enthalpy of unfolding, reports on the overall thermodynamic stability of the protein. Comparable DSC thermograms between biosimilar and RP — including the same number of unfolding transitions at the same temperatures, indicating the same domain unfolding sequence — are strong evidence that the two molecules have the same three-dimensional fold and comparable stability under thermal stress.
Key Takeaway (Section 3.3): The FDA-aligned minimum HOS package for a monoclonal antibody includes far-UV and near-UV CD, FTIR, and 2D-NMR or HDx-MS. Programs that invest in all five methods, with proper sample preparation and statistical comparison across multiple RP lots, build the strongest case for waiving the apex clinical study. Programs that cut corners on HOS characterization face the highest risk of regulatory deficiency letters and forced clinical escalation.
3.4 Glycosylation: The Hardest CQA to Match
Of all the structural differences that can exist between a biosimilar and its reference product, glycosylation differences are the most consequential and the most difficult to control. Glycosylation is not encoded in the DNA; it is determined by the host cell’s enzymatic glycosylation machinery, which is in turn exquisitely sensitive to cell culture conditions. Two CHO cell clones expressing the same protein sequence will produce different glycan profiles. The same clone grown in different media or at different pH will produce different glycan profiles. Different bioreactor geometries will produce different glycan profiles. This is the most concrete manifestation of ‘the process is the product.’
For an IgG1 monoclonal antibody, N-linked glycosylation occurs at Asn297 in the Fc region. The glycan at this position is not a single structure but a heterogeneous mixture of biantennary complex-type oligosaccharides that differ in their terminal galactose content (G0, G1, G2), fucose content (afucosylated vs. core-fucosylated), and sialic acid content. This glycoform distribution directly affects the antibody’s effector functions. Afucosylated glycoforms, for example, bind the FcgammaRIIIa receptor on NK cells with dramatically higher affinity than core-fucosylated forms, resulting in substantially enhanced ADCC activity. A biosimilar with a higher proportion of afucosylated glycans than its RP would be expected to have stronger ADCC activity — a functional difference that could be clinically relevant for an oncology antibody.
Glycan analysis requires a multi-level approach. Released glycan profiling by HILIC-FLR-MS is the standard method: glycans are enzymatically released from the protein (PNGase F for N-glycans), fluorescently labeled, separated by hydrophilic interaction chromatography with fluorescence detection, and identified by mass spectrometry. This provides a quantitative profile of the relative abundance of each glycoform species. Site-specific glycan analysis by LC-MS/MS peptide mapping of non-reduced and reduced samples provides orthogonal confirmation of glycosylation site occupancy and glycoform distribution at each specific site. For proteins with multiple glycosylation sites — some glycoprotein hormones and fusion proteins carry four to eight sites — site-specific analysis is essential because glycan profiles can differ substantially between sites.
Matching the glycan profile of an innovator biologic is the primary process development challenge in most biosimilar programs. CHO cell line selection, specifically choosing a clone whose basal glycosylation machinery produces a glycoform distribution closest to the RP target, is the first lever. Cell culture media supplementation with specific sugars, ammonium, manganese, or other modulators is the second lever. Bioreactor pH, temperature, and dissolved oxygen control affect glycosyltransferase activity and therefore glycan processing. Downstream processing steps, including the choice of viral inactivation pH and hold conditions, can modify glycan structures post-biosynthesis. Getting all these levers right simultaneously, consistently, and at manufacturing scale is what separates the companies with genuine biosimilar manufacturing capability from those who cannot clear the technical barrier.
Key Takeaway (Section 3.4): Glycan profile matching is the rate-limiting technical step in most monoclonal antibody biosimilar programs. Programs should budget for extensive cell line screening, media optimization, and DOE-based process characterization studies specifically targeting Fc glycan distribution control before initiating formal comparability studies. The cost of this upstream work is orders of magnitude less than the cost of a failed comparability package.
3.5 Impurity Profiling: Aggregates, Fragments, and Host Cell Proteins
The purity profile of a biosimilar must be comparable to that of the reference product. Impurities in a biologic therapeutic fall into two categories: product-related variants and process-related impurities. Both carry safety implications, particularly for immunogenicity.
Product-related aggregates are the primary size-variant concern. Protein aggregates — dimers, oligomers, and higher-order species — are immunogenic because they present repetitive epitopes that can cross-link B-cell receptors and drive antibody responses, including potentially neutralizing responses to the therapeutic protein and, in worst cases, to the patient’s own endogenous protein. The standard analytical tool for aggregate quantitation is size-exclusion chromatography with UV and multi-angle light scattering detection (SEC-MALS). SEC provides a rapid, high-throughput measurement of the monomer, dimer, and higher oligomer content of a sample. Its limitation is the potential for on-column artifacts: protein-column interactions can cause aggregates to dissociate during analysis, underestimating the true aggregate content in the formulation.
Analytical ultracentrifugation (AUC) is the orthogonal method mandated by FDA and EMA guidance for aggregate characterization. AUC separates molecules by their sedimentation coefficient, a function of molecular mass and shape, in the ultracentrifuge’s gravitational field. Because there is no column matrix and the analysis occurs in the actual sample buffer, AUC avoids the on-column interaction artifacts of SEC and provides a more accurate measurement of the aggregate population, particularly for small, reversible aggregates that SEC might miss. The regulatory expectation is that both SEC and AUC are applied and that their results are interpreted together. A discrepancy — for example, SEC showing 0.5% aggregate but AUC showing 3% — is diagnostically important and must be investigated.
Protein fragments — generated by non-enzymatic peptide bond cleavage, particularly in flexible hinge regions or linker sequences — reduce potency proportionally to their abundance and may expose new epitopes. SEC and, for smaller fragments, capillary electrophoresis under non-reducing and reducing conditions (non-reducing to distinguish between fragments and partially reduced intact chains) are standard characterization tools.
Host cell proteins (HCPs) are the residual proteins derived from the CHO or other host cell system used to manufacture the therapeutic. Despite extensive purification, trace levels of HCPs persist in the final drug product. These are a safety concern because they are, by definition, foreign proteins with immunogenic potential. HCP profiles differ between manufacturing processes — the specific HCPs present and their relative abundance depend on the host cell line, the culture conditions, and the purification train — meaning a biosimilar and its RP will have quantitatively and qualitatively different HCP impurity profiles. Demonstrating that the biosimilar’s HCP level is within the range of the RP, and that the HCP profile does not contain any specific immunogenic proteins not present in the RP at comparable levels, requires a combination of enzyme-linked immunosorbent assay (ELISA) with process-specific HCP antibodies and orthogonal mass spectrometry-based HCP identification.
Key Takeaway (Section 3.5): The orthogonal characterization table below is the practical tool for designing the analytical comparability package. Developers should treat it as a minimum specification, not a ceiling.
