Drug repurposing has moved well past the era of accidental bedside observations. Today, systematic computational platforms screen millions of compound-indication pairs per week, feeding a pipeline that accounts for roughly 30 percent of newly marketed drugs in the United States. The economics are compelling, the patent strategies are complex, and the translational risks are real. This guide covers all three dimensions in technical depth, written for IP counsel, R&D leadership, and institutional investors who need more than a summary.
PART I: Fundamentals, Economics, and the IP Framework
What Drug Repurposing Actually Means
Drug repurposing, sometimes called drug repositioning, is the clinical and commercial strategy of identifying new therapeutic indications for compounds that have already cleared at least one stage of human evaluation. The practical boundary matters: a compound with Phase I safety data but no approved indication occupies a meaningfully different regulatory position than a fully approved drug. Most repurposing activity clusters around approved drugs because their established pharmacokinetic and safety profiles allow sponsors to skip Phase I in new indication programs, compressing the path to a Phase II proof-of-concept study.
The distinction between ‘repurposing’ and ‘repositioning’ is mostly semantic in the current literature, with both terms describing the same strategic act. Where the terms diverge in practice is in IP structuring: repositioning often refers to NDA label expansion under 21 CFR 314.50(b), while repurposing sometimes encompasses broader off-label and compassionate-use strategies that do not require a separate regulatory filing. For IP teams, this distinction matters because the exclusivity protections available to each pathway differ substantially.
Historical Arc: From Accidental Findings to Algorithmic Pipelines
The history of drug repurposing splits cleanly into two eras. Before the genomics revolution, most repositioning discoveries were serendipitous. Physicians noticed unexpected clinical benefits in patients on existing medications, hypotheses were formed retrospectively, and new indications were occasionally filed. Sildenafil is the most frequently cited example: Pfizer’s compound for angina produced a side effect profile that pointed to phosphodiesterase-5 inhibition in vascular smooth muscle, leading to the Viagra NDA in 1998. The compound’s core phosphodiesterase-5 composition-of-matter patent had already issued, but Pfizer’s decision to file new use patents on its erectile dysfunction indication extended effective commercial exclusivity for additional years.
The second era began roughly with the publication of the first large-scale gene expression compendia in the early 2000s. The Connectivity Map project at the Broad Institute, which cataloged transcriptional responses to approximately 1,300 bioactive small molecules across cancer cell lines, gave researchers their first structured tool for asking computational repurposing questions at scale. From there, the methodological stack expanded steadily through network biology, machine learning, and, most recently, large language model-assisted literature mining. The defining characteristic of this second era is that hypothesis generation has become cheap enough to run in parallel across thousands of indication pairs simultaneously.
The Full Economic Case: Costs, Timelines, and Failure Rates
The canonical cost comparison positions novel drug development at approximately $2.6 billion per approved compound, incorporating the cost of failures across a development portfolio. Repurposing estimates vary by source and scope of data included, but the most widely cited figure runs around $300 million with a development timeline of approximately six years from repurposing hypothesis to approval. These figures reflect programs where the candidate has an existing IND and acceptable safety data in hand.
The failure-rate picture is more nuanced than cost figures alone suggest. Although repurposed drugs enter Phase II with a pharmacokinetic and toxicology head start, their probability of technical success from Phase II to approval is not dramatically higher than that of novel compounds across all therapeutic areas. The advantage is concentrated in indications where the drug’s mechanism of action has a clear and direct rationale in the new disease context, such as baricitinib’s JAK-STAT inhibition applied to COVID-19-associated hyperinflammation. Repurposing programs that depend on polypharmacology or off-target effects show Phase II success rates more in line with the broader industry average.
~$300M cost; ~6 year timeline; Phase II-to-approval rate varies by indication class
505(b)(2) repurposing NDA
Partial reliance on existing safety data; typically 3-6 year timeline; reduced Phase I requirements
Supplemental NDA (sNDA) for new indication
Fastest pathway; requires no new IND; typically 2-4 year development cycle for well-characterized mechanism
Orphan Drug designation overlay
7-year market exclusivity in US; 10-year in EU; tax credit on qualified clinical trial costs (25% as of 2026 IRA adjustments)
The IP Framework for Repurposed Drugs
Intellectual property protection for repurposed drugs requires a different analytical lens than for novel compounds. When a drug’s composition-of-matter patent has already issued or expired, the available IP levers shift to method-of-use patents, formulation patents, dosing regimen patents, and, where applicable, patient population selection patents. Each of these carries different claim strength, different vulnerability to inter partes review (IPR), and different duration relative to the commercial exclusivity window.