Orthogonal Analytical Characterization Blueprint
Quality Attribute
Primary Method
Orthogonal Method(s)
Rationale
Primary Sequence
LC-MS/MS Peptide Mapping (trypsin)
LC-MS/MS Peptide Mapping (Lys-C, Asp-N); Intact Mass by ESI-MS
Multiple digestion enzymes close coverage gaps; intact mass confirms overall molecular identity
Secondary Structure
Far-UV Circular Dichroism
FTIR (Amide I band analysis)
CD measures polarized light absorption by backbone; FTIR measures amide bond vibrations; orthogonal to artifacts from buffer interference
NMR provides atomic-resolution fingerprint; CD and fluorescence probe aromatic environments; HDx-MS maps solvent accessibility dynamics
Quaternary Structure / Disulfide Bonds
Non-Reduced Peptide Mapping (LC-MS/MS)
SEC-MALS (stoichiometry); DSC (domain stability)
Peptide mapping locates specific disulfide pairs; SEC-MALS confirms subunit stoichiometry; DSC reports on domain-level stability
N-Glycan Profile
Released Glycan HILIC-FLR-MS
Site-Specific Glycan LC-MS/MS; Intact/Subunit MS
Released glycan analysis quantifies total glycoform pool; peptide mapping provides site-specific glycoform distribution; intact mass reveals major glycoforms on the intact protein
Charge Variants
Cation-Exchange Chromatography (CEX)
Capillary Isoelectric Focusing (cIEF); Image cIEF
CEX separates by charge interaction with stationary phase; cIEF separates by isoelectric point in pH gradient — different selectivities reveal different variant populations
SEC-MALS quantifies and sizes peaks; AUC-SV avoids column artifacts for reversible aggregates; DLS detects sub-visible particle populations
Fragments
SEC; Non-Reducing CE-SDS
Reducing CE-SDS; LC-MS of intact subunits
SEC separates by size; CE-SDS separates under denaturing conditions; comparing non-reducing vs. reducing CE-SDS distinguishes inter- vs. intra-chain fragments
PTMs (Oxidation, Deamidation, Pyroglutamate)
LC-MS/MS Peptide Mapping
Peptide Mapping under different digestion conditions
Site-specific quantitation of each modification; orthogonal digestion confirms findings and closes peptide coverage gaps
Host Cell Proteins
Process-Specific HCP ELISA
LC-MS/MS HCP Identification
ELISA provides total HCP quantitation with process-specific antibody coverage; MS identifies specific HCP species for safety assessment
Section 4: From Structure to Action: Functional Equivalence
4.1 The Structure-Function Relationship as the Scientific Rationale for the Biosimilar Pathway
Every analytical similarity argument rests on the structure-function relationship: the principle that biological activity is determined by molecular structure. If the biosimilar’s structure is highly similar to the reference product’s structure, its function should be highly similar. The functional assay panel tests this hypothesis directly by measuring biological activity in systems that replicate the drug’s mechanism of action.
Functional data serves two roles in the regulatory package. First, it provides independent evidence of similarity that is more biologically relevant than any structural measurement. Second, it contextualizes structural differences. If a glycan profile comparison reveals that the biosimilar has a slightly higher proportion of afucosylated Fc glycoforms than the RP, the ADCC bioassay result answers the consequential question: does this difference translate into a measurable functional difference? If the ADCC activity of the biosimilar is within the range of the RP across multiple RP lots, the structural difference is likely within the range of natural RP variability and can be characterized as clinically inactive. If the ADCC activity is demonstrably elevated, process development intervention is required.
This iterative feedback loop — from structural difference to functional assay to process parameter adjustment — is the core operational logic of late-stage cell culture and downstream process development in a biosimilar program. The analytical and functional teams must operate in close coordination throughout this phase, which is one reason why programs that outsource these functions to separate CDMOs without integrated data management face higher risk of miscommunication and delayed course correction.
Key Takeaway (Section 4.1): The functional assay panel is the bridge between analytical characterization and clinical translatability. It is the strongest regulatory argument for waiving the apex clinical study, because a well-validated, sensitive bioassay panel detecting equivalent activity across all mechanisms of action makes the clinical trial redundant from an evidentiary standpoint.
4.2 Binding Assays: Kinetics, Affinity, and the Fc Receptor Panel
Binding assays measure the fundamental interaction between the therapeutic protein and its molecular targets. For a monoclonal antibody, the binding panel covers the therapeutic target (e.g., TNF-alpha for adalimumab, VEGF for bevacizumab, HER2 for trastuzumab), the neonatal Fc receptor (FcRn, which governs half-life), FcgammaRIIIa (which mediates ADCC), FcgammaRIIb (an inhibitory Fc receptor), C1q (complement), and FcgammaRIIa (relevant for platelet function and some inflammatory mechanisms).
Surface plasmon resonance (SPR) is the standard platform for kinetic characterization. The reference product or binding partner is immobilized on a sensor chip surface. Analyte in solution flows over the chip surface, and binding is detected in real time as a change in the refractive index at the chip surface, reported as response units (RU). The resulting sensorgrams are fit to a kinetic model to extract the association rate constant (ka), dissociation rate constant (kd), and equilibrium dissociation constant (KD = kd/ka). Comparing ka, kd, and KD values for the biosimilar and RP at each target provides a quantitative kinetic similarity assessment. SPR is sensitive to small differences in binding kinetics, making it a capable tool for detecting differences that would not be apparent from endpoint binding assays.
Bio-layer interferometry (BLI) provides an orthogonal kinetic measurement using a different physical principle: interference of white light reflected from the biosensor tip surface changes as analyte binds and dissociates. BLI is particularly useful for rapid screening of multiple concentration points and is increasingly applied as the orthogonal confirmation of SPR results in the regulatory package.
Key Takeaway (Section 4.2): The complete Fc receptor binding panel — FcRn, FcgammaRIIIa (both V/V and F/F158 allotypes), FcgammaRIIa, FcgammaRIIb, and C1q — is a regulatory expectation for monoclonal antibody biosimilars, not optional characterization. Shortcutting the panel creates regulatory risk. Both V/V and F/F allotypes of FcgammaRIIIa must be tested because the receptor’s binding affinity and the glycan sensitivity of that binding differ between allotypes.
4.3 Cell-Based Potency Assays: Capturing Clinical Mechanism In Vitro
Cell-based bioassays are the most clinically relevant functional measurements in the comparability package. They measure the downstream consequence of target binding in living cells: cell killing, cytokine neutralization, proliferation inhibition, receptor signaling modulation, or apoptosis induction. The specific assays required depend on the reference product’s mechanism of action.
For antibodies with ADCC as a mechanism of action — trastuzumab, rituximab, obinutuzumab — the ADCC assay is a critical quality attribute. The current regulatory-preferred method uses an engineered Jurkat cell line expressing FcgammaRIIIa (the ‘Jurkat-ADCC reporter’ assay), which produces a luciferase signal proportional to receptor activation when the antibody crosslinks FcgammaRIIIa on the effector cell to the target cell’s surface antigen. This assay is more reproducible than classical NK cell-based ADCC assays using primary donor NK cells, which introduce substantial donor-to-donor variability. The reproducibility advantage is important for demonstrating statistical equivalence across the biosimilar and multiple RP lots.
Complement-dependent cytotoxicity (CDC) assays are relevant for antibodies that fix complement — rituximab being the canonical example. CDC assays use human complement serum and antibody-coated target cells; cell viability is measured as the readout. CDC is more difficult to quantify precisely than ADCC because complement activity in donor serum is variable and the cell lysis endpoint has a narrower dynamic range. Careful assay development and donor serum qualification are prerequisites for a CDC assay of sufficient quality for regulatory submission.
For cytokine-neutralizing antibodies (adalimumab, etanercept, infliximab), the primary bioassay is a cell-based neutralization assay measuring the antibody’s ability to block cytokine-induced cellular signaling, typically TNF-alpha-induced cytotoxicity in a TNF-sensitive cell line or reporter gene activation. Potency is expressed as the IC50 or the relative potency compared to a reference standard, with the RP used as the assay calibrator.
Key Takeaway (Section 4.3): Regulatory submissions increasingly expect bioassay results to be presented with full relative potency statistics, including 95% confidence intervals, parallelism testing results, and a comparison against multiple RP lots rather than a single lot. Programs that run their biosimilar against only one or two RP lots in bioassay comparisons have an incomplete package and face requests for additional data.
Section 5: The Science of Replication: Manufacturing, Bioprocess Engineering, and Formulation IP
5.1 Cell Line Development: The Founding Decision
Every element of the biosimilar’s quality attribute profile flows from the producing cell line. The choice of expression system, the selection of the specific production clone, and the development of a stable, high-producing cell bank are the founding decisions of the manufacturing program, with implications that extend through process characterization, scale-up, regulatory filing, and patent strategy.
Chinese Hamster Ovary (CHO) cells are the standard mammalian expression system for complex glycoproteins, including nearly all commercially relevant monoclonal antibodies. CHO cells are capable of human-compatible N-glycosylation and have a long history of regulatory acceptance. The specific CHO subclone — CHO-K1, CHO-S, CHO-DG44, or proprietary variants developed by CDMOs and major manufacturers — influences the basal glycosylation enzyme expression profile and therefore the glycan distribution produced on the target protein. A developer seeking to match a reference product with a specific glycan profile will screen multiple CHO subclones to identify the one whose basal glycosylation machinery produces the closest starting point.
Clone selection from a transfection pool involves screening hundreds to thousands of individual cell clones for both productivity (measured as specific productivity, qp, in picograms per cell per day) and quality profile (measured by the CQA panel, with glycosylation as the primary focus). The challenge is that high-producing clones do not always produce the best-matching quality profile, and vice versa. The selection process therefore requires optimizing a multi-dimensional objective function, not a single variable. Proprietary high-throughput screening platforms — including microfluidic droplet sorting (e.g., Berkeley Lights Beacon, Sphere Fluidics Cyto-Mine), image-based screening (Sartorius Incucyte), and miniaturized fed-batch production models in 96-well or 24-well plates — have compressed the clone selection timeline from twelve to eighteen months (the historical benchmark) to three to six months in leading programs.
Key Takeaway (Section 5.1): Cell line development is on the critical path for every biosimilar program and should be resourced as such. Programs that underinvest in clone screening depth — selecting from too few candidates — accept higher risk of quality attribute mismatch that becomes apparent only in later process characterization studies, at a much higher cost to remediate.
5.2 Bioprocess Development: From Shake Flask to Commercial Scale
Once a production clone is selected, the upstream bioprocess must be developed and optimized to consistently produce the target molecule within the defined CQA ranges. This means developing a fed-batch or perfusion bioreactor process with defined media composition, feeding strategy, pH, temperature, and dissolved oxygen control — and demonstrating that the process remains within control limits from the smallest development scale through commercial manufacturing.
Scale-up from development scale (1-50L bioreactors) to clinical manufacturing scale (200-2,000L) to commercial scale (2,000-15,000L) is one of the highest-risk transitions in biopharmaceutical manufacturing. The physics of large-scale bioreactors differ fundamentally from those of small-scale systems. Oxygen and carbon dioxide mass transfer, nutrient and pH gradients within the bioreactor volume, hydrodynamic shear stress from agitation, and foam formation all behave differently at scale. These differences affect cell viability, specific productivity, and, critically, the quality attribute profile of the product — particularly glycosylation. A process that produces a perfect glycan match to the RP at 50L may produce a shifted glycan profile at 2,000L if the scale-up does not account for the changed environmental conditions experienced by the cells.
Mechanistic and computational fluid dynamics (CFD) modeling of bioreactor hydrodynamics is increasingly used to predict scale-dependent effects and design scale-down models that accurately represent the commercial-scale environment. Scale-down models — small-scale bioreactors whose operating parameters are designed to mimic the mass transfer and mixing characteristics of the commercial-scale vessel — are essential tools for process characterization studies performed at scale-down during regulatory development. FDA and EMA expect that scale-down model qualification data be included in the manufacturing section of the biosimilar application.
Downstream processing — the purification train that takes the crude cell culture harvest and produces the purified drug substance — is also a source of quality attribute variability. Protein A affinity chromatography (for IgG antibodies) is nearly universal and provides the primary capture step. Subsequent ion-exchange chromatography steps (cation-exchange and/or anion-exchange, depending on the target protein’s charge properties) remove HCPs, DNA, aggregates, and other process-related impurities. Hydrophobic interaction chromatography provides additional aggregate and HCP clearance. Viral inactivation (typically by low-pH incubation) and viral filtration are mandatory biosafety steps. Each of these steps affects the final product’s quality attribute profile, and the parameters of each step (pH, salt concentration, column loading density, wash and elution conditions) must be characterized to establish a design space that reliably produces a product within specification.
Key Takeaway (Section 5.2): Manufacturing development and process characterization represent approximately 30-40% of total biosimilar program cost. Programs that treat this phase as a cost center to be minimized, rather than as the foundation of the entire regulatory and commercial strategy, face a high probability of comparability failures that are far more expensive to remediate than to prevent.
5.3 Formulation: Stability, Patient Experience, and IP Navigation
The drug product formulation — the combination of the purified therapeutic protein with buffers, stabilizers, surfactants, and tonicity agents — is the final determinant of the molecule’s physical stability over its shelf life and of the patient experience during administration.
A biosimilar developer may choose to replicate the innovator’s formulation or develop a novel one. The innovator’s formulation is published in the prescribing information and is generally not independently patent-protectable as a trade secret, but specific formulation compositions and stabilization approaches are frequently the subject of secondary patents. AbbVie’s Humira, for example, is formulated in a citrate-containing buffer at high concentration. AbbVie holds patents on the citrate-free, high-concentration formulation introduced later in the product’s lifecycle (marketed as the low-volume auto-injector), as citrate is the primary cause of injection site pain in adalimumab formulations. Several adalimumab biosimilar developers chose to develop citrate-free formulations not only to improve patient experience but specifically to navigate AbbVie’s citrate-free formulation patent.
This is a textbook example of formulation innovation as freedom-to-operate strategy. A novel formulation that demonstrably improves on the reference product’s patient experience — reduced injection volume, reduced injection site pain, improved stability allowing room-temperature storage — simultaneously provides a commercial differentiation point, a potential IP position, and potentially earlier market entry by avoiding a specific patent claim. The commercial, IP, and formulation development teams must coordinate on this decision early, as it drives downstream analytical work (demonstrating comparability in the new formulation), regulatory strategy (justifying the formulation difference to regulators), and marketing positioning.
High-concentration formulations present a specific technical challenge: protein-protein interactions become significant at concentrations above 50-100 mg/mL, and viscosity increases non-linearly. High viscosity impedes syringeability, requiring injection forces that can be problematic for auto-injectors designed for patient self-administration. Formulation scientists use a combination of excipient screening (amino acids, sugars, polyols, specific surfactants) and computational modeling of protein-protein interaction potentials (B22, kD parameters measured by DLS and SEC) to develop low-viscosity, high-concentration formulations. This is an active area of formulation science where academic literature and proprietary know-how intersect.
Key Takeaway (Section 5.3): Formulation strategy decisions should be made in the context of a comprehensive patent landscape review conducted before preclinical development begins, not after. Late-stage discovery of a blocking formulation patent is a high-impact, often avoidable program risk.
Section 6: Clinical Confirmation, Global Regulatory Pathways, and the Interchangeability Calculus
6.1 The Clinical Program: Confirmation, Not Re-Demonstration
The biosimilar clinical development program has a specific, narrow evidentiary goal: confirm that there are no clinically meaningful differences between the biosimilar and the reference product. This is a different objective from the innovator’s clinical program, which establishes the clinical benefit of a new molecule. The biosimilar’s clinical program is a comparative exercise, not an exploratory one.