Method-of-use patents covering a new therapeutic indication typically enjoy a 20-year term from filing date. If the composition-of-matter patent on the underlying compound issued 15 years ago, a new method-of-use patent filed today could theoretically extend effective protection well beyond the point at which the original patent expires. This is the core of secondary patent strategy in repurposing. The key legal vulnerability is that method-of-use claims must clear the obviousness bar: if the new indication was predictable from the drug’s known mechanism, the claim may not survive IPR. Compound patents with broad mechanism-of-action disclosures create particular risk for downstream repurposing patent applicants.
A Paragraph IV certification challenge (21 USC 355(j)(2)(A)(vii)(IV)) remains the primary generic entry mechanism for any repurposed drug still listed in the FDA Orange Book. When a generic manufacturer believes a new-use or formulation patent is invalid or will not be infringed by the generic’s label, it files a Paragraph IV certification, triggering a 30-month stay if the brand-side NDA holder sues within 45 days. Repurposing patent claims around dosing regimens and patient selection criteria have shown mixed results in district court, with courts scrutinizing whether the claims represent genuine invention or mere optimization of a known drug.
KEY TAKEAWAYS
1. Repurposing accounts for ~30% of newly marketed US drugs, but the cost and timeline advantages are sharpest when the drug’s mechanism maps directly to the new indication.
2. IP protection for repurposed drugs depends on method-of-use, formulation, and dosing regimen patents, all of which carry higher obviousness risk than composition-of-matter claims.
3. A 505(b)(2) NDA allows partial reliance on existing safety data, but does not by itself create exclusivity beyond what the underlying patents and regulatory exclusivity protections already provide.
4. Orphan Drug Designation overlaid on a repurposing program generates seven years of US market exclusivity independent of patent status, making it a high-value strategic tool for rare-disease repositioning.
5. Paragraph IV certification risk applies to any repurposed drug with Orange Book-listed patents; method-of-use claims covering dosing regimens and patient selection have shown mixed durability in district court litigation.
PART II: Computational Methodology in Technical Depth
Computational drug repurposing is not a single method. It is a collection of distinct algorithmic and data-driven strategies, each with specific data requirements, output types, and validation demands. The field organizes broadly around three orienting perspectives: disease-centric, target-centric, and drug-centric. In practice, the most productive programs combine methods from two or more categories.
Disease-Centric Approaches
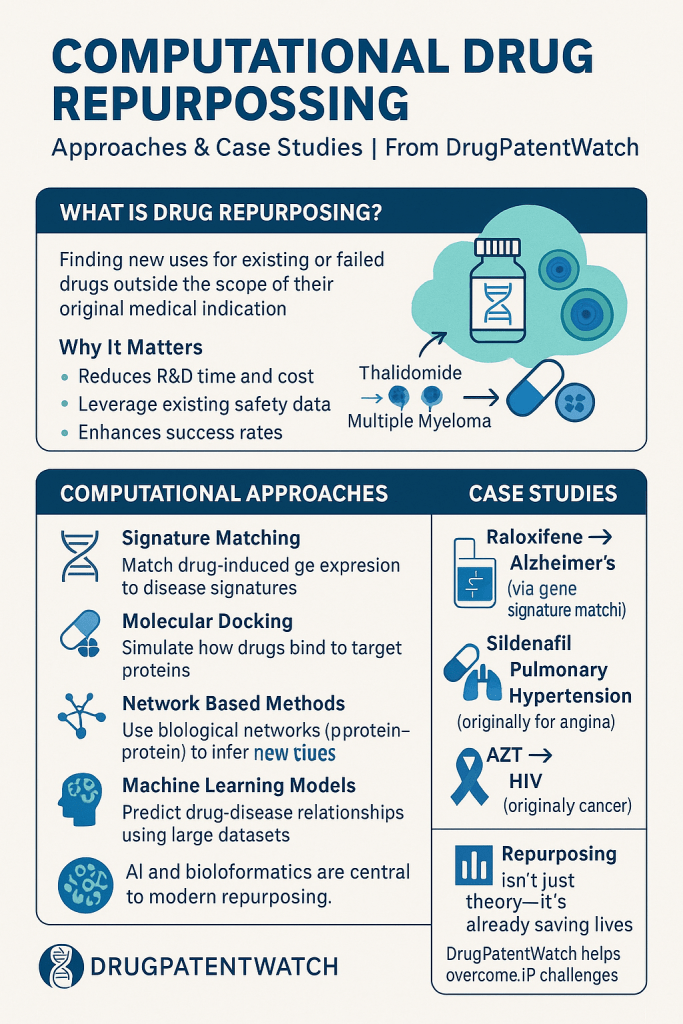
Disease-centric repurposing starts from a specific pathology and asks which approved compounds could reverse or modify its molecular signature. The methodological workhorse for this approach is differential gene expression analysis, where transcriptomic data from diseased tissue is compared to healthy tissue, and the resulting gene expression signature is matched against drug-induced expression signatures from resources like the Connectivity Map (CMap) at the Broad Institute.