The cornerstone is the comparative pharmacokinetic (PK) study. For most systemic biologics, this study is conducted in healthy volunteers using a crossover design (single dose of biosimilar, then single dose of RP, or vice versa, in the same subjects) or a parallel design (separate cohorts receiving biosimilar and RP). The primary endpoints are AUC (area under the concentration-time curve, measuring total drug exposure) and Cmax (maximum concentration). For the study to support a biosimilarity conclusion, the 90% confidence intervals for the geometric mean ratios of AUC and Cmax must fall within 80-125%, the standard bioequivalence interval. These PK parameters are measured using a validated immunoassay or LC-MS/MS assay for the drug.
Where a pharmacodynamic biomarker exists that is mechanistically linked to the drug’s effect and is measurable in a short-term study, comparative PD data strengthens the clinical package. For TNF-alpha inhibitors, soluble TNF-alpha levels or C-reactive protein suppression can serve as PD markers. For VEGF inhibitors, circulating VEGF levels may be measured. PD data does not replace the PK study; it supplements it.
The comparative immunogenicity assessment is mandatory and involves monitoring for anti-drug antibodies (ADA) at defined timepoints throughout the clinical study. The ADA assay strategy must include a sensitive screening assay, a confirmatory assay to distinguish true positives from non-specific binding, and a neutralizing antibody (NAb) assay to determine whether detected ADAs inhibit drug activity. Comparable ADA incidence, ADA titers, and NAb rates between the biosimilar and RP groups are required to support a biosimilarity conclusion.
Key Takeaway (Section 6.1): The clinical PK study in healthy volunteers is the rate-limiting step for most biosimilar programs in terms of timeline. It requires regulatory submission and approval of a clinical trial application, recruitment, dosing, sample collection, bioanalytical assay execution, and statistical analysis — typically adding twelve to eighteen months to the program after the manufacturing and analytical package is ready. Delays in bioanalytical assay development are a common but underappreciated cause of clinical study timeline extension.
6.2 The Confirmatory Clinical Efficacy Study: Science, Cost, and the Case for Waiver
The confirmatory comparative clinical efficacy study represents the largest single cost item in most biosimilar programs. A Phase 3 equivalence study in, for example, rheumatoid arthritis patients comparing an adalimumab biosimilar to Humira across 500-800 patients over 52 weeks costs approximately $80M-$150M. This estimate covers investigator fees, site costs, drug supply, contract research organization fees, and regulatory interactions. It does not include the opportunity cost of the manufacturing and analytical team’s time spent supporting the study.
The scientific justification for this cost is increasingly weak. Statistical power calculations for these studies are typically powered to detect a difference of 15% or more in the primary efficacy endpoint — for example, the proportion of patients achieving ACR20 response at week 24 in RA. A product with the structural and functional similarity demonstrated by a comprehensive analytical and bioassay package is expected to produce ACR20 response rates differing from the RP by less than 2-3%, well within the noise of clinical trial variability. The probability that such a study will detect a clinically meaningful difference when the analytical evidence is strong approaches zero.
The EMA reached a version of this conclusion in its 2023 Reflection Paper on Tailored Clinical Approaches in Biosimilar Development. The paper describes a framework in which the confirmatory clinical study may be waived or replaced by extended clinical PK/PD data when the analytical similarity evidence is robust and the molecule is well-characterized. Multiple EMA biosimilar approvals since 2020 have proceeded without a confirmatory efficacy study, relying on analytical, functional, and PK/PD data alone. Omnitrope (somatropin), Benepali (etanercept), and several more recent antibody biosimilars have followed this streamlined pathway in the EU.
FDA has been more conservative, but the direction is clear. The agency’s 2023 draft guidance on statistical approaches to demonstrating biosimilarity explicitly discusses the use of analytical similarity data as a primary evidentiary element. Several FDA advisory committee meetings have featured expert panels arguing that well-characterized biosimilars with comprehensive analytical packages should not require confirmatory clinical studies. No FDA policy change has formally codified this position, but the advisory committee record is a leading indicator.
Key Takeaway (Section 6.2): Programs that invest in building the strongest possible analytical and functional similarity package — broader than the regulatory minimum, with statistical comparison across the maximum feasible number of RP lots — are building the evidentiary case for clinical study waiver. The ROI on incremental analytical investment is potentially 10-20x in avoided clinical costs if waiver is achieved.
6.3 FDA vs. EMA: The Regulatory Divergence Table
Feature
FDA (United States)
EMA (European Union)
Statutory Pathway
Section 351(k) of the PHS Act (BPCIA, 2009)
EU Directive 2001/83/EC, Article 10(4); first guidelines 2005
First Biosimilar Approved
Zarxio (filgrastim-sndz), 2015
Omnitrope (somatropin), 2006
Biosimilarity Standard
‘Highly similar’ with ‘no clinically meaningful differences’ in safety, purity, potency
‘Highly similar’ in quality, safety, and efficacy
Totality of Evidence
Yes — explicit regulatory standard
Yes — explicit regulatory standard
Reference Product Sourcing
Requires bridging to US-licensed RP; 3-way comparative studies typically needed for global programs
Accepts non-EU RP with scientific bridging justification
Interchangeability Designation
Separate statutory designation requiring switching study data; permits pharmacy-level substitution subject to state law
No separate designation; all approved biosimilars considered scientifically interchangeable; substitution policy determined by member state
Innovator Data Exclusivity
12 years from first approval
8 years data exclusivity + 2 years market protection (+1 year for new indication)
Clinical Efficacy Study Trend
Required for complex molecules; waiver increasingly discussed but not yet formally codified
Waiver increasingly accepted per 2023 Reflection Paper; multiple approvals without confirmatory study since 2020
6.4 Interchangeability: The US-Specific High-Stakes Calculation
Biosimilar interchangeability in the US is a statutory designation with direct commercial consequences. An interchangeable biosimilar can be substituted for the reference product at the pharmacy without prescriber intervention, in any US state with a pharmacy substitution law (currently nearly all states). This is the mechanism that drives generic drug market penetration to 80-90% within twelve months of launch, and it is the commercial prize that many biosimilar developers are pursuing.
The regulatory requirements for interchangeability go beyond standard biosimilarity. The developer must demonstrate that the product can be expected to produce the same clinical result in any given patient and, for products administered more than once, that alternating or switching between the interchangeable product and the RP does not produce greater safety or immunogenicity risk than continued use of the RP. This second requirement is the practical barrier: it demands a dedicated switching study in which patients transition between the RP and the proposed interchangeable product across multiple switch cycles.
The switching study design is complex and expensive. FDA guidance describes a study design in which subjects are alternated between RP and biosimilar (or vice versa) across three to five switch cycles, with immunogenicity and PK monitoring at each transition. The study must be powered to demonstrate that the switch groups are not inferior to the non-switch group (RP only) on safety and immunogenicity endpoints. Depending on the molecule and the target population, such a study may require 200-600 subjects and twelve to eighteen months of follow-up, at a cost of $30M-$80M on top of the standard biosimilar program cost.
The commercial logic for pursuing interchangeability is straightforward in theory but complicated in practice. For high-volume, subcutaneously self-administered products — insulin, adalimumab, etanercept — pharmacy-level substitution is a powerful driver of market penetration and does not require the sales force investment needed to convince individual physicians to switch stable patients. For hospital-administered infusion products — infliximab, rituximab, bevacizumab, trastuzumab — the purchasing decision is made by hospital pharmacy and therapeutics (P&T) committees and group purchasing organizations (GPOs), and formulary positioning matters more than interchangeability. Several biosimilar developers for infusion products have concluded that the cost of the switching study exceeds the commercial benefit of the interchangeability designation and have not pursued it.
Key Takeaway (Section 6.4): The interchangeability calculation should be made early in program design and should incorporate a bottom-up forecast of the commercial premium attributable to pharmacy-level substitution versus the fully loaded cost of the switching study, including the opportunity cost of the delayed launch timeline if the switching study extends the development period.
Section 7: The IP Battlefield: Patent Thickets, the Patent Dance, and Freedom-to-Operate
7.1 The Patent Thicket: Anatomy of an Evergreening Strategy
The ‘patent thicket’ is the innovator’s primary tool for extending commercial exclusivity beyond the primary composition-of-matter patent. For a biological drug, the primary patent covers the molecule itself — the amino acid sequence of the therapeutic protein, the nucleic acid encoding it, or the method of producing it using a specific cell line. The primary patent typically expires twelve to twenty years after filing, though patent term restoration provisions under the Hatch-Waxman Act can extend the effective exclusivity period.
Secondary patents, filed at any point during the product’s commercial life, cover the periphery. For a therapeutic antibody, the secondary patent landscape may include formulation patents (specific excipient compositions, buffer systems, protein concentrations), manufacturing process patents (cell culture media compositions, bioreactor control parameters, purification methods), device patents (auto-injector design, needle gauge, dose volume), dosing regimen patents (specific dose amounts, dosing intervals, treatment schedules), and method-of-use patents (treatment of specific patient subpopulations, combination therapies, biomarker-guided treatment selection).
AbbVie’s Humira portfolio is the most thoroughly documented example of this strategy. At peak, the Humira patent estate encompassed more than 250 patents. The primary composition-of-matter patent expired in 2016, but AbbVie’s formulation and device patents extended effective exclusivity in the US market until 2023, seven years longer. EU biosimilar entry occurred in 2018, five years before US entry. This temporal gap, entirely attributable to the asymmetric patent landscape between jurisdictions, cost the US healthcare system an estimated $20 billion in excess drug spending over that five-year period according to RAND Corporation analysis.
AbbVie’s strategy has been replicated, with varying degrees of effectiveness, by Janssen (infliximab/Remicade), Amgen (etanercept/Enbrel), Roche (trastuzumab/Herceptin, bevacizumab/Avastin, rituximab/Rituxan), Regeneron (aflibercept/Eylea), and Johnson & Johnson (ustekinumab/Stelara). The pattern is consistent: primary patent expiration triggers a wave of secondary patent filings designed to create the maximum legal uncertainty for potential biosimilar entrants.
A 2022 study in PLOS Medicine found that the US patent system, on average, supports nine to twelve times more patents per biologic than comparable systems in Canada and the UK. The authors attributed this to the US continuation patent system (allowing new claims to be filed on pending applications, effectively ‘evergreening’ the filing date), the absence of a robust ‘obvious’ standard for secondary pharmaceutical patents, and aggressive PTO practice in prosecuting pharmaceutical claims. This structural difference in patent density is the primary explanation for why biosimilar adoption in the US lags EU adoption by years for most products.
Key Takeaway (Section 7.1): Freedom-to-operate analysis for a biosimilar program must cover the full secondary patent landscape, not just the primary composition-of-matter patent. Programs that identify the primary patent expiration and assume commercial freedom to market from that date are making a potentially catastrophic planning error. The secondary patent review is not a legal formality; it is a core strategic input to the program initiation decision.
7.2 The Patent Dance: A Step-by-Step Operational Guide
The BPCIA’s patent information exchange procedure — the ‘patent dance’ — is the US-specific mechanism for managing pre-launch patent disputes between biosimilar applicants and reference product sponsors. It is structured to surface patent issues early, ideally resolving them before commercial launch through negotiation or early-stage litigation, rather than triggering a broad injunction battle at the moment of market entry.
Engagement with the patent dance is technically optional: the Supreme Court’s 2017 ruling in Sandoz Inc. v. Amgen Inc. confirmed that a biosimilar applicant may decline to provide its application to the reference product sponsor (Step 1 of the dance). However, declining triggers its own risks. Under the BPCIA, a sponsor who does not receive the biosimilar application may seek immediate injunctive relief in federal court based on artificial act of infringement under 35 U.S.C. Section 271(e)(2)(C). The injunction risk is broader and less predictable outside the structured dance framework.
The 180-day notice of commercial launch (Step 7 below) is not contingent on completing the earlier steps of the dance. Regardless of whether the application exchange occurred, a biosimilar applicant must provide 180 days’ advance commercial launch notice to the reference product sponsor. This notice triggers the second wave of litigation on patents not yet litigated in the first wave, and it defines the launch date for market access planning.
Step-by-Step Patent Dance Reference
Step
Statutory Timeline
Biosimilar Applicant Action
Reference Product Sponsor Action
1
Within 20 days of FDA accepting BLA for review
Provides FDA-accepted BLA and manufacturing information to sponsor under confidentiality
—
2
Within 60 days of receiving Step 1 package
—
Provides list of patents believed to be infringed; identifies patents willing to license
3
Within 60 days of receiving Step 2 list
Provides claim-by-claim statement of invalidity, unenforceability, or non-infringement for each listed patent
—
4
Within 60 days of receiving Step 3 statement
—
Provides claim-by-claim rebuttal to applicant’s Step 3 positions
5
15-day negotiation period after Step 4
Engages in good-faith negotiation to agree on patents for immediate litigation
Engages in good-faith negotiation to agree on patents for immediate litigation
6
30 days after negotiation period
—
Files suit on agreed patents (or, if no agreement, on a number of patents determined by an exchange of lists per statutory formula)
7
Not less than 180 days before commercial marketing
Provides notice of intent to commercially market
May file suit on patents identified in Step 2 but not litigated in Step 6
Key Takeaway (Section 7.2): Patent dance strategy should be designed by litigation counsel with specific biosimilar BPCIA experience before the BLA is filed. The decisions made at each step — what to include in the manufacturing information disclosure, how to frame the invalidity arguments in Step 3, which patents to concede to an early litigation list — have material consequences for the speed of market entry and the scope of patent exposure.
7.3 Freedom-to-Operate: The Proactive Defense
Freedom-to-operate (FTO) analysis is not a one-time legal exercise; it is a continuous intelligence function that should be embedded in the development program from initiation to commercial launch. The FTO landscape changes throughout development as new patents are granted, continuation applications mature, and the biosimilar’s own process and formulation design evolve.
An initial FTO review at program initiation should map all granted patents and published applications in the primary filing jurisdictions (US, EU, Japan, China, Canada, Australia) with claims that could potentially cover the reference molecule, its manufacture, its formulation, or its administration. This review should be performed by patent counsel with relevant technical expertise, not by generalist attorneys unfamiliar with protein chemistry or biomanufacturing.
Process design-around strategies are the most technically demanding FTO response. If a key manufacturing step — for example, a specific Protein A chromatography elution condition — is covered by an innovator patent, the development team must identify an alternative process that achieves equivalent product quality without infringing the claim. This requires close collaboration between the patent counsel and the process development scientists, with each proposed design-around being evaluated for both technical feasibility and legal defensibility.
Paragraph IV-equivalent challenges are available in the biosimilar context through the inter partes review (IPR) and post-grant review (PGR) procedures at the Patent Trial and Appeal Board (PTAB). IPR petitions challenging the validity of specific patent claims have become a standard element of biosimilar IP strategy. PTAB has a higher rate of claim cancellation than district courts for the same patents, making IPR a cost-effective tool for eliminating weak secondary patents that are blocking or delaying market entry. Coordinating IPR petitions with the patent dance timeline requires careful planning, as PTAB and district court proceedings run on different schedules and the strategic interaction between them is complex.