The CMap database, now expanded through the LINCS (Library of Integrated Network-Based Cellular Signatures) project to over 1.5 million compound-cell-dose-time perturbation profiles, is the reference data source for signature-matching repurposing. The LINCS L1000 assay measures approximately 1,000 landmark genes across perturbation conditions, then imputes the remaining transcriptome using a computational model. The resulting ‘landmark-to-inferred’ approach reduces assay cost by roughly 90 percent relative to whole-transcriptome profiling, but introduces measurement noise that must be accounted for in downstream analyses.
From a disease-centric signature match, the output is a ranked list of compounds whose transcriptional effect is anti-correlated with the disease signature. This anti-correlation hypothesis rests on the assumption that reversing the disease’s gene expression pattern represents a therapeutic intervention. The assumption holds well for diseases with clear, consistent molecular signatures but breaks down in heterogeneous diseases like Alzheimer’s or schizophrenia, where no single transcriptional signature captures the full patient population. Disease stratification by molecular subtype is the practical workaround, and it also creates an IP opportunity: a method-of-use patent claiming a repurposed drug in a specific, molecularly defined patient subset.
Target-Centric Approaches
Target-centric repurposing identifies disease-relevant protein targets through genomic analysis, GWAS results, or mechanistic pathway studies, then screens existing drugs for binding activity at those targets. The primary computational tools are structure-based virtual screening, pharmacophore modeling, and binding affinity prediction models trained on known drug-target interaction databases.
Structure-based virtual screening requires a high-quality three-dimensional structure of the target protein, typically from X-ray crystallography, cryo-electron microscopy, or AlphaFold2 prediction. AlphaFold2, released by DeepMind in 2021, generated predicted structures for over 200 million proteins and has substantially expanded the tractable target space for computational repurposing. The limitation of AlphaFold2 structures in repurposing workflows is that they represent static ground-state conformations and may not capture allosteric sites or induced-fit binding pockets that are relevant to drug binding. Ensemble docking against multiple conformations extracted from molecular dynamics simulations partially addresses this limitation.
Pharmacophore modeling abstracts the essential geometric and chemical features required for a molecule to bind a target, then screens drug libraries for compounds matching those features. This approach is computationally faster than structure-based docking but generates more false positives and requires experimental validation at a higher rate. The best repurposing workflows use pharmacophore modeling as a rapid first-pass screen to reduce a large drug library to a manageable candidate set, which is then subjected to more computationally intensive structure-based methods.
Binding affinity prediction models, trained on databases like ChEMBL (over 17 million bioactivity records as of 2025) and BindingDB (more than 2.8 million measured binding affinities), use machine learning to predict the strength of drug-target interactions without explicit molecular docking. Random forest and gradient boosting models were the standard approach through the mid-2010s. Graph neural network (GNN) architectures, which represent molecules as graphs where atoms are nodes and bonds are edges, now outperform classical ML approaches on most standard binding affinity benchmarks.
Drug-Centric Approaches and Polypharmacology
Drug-centric repurposing starts with a known compound and asks what additional diseases or targets it might engage. The biological basis for this approach is polypharmacology: most small-molecule drugs bind multiple protein targets, even if only one of those interactions was exploited in the original indication. Systematic mapping of off-target binding profiles, through affinity mass spectrometry, proteome-wide thermal shift assays (thermal proteome profiling), or computational prediction, produces a drug-protein interaction network that can be queried against disease biology databases to identify potential new indications.
Drug-centric approaches are particularly well-suited to first-generation kinase inhibitors, which were developed before selectivity profiling was standard practice and often bind dozens of kinases in addition to their primary target. Imatinib (Gleevec), originally developed for BCR-ABL-positive CML, has been computationally and clinically explored across multiple rare sarcomas and systemic mastocytosis based on its off-target activity against KIT and PDGFR. The Novartis IP estate around imatinib illustrates what an aggressive method-of-use patent strategy looks like in practice: the company filed patents covering numerous specific indications and patient subpopulations, some of which generated significant legal controversy over the propriety of secondary patents on known compounds.
Network Biology and Systems-Level Repurposing
Network-based approaches model biological systems as heterogeneous graphs in which nodes represent biological entities (genes, proteins, drugs, diseases, metabolites) and edges represent experimentally or computationally inferred relationships between them. The most widely used network resource for repurposing work is the human protein-protein interaction (PPI) network, augmented with gene regulatory networks, metabolic networks, and disease-gene association networks derived from GWAS and phenome-wide association studies (PheWAS).
The network proximity hypothesis, formalized in work by the Barabasi group at Northeastern University, posits that a drug’s therapeutic targets and the genes associated with a disease should cluster in the same network neighborhood if the drug is effective against that disease. Conversely, drugs whose targets are distant from disease genes in the network are unlikely to be therapeutic. This hypothesis has been validated retrospectively on known drug-disease pairs and has prospective predictive value as a repurposing filter, though it is not predictive at the individual compound level with sufficient specificity for direct clinical translation without additional experimental validation.
Knowledge graph embeddings have extended network-based repurposing into the era of deep learning. Algorithms like TransE, RotatE, and their successors learn low-dimensional vector representations of entities in a knowledge graph such that the geometric relationships between vectors encode biological relationships. A drug whose embedding vector is close in representation space to a disease embedding vector is predicted to be a candidate for that indication. Commercial implementations of this approach, offered by companies including Insilico Medicine, BenevolentAI, and Recursion Pharmaceuticals, have generated repurposing candidates that have entered clinical development.
Machine Learning and Deep Learning in Repurposing: A Technology Roadmap
The machine learning stack in drug repurposing has evolved through several distinct generations. Understanding where each method sits on the maturity curve is important for R&D teams allocating resources and for investors evaluating platform claims.
Technology Roadmap: Generation 1 (2005-2015): Classical ML on Molecular Descriptors
Fingerprint-based descriptors (Morgan fingerprints, ECFP4) fed into SVMs, naive Bayes, and random forests. Good on retrospective benchmarks; poor scaffold-hopping. Primary data sources: ChEMBL, DrugBank, SIDER.
The first wave of ML-based repurposing used fingerprint-based molecular descriptors (Morgan fingerprints, ECFP4, topological torsion descriptors) as input features for classical algorithms including SVMs, naive Bayes classifiers, and random forests. These methods performed well on retrospective benchmarks but struggled with scaffold hopping, where structurally dissimilar compounds with similar biological activity were missed. The primary data sources were ChEMBL, DrugBank, and SIDER (side effect database), all of which remain in active use.
Technology Roadmap: Generation 2 (2015-2020): Deep Learning on Raw Molecular Representations
CNN/GNN architectures on SMILES strings and molecular graphs. MPNN framework (Gilmer et al. 2017) outperformed fingerprint methods on drug-target interaction prediction without hand-engineered features.
Convolutional neural networks applied to string-based molecular representations (SMILES notation) and graph neural networks applied to molecular graphs defined the second generation. GNN architectures, particularly the message-passing neural network (MPNN) framework described by Gilmer et al. in 2017, outperformed fingerprint-based methods on drug-target interaction prediction by learning molecular representations from raw atomic connectivity without hand-engineered features. This generation also produced the first successful graph convolutional approaches to predicting drug-drug interactions and side effect profiles.
Technology Roadmap: Generation 3 (2020-2024): Transformer Models and Multi-Modal Integration
Protein language models (ESM series), molecular BERT variants, and shared embedding spaces for chemical, genomic, and clinical data. Zero-shot protein function prediction expanded tractable target space.
Transformer architectures, originally developed for natural language processing, reached drug discovery through protein sequence modeling (ESM series from Meta AI), molecular property prediction (ChemBERTa, MolBERT), and multi-modal integration platforms that jointly represent chemical, genomic, and clinical data in a shared embedding space. The protein language models are particularly relevant for repurposing because they enable zero-shot prediction of protein function for targets with limited experimental data, expanding the set of tractable targets for in silico screening.
Technology Roadmap: Generation 4 (2024-Present): Generative AI, Causal Modeling, and Foundation Models
Biological foundation models (scGPT, scFoundation) predict cell-type-specific drug perturbation responses. Causal inference frameworks address confounding in observational drug-disease associations.