Key Takeaway (Section 7.3): Companies with systematic IP monitoring programs, integrated patent analysis tools (e.g., DrugPatentWatch, PatSnap, Derwent Innovation), and deep biosimilar litigation experience build sustainable competitive advantages in market entry timing. This is a capability that compounds over time — each program generates legal intelligence that improves the next program’s FTO strategy.
Section 8: Reference Product IP Valuation — AbbVie, Amgen, and Roche as Case Studies
8.1 AbbVie/Humira (Adalimumab): The $20 Billion Patent Fortress
Humira’s IP architecture is the definitive case study in biologic patent thicket construction. AbbVie’s strategy, executed over more than a decade, converted a composition-of-matter patent cliff into a seven-year US market exclusivity extension worth an estimated $20 billion in incremental US revenue.
At its core, Humira is a fully human anti-TNF-alpha IgG1 monoclonal antibody. The primary composition-of-matter patents expired by 2016. AbbVie had, however, built a secondary patent portfolio covering the high-concentration (100 mg/mL), citrate-free formulation used in the prefilled syringe and auto-injector devices marketed from 2012 onward, the manufacturing process for achieving high-concentration, low-viscosity formulations of adalimumab, the specific device design of the Humira auto-injector, and dosing regimens for specific indications including plaque psoriasis and hidradenitis suppurativa. EU biosimilar entrants (Amgevita, Hyrimoz, Imraldi, and others) launched in October 2018 after negotiating licenses or designing around the relevant EU patents. US biosimilar entry was delayed until January 2023 due to a broader and more defensible US patent estate, specifically the suite of formulation and manufacturing process patents.
AbbVie’s Humira patent estate generated an estimated $200M+ per patent per year in extended US revenue during the 2018-2023 exclusivity extension period. From an IP valuation standpoint, the formulation and device patents accounted for the majority of this value — a compelling case that secondary IP, intelligently constructed and defended, is worth a substantial fraction of the composition-of-matter IP for a major biologic.
For biosimilar developers targeting the post-2023 US market: the adalimumab market is now genuinely competitive, with seven FDA-approved biosimilars including Amgevita (Amgen), Cyltezo (Boehringer Ingelheim — the first US-designated interchangeable adalimumab), Hadlima (Samsung Bioepis/Organon), Hyrimoz (Sandoz), Hulio (Mylan/Viatris), Yusimry (Coherus), and Simlandi (Alvotech/Teva). Commercial competition is now determined by contracting, rebate offers, GPO positioning, and payer formulary access — not patent litigation. Gross-to-net spread analysis for this market suggests WAC discounts of 5-20% with substantial rebate obligations, compressing net realized price significantly.
Key Takeaway (Section 8.1): The Humira case teaches that the maximum IP value in a biologic program lies not in composition-of-matter exclusivity but in the architecture of secondary patents built during the commercial phase. Innovators who invest in continuous patent portfolio construction during the product lifecycle generate substantially higher terminal asset values than those who rely on primary patent exclusivity alone.
8.2 Amgen/Enbrel (Etanercept): The Process Patent Moat
Etanercept (Enbrel), a TNF-alpha inhibitor fusion protein combining the extracellular domain of the p75 TNF receptor with the Fc region of human IgG1, illustrates a different IP architecture. Amgen’s primary moat for Enbrel in the US was not a formulation patent but a suite of manufacturing process patents covering the specific CHO cell culture and purification methods used to produce etanercept at commercial scale.
Etanercept is structurally simpler in some respects than a full-length monoclonal antibody but more demanding in its glycosylation requirements. It carries multiple N- and O-glycosylation sites, and its effector function profile is highly sensitive to glycan composition. Reproducing the reference product’s glycan profile requires either a very similar CHO cell culture process or an alternative process with equivalent glycosylation control. Amgen’s process patents covered specific aspects of the CHO cell culture conditions that produce the characteristic etanercept glycan profile.
Samsung Bioepis (developing Eticovo/Benepali) and Sandoz (developing Erelzi) were the primary US biosimilar challengers. Both companies had to develop manufacturing processes that produced an etanercept with comparable glycan profiles to Enbrel without infringing Amgen’s process patents — a classic design-around problem. Both achieved US approval (Samsung Bioepis in 2016, Sandoz in 2016), but US commercial launch was delayed by patent litigation. Sandoz launched Erelzi in the US in 2019 after a patent settlement. Eticovo launched in the US in 2019 as well.
The key lesson for IP strategy from the etanercept case: process patents, while theoretically harder to defend than composition-of-matter patents (since the ‘product’ claim often has broader coverage), can be highly effective when they cover the specific process conditions required to achieve the target quality attribute profile for a complex molecule. A biosimilar developer who must use a process ‘close to’ the innovator’s to match the quality attributes may find that ‘close to’ is, in fact, infringing.
Key Takeaway (Section 8.2): For biosimilar developers targeting etanercept, adalimumab, and structurally complex molecules with demanding glycosylation requirements, process patent clearance is as important as composition-of-matter clearance. The process development team and the patent counsel must work in close collaboration throughout upstream development to ensure that every process parameter decision is evaluated for patent exposure.
8.3 Roche Oncology Portfolio (Trastuzumab, Bevacizumab, Rituximab): The Global Interchangeability Race
Roche’s HER2, VEGF, and CD20 biologic portfolio represents the largest oncology biosimilar opportunity in pharmaceutical history. Herceptin (trastuzumab), Avastin (bevacizumab), and Rituxan (rituximab) generated a combined $20 billion+ in annual global revenue at peak. All three have seen primary patent expiration across major markets and the subsequent entry of multiple biosimilars.
The oncology biosimilar market differs from the autoimmune/immunology biosimilar market in several important ways. Hospital P&T committees and oncology pharmacies are the primary decision-makers. Physician familiarity with the specific biologic and the availability of clinical data in the specific tumor type and line of therapy drive prescribing decisions. Automatic pharmacy substitution is less relevant for IV infusion products administered in clinical settings. The commercial battleground is formulary positioning at large health systems, GPO contracts, and coverage decisions by major payers.
Roche has responded to biosimilar competition with two strategies. The first is next-generation molecule development: obinutuzumab (Gazyva) is a glycoengineered anti-CD20 antibody with enhanced ADCC activity compared to rituximab, and pertuzumab (Perjeta) and ado-trastuzumab emtansine (Kadcyla) extend the HER2 franchise beyond trastuzumab’s intellectual property. The second is biosimilar co-commercialization in markets where biosimilar entry is inevitable, accepting margin erosion in exchange for volume retention.
For institutional investors, the Roche oncology biosimilar dynamic illustrates a critical portfolio valuation principle: when a blockbuster biologic faces biosimilar entry, the net present value (NPV) of the franchise is not simply the NPV of the reference product minus the biosimilar discount. The innovator’s response — in new molecule development, in combination therapy regimens, in market access strategies — materially affects the residual franchise value. Modeling the innovator’s strategic response to biosimilar entry is as important as modeling the biosimilar’s market penetration curve.
Key Takeaway (Section 8.3): Oncology biosimilars operate in a ‘specialist pharmaceutical’ commercial model where GPO contracting, hospital formulary positioning, and evidence generation in specific indications determine market share. The commercial infrastructure required to compete effectively in this market is substantially larger than for subcutaneous autoimmune biosimilars.
Section 9: The Future State: Multi-Attribute Methods, AI, and the Post-Clinical Trial Era
9.1 Multi-Attribute Methods: One Assay, Many Answers
The biosimilar analytical paradigm is moving toward platform methods that measure multiple critical quality attributes simultaneously, reducing the number of separate assays required and providing richer, more integrated data. The Multi-Attribute Method (MAM) is the most advanced expression of this trend.
MAM is a high-resolution LC-MS/MS-based workflow that monitors a predefined panel of peptide-level attributes in a single chromatographic run. Rather than performing separate peptide mapping experiments for sequence confirmation, PTM quantitation, and charge variant assessment, MAM integrates these into one validated method. A typical MAM panel for a monoclonal antibody monitors deamidation at each asparagine site, oxidation at each methionine site, glycation at each lysine site, sialic acid modifications, and specific glycan attachment sites — all from a single tryptic digest analyzed in one LC-MS/MS run.
The regulatory acceptance of MAM as a release and stability assay is advancing. FDA’s Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER) have both published on MAM implementation, and several MAM-based release methods are already in approved BLAs. The efficiency gain is substantial: a MAM-based quality control panel can replace four to six separate conventional assays, reducing per-lot testing time by 30-50% and generating more comprehensive attribute data than the conventional panel.
The biosimilar-specific application of MAM is particularly powerful in comparability studies. Running MAM on biosimilar and RP lots in parallel generates a comprehensive, quantitative attribute comparison in a single experimental workflow, with statistical analysis of each attribute’s comparability built into the data processing pipeline. This accelerates comparability assessment during process development and provides a more analytically complete picture for the regulatory submission.
Key Takeaway (Section 9.1): MAM implementation in biosimilar programs is transitioning from an advanced capability to a competitive expectation. Programs that establish validated MAM workflows early gain analytical efficiency and generate richer comparability data. Programs without MAM capability face higher per-lot analytical costs and potentially less comprehensive comparability packages.
9.2 Artificial Intelligence in Biosimilar Development: Current Applications and Realistic Expectations
Artificial intelligence and machine learning are being applied across the biosimilar development pipeline, though with varying levels of technical maturity and demonstrated ROI. Realistic assessment of where AI currently adds value and where the hype exceeds the reality is essential for development teams evaluating technology investments.
In cell line development, machine learning models trained on historical clone screening data can predict the productivity and quality attribute profile of individual clones from early-passage screening data, enabling earlier prioritization of high-value candidates and reducing the number of clones that need to progress to full production evaluation. Sartorius, Lonza, and several CDMOs have published on ML-assisted clone selection workflows.
In bioprocess development, machine learning models trained on bioreactor process data can identify the relationships between process parameters and quality attribute outcomes. These models support design-of-experiment (DoE) planning by predicting which parameter combinations are most likely to yield a quality attribute profile within the comparability acceptance criteria, reducing the number of experimental runs required to define the process design space. This has been demonstrated in academic and industry publications for glycan profile control in CHO cell culture.
In formulation development, computational models of protein-protein interactions — specifically models that predict aggregation propensity and viscosity at high concentration from amino acid sequence and structural data — are used to guide excipient selection and concentration optimization. These models are not yet accurate enough to replace experimental screening, but they can reduce the experimental design space by eliminating clearly unfavorable excipient combinations before bench work begins.
In patent landscape analysis, natural language processing (NLP) models trained on patent text can accelerate FTO reviews by automatically classifying patent claims by subject matter, identifying claims with potential relevance to the biosimilar’s product and process characteristics, and tracking new filings in real time. These tools accelerate the attorney review process but do not replace the legal judgment required for FTO assessment.
What AI cannot yet do reliably: predict clinical outcomes from structural data, generate novel process parameters that are beyond the training data distribution, or substitute for physical experimentation in process characterization and validation. The biosimilar development community should treat AI as a tool that accelerates and enhances human expertise, not as a replacement for it.
Key Takeaway (Section 9.2): AI investment in biosimilar development generates the highest ROI in clone selection screening, process DoE design, and IP monitoring — three areas where the decision space is large, the data is rich, and the cost of experimental exploration is high. Teams evaluating AI vendors should require demonstration data from actual biosimilar programs, not theoretical performance benchmarks.
9.3 The Post-Clinical Trial Era: Regulatory Science Roadmap
The regulatory trajectory for biosimilars points toward a future in which a comprehensive analytical and functional similarity package, combined with comparative PK data in humans, is sufficient for full biosimilar approval without a confirmatory clinical efficacy study for most products. This shift is not hypothetical; it is already occurring in the EU and is advancing in the US.
The scientific foundation for this shift is well-established. Analytical methods are more sensitive than clinical trials for detecting structural and functional differences. Well-characterized molecules with documented structure-function relationships provide a scientifically grounded basis for extrapolating analytical similarity to clinical similarity. The clinical trial, when performed in a patient population that is variable, on a background of other therapies, with endpoints that have clinical and statistical noise, is a less sensitive instrument than the analytical and functional assay panel.
The regulatory science community is actively developing the framework for operationalizing this transition. Key elements include standardized, quantitative analytical similarity scoring frameworks that provide a transparent, reproducible basis for evaluating analytical similarity across the attribute panel; validated clinical relevance thresholds for each CQA — the defined range of variation that can be considered clinically inactive based on mechanistic understanding and historical clinical data; and enhanced post-market pharmacovigilance frameworks that monitor biosimilar performance at population scale after approval, providing real-world confirmation of the analytical similarity conclusion.
FDA’s Biosimilar Action Plan, updated in 2023, explicitly commits the agency to ‘developing and using scientific advances to inform biosimilar development,’ which is agency language for the streamlining trend. The Plan identifies MAM, improved immunogenicity prediction models, and analytical similarity quantitation frameworks as priority scientific development areas.
Key Takeaway (Section 9.3): The biosimilar programs initiated in 2025 and beyond will increasingly be designed for streamlined approval without a confirmatory clinical study, relying on analytical-first regulatory strategies. Companies whose development infrastructure and regulatory expertise are built around the full-pyramid model face an efficiency disadvantage versus those who are building for the analytically-driven paradigm now.
Section 10: Investment Strategy for Portfolio Managers and Institutional Analysts
10.1 Valuing a Biosimilar Pipeline: Key Financial Parameters
Institutional investors evaluating biosimilar pipeline companies or biosimilar programs within larger pharmaceutical portfolios need a consistent analytical framework. The financial parameters that determine biosimilar program value are qualitatively different from small-molecule generics and require a separate modeling approach.
Development cost per approved product: industry benchmarks put total development cost at $100M-$300M for a monoclonal antibody biosimilar following the full regulatory pathway. Streamlined programs targeting EMA approval without a confirmatory clinical study can potentially achieve approval at $80M-$150M. These figures include discovery and cell line development, analytical characterization, process development and scale-up, non-clinical studies, clinical studies (PK and immunogenicity; confirmatory efficacy if required), regulatory submission preparation, and quality systems buildout. They exclude manufacturing facility capital expenditure, which is a separate line item of $200M-$500M for a new 2,000-10,000L GMP biologics facility.
Development timeline: from program initiation (cell line development start) to FDA approval typically runs eight to twelve years. EMA timelines are comparable. The rate-limiting steps are cell line development and process development (two to four years), analytical package development (parallel, two to four years), and clinical studies (two to four years). Programs that achieve strong analytical similarity early, enabling reduced clinical requirements, can compress the timeline by one to two years.
Market penetration assumptions: US market penetration for non-interchangeable biosimilars at three years post-launch has ranged from 5% to 40% across approved products, with a median around 20-25%. European market penetration is faster and deeper, with centralized procurement in several member states driving 60-80% penetration within two years. US interchangeable biosimilars have achieved higher penetration rates in the pharmacy channel (30-50% within two to three years for retail pharmacy products).
Pricing assumptions: US biosimilar WAC pricing has typically been set at 10-30% below the reference product’s WAC, but the net realized price after rebates and GPO contracting discounts is substantially lower. Net price realizations of 50-70% of the reference product’s net price are common in competitive market segments. European tender pricing can reach 40-60% discounts from reference product prices in centralized tender markets.
Key Takeaway (Section 10.1): Biosimilar program NPV analysis should model probability of technical success (typically 60-75% for programs with experienced developers), probability of regulatory approval (conditional on technical success, approximately 80-90%), probability of commercial success (conditional on approval, highly dependent on IP clearance, competitor count, and market access strategy), and residual value of manufacturing assets if the program is discontinued.
10.2 Identifying the Best Risk-Adjusted Opportunities
Not all biosimilar targets are created equal. The most attractive opportunities combine a large addressable market, a manageable patent thicket, a technically feasible comparability exercise, and a competitive landscape with few well-capitalized entrants at program initiation.
Molecule complexity is an underappreciated risk factor. Monoclonal antibodies are the most commercially significant targets and have the most established development playbook, but fusion proteins, bispecific antibodies, and antibody-drug conjugates present substantially greater technical challenges and higher development cost. Programs targeting these more complex molecules should require a higher technical risk premium in the discount rate.
Patent thicket density is the most important commercial risk factor for US market opportunities. Programs where the primary patent expires within two to three years and secondary patent coverage is thin, expired, or vulnerable to IPR challenge offer the highest risk-adjusted returns. Programs where the innovator has a dense secondary portfolio with multiple pending continuation applications — where new claims may emerge during the development timeline — carry structural commercial risk that is difficult to quantify ex ante.
Competitive landscape at program initiation is predictive of steady-state pricing and market share. Programs where fewer than four developers have disclosed active development at the time of program initiation typically yield better pricing outcomes than programs with eight or more declared entrants. The biosimilar competitive intelligence landscape — program disclosures in clinical trial registries, FDA BLA filing notifications, patent challenge filings — provides real-time data on competitive positioning.
Key Takeaway (Section 10.2): The best-positioned biosimilar companies are those with integrated analytical, manufacturing, regulatory, and legal capabilities that allow them to execute development programs faster and cheaper than less capable competitors, creating first-mover advantages in a market where launch timing directly determines commercial outcomes.
10.3 Strategic M&A and Partnership Considerations
Biosimilar development capabilities are increasingly concentrated among a relatively small number of companies with the full-stack infrastructure required to execute programs from cell line development through commercial manufacturing. This concentration creates specific M&A and partnership dynamics.
CDMO relationships are a critical strategic variable. Companies without GMP biologics manufacturing infrastructure must partner with CDMOs for drug substance production. The choice of CDMO — technical capability, batch success rate, regulatory inspection history, capacity availability — directly affects program timeline and cost. Leading CDMOs with biosimilar-specific experience include Lonza, Samsung Biologics, WuXi Biologics, Boehringer Ingelheim Pharma, and Fujifilm Diosynth Biotechnologies. CDMO capacity constraints, particularly at the 2,000L+ commercial scale, have been a limiting factor for several biosimilar programs during peak development periods.
Licensing and settlement agreements between biosimilar developers and innovators are common. Most Humira biosimilar developers settled with AbbVie and received negotiated launch dates and royalty obligations rather than litigating to final judgment. Rituximab biosimilar developers similarly settled with Roche/Biogen. These settlements define the commercial parameters of market entry and are financially material events that must be modeled explicitly in program valuation.
Portfolio strategy for mid-size biosimilar companies: a portfolio of four to six programs across different molecular targets and market segments distributes technical risk and creates commercial manufacturing scale that is not achievable with one or two programs. Programs with overlapping manufacturing requirements (all monoclonal antibodies in CHO cells using Protein A purification) are natural portfolio complements, enabling manufacturing platform leverage. Programs with overlapping commercial targets (all autoimmune subcutaneous products sold to rheumatology practices) enable commercial infrastructure leverage.
Key Takeaway (Section 10.3): Investors evaluating biosimilar companies should assess the depth and integration of the company’s analytical, manufacturing, regulatory, legal, and commercial capabilities as a primary valuation driver, alongside the portfolio’s IP clearance status and market timing. Companies with platform capabilities that apply across multiple programs generate compounding efficiency advantages that narrow-focus developers cannot match.
Master Key Takeaways
The following summarizes the most actionable intelligence from this analysis, organized by functional audience.
For R&D and analytical teams: The analytical package is the program. Every dollar invested in comprehensive, orthogonal characterization of the reference product and the biosimilar candidate reduces downstream regulatory risk, generates the basis for clinical study waiver, and provides the factual foundation for IP design-around and litigation. Multi-attribute method implementation and HDx-MS for HOS characterization are now expected elements of a competitive analytical package, not optional enhancements.
For regulatory affairs teams: The EMA’s 2023 tailored clinical approach framework, combined with FDA’s explicit commitment to science-driven development pathways in the 2023 Biosimilar Action Plan, creates a real pathway to approval without a confirmatory clinical efficacy study for well-characterized molecules with strong analytical and PK/PD packages. Programs initiated today should be designed for this streamlined pathway as the base case, not as a stretch goal.
For IP and legal teams: The patent dance is a strategic exercise, not a procedural formality. The disclosure, timing, and framing of positions at each step have material consequences for market entry timing and litigation exposure. IPR petitions at PTAB have a higher claim cancellation rate than district court litigation for the same patents and should be part of every active biosimilar IP strategy. FTO analysis must be a continuous program function, not a one-time initial assessment.
For commercial and market access teams: US biosimilar market dynamics resemble specialty pharmaceutical competition, not generic drug competition. Formulary positioning, GPO contracting, and payer rebate negotiations — not automatic substitution — drive market penetration for most products. Interchangeability status is commercially valuable for retail pharmacy products but may not justify the switching study cost for hospital-administered products. Build the commercial strategy around active physician and payer engagement from day one.
For portfolio managers and institutional investors: Biosimilar program NPV is a function of development cost, timeline, IP clearance probability, market penetration curve, and pricing assumptions — all of which differ substantially from small-molecule generic programs. Programs targeting molecules with dense secondary patent landscapes, many disclosed competitors, and uncertain regulatory pathways require a higher risk premium. The most attractive opportunities are well-characterized molecules with thin or expiring secondary patent coverage, fewer than four active developers at program initiation, and large, payer-motivated markets with established biosimilar adoption history.
The biosimilar industry is moving toward an analytically-led development paradigm in which the laboratory, not the clinic, is the primary arena for proving equivalence. Companies that build superior analytical capability, integrate it with smart regulatory and IP strategy, and execute manufacturing development with bioprocess engineering discipline will capture the largest share of the $200 billion biologic patent cliff opportunity over the next decade.
This analysis draws on public regulatory guidance, peer-reviewed literature, patent database analysis, and industry-reported development program disclosures. It does not constitute legal advice or investment advice. Readers should consult qualified legal counsel for IP assessments and registered investment advisors for portfolio decisions.
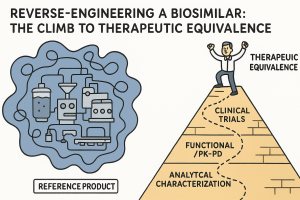

















")