The current frontier of computational repurposing incorporates large biological foundation models trained on multi-modal datasets spanning genomics, proteomics, imaging, and electronic health records. Genentech’s scFoundation, the Broad Institute’s scGPT, and similar models learn cell-type-specific biology at single-cell resolution and can be queried to predict how a given drug perturbation will alter gene expression in a specific cell type within a specific tissue context. This generation of models also incorporates causal inference frameworks to distinguish correlation from causation in drug-disease associations, directly addressing one of the field’s most persistent failure modes: confounding in observational data.
Text Mining and Literature-Scale Evidence Synthesis
Biomedical literature represents one of the largest structured knowledge repositories available for repurposing research, and text mining approaches have scaled to extract drug-disease relationships from tens of millions of PubMed abstracts and full-text articles. Named entity recognition (NER) pipelines identify drug and disease mentions in text; co-occurrence analysis establishes statistical associations; and relation extraction models classify the nature of the relationship (therapeutic, adverse, mechanistic).
The PubTator system from NCBI, the SemMedDB semantic relationship database, and the OpenTargets Evidence string all aggregate text-mined drug-disease associations at scale. The key limitation of text mining for repurposing is publication bias: positive results are published at higher rates than negative results, creating a systematically skewed training corpus for supervised relation extraction models. Calibration against clinical trial registries, where both positive and negative outcomes are recorded, partially corrects for this bias but is still an open methodological problem.
Large language models fine-tuned on biomedical text, including BioMedLM, Galactica derivatives, and GPT-4 applied with domain-specific prompting, have demonstrated capability in zero-shot repurposing hypothesis generation. The clinical utility of these models as standalone repurposing tools is debated; their primary value in current workflows is as evidence synthesis engines that consolidate mechanistic rationale from the literature into a structured format that can be reviewed by domain experts before committing resources to experimental validation.
KEY TAKEAWAYS
1. Disease-centric repurposing via CMap/LINCS signature matching works best for diseases with consistent, reproducible transcriptional signatures across patient samples.
2. AlphaFold2 has expanded the tractable target space for target-centric virtual screening, but structural limitations require ensemble docking or molecular dynamics to capture drug-relevant conformations.
3. Network proximity analysis provides a coarse but scalable filter for prioritizing drug-disease pairs; it is most useful as a pre-screening step, not as a primary evidence source.
4. GNN architectures now set the benchmark for drug-target interaction prediction, outperforming classical fingerprint-based ML on most publicly available benchmarks.
5. The current generation of biological foundation models (scGPT, scFoundation) enables cell-type-specific perturbation prediction, which is qualitatively more informative than bulk transcriptomic signature matching.
6. Text mining produces systematic false positives due to publication bias; calibration against clinical registry data is necessary before using literature-derived associations as primary repurposing evidence.
Does the platform have proprietary patient-level data or rely only on public databases?
Proprietary EHR or proteomic data is a genuine moat; public-data-only platforms have thin differentiation
Prospective vs. retrospective validation
Has the platform predicted a novel indication that subsequently succeeded in an IIT or sponsored trial?
Retrospective hits on known drug-disease pairs are necessary but insufficient; prospective clinical validation is the real proof point
IP on computational methods
Does the company hold patents on its core algorithms, or only on individual repurposed compounds?
Method patents on AI/ML repurposing workflows face 35 USC 101 eligibility risk; compound-level IP is more defensible but narrower
Pipeline depth beyond lead compound
How many active repurposing programs has the platform generated internally?
Single-asset AI repurposing companies carry binary clinical risk; platform value requires multiple validated outputs
Partner validation
Have pharma partners paid for access or co-development rights to platform-derived candidates?
Cash-paying pharma collaborations are the most credible third-party validation of platform quality
PART III: The Validation Pipeline from Algorithm to Clinical Signal
A computational repurposing hypothesis is worth exactly as much as the experimental evidence supporting it, which at the moment of generation is nothing. The validation pipeline converts probabilistic algorithmic output into graded evidence that can justify human clinical exposure. Each stage of that pipeline has its own failure modes, and understanding them is essential for both R&D teams planning experiments and investors modeling probability-of-success assumptions.
Computational Validation: Statistical Rigor Before the Lab
Computational validation evaluates whether an algorithmic repurposing prediction is statistically robust and biologically plausible before any wet-lab resources are committed. The standard metrics are AUROC (area under the receiver operating characteristic curve) and AUPR (area under the precision-recall curve). AUROC measures the model’s ability to rank known drug-disease pairs above randomly selected pairs; AUPR is more informative when known positive drug-disease pairs are sparse relative to unknowns, which is the norm in repurposing datasets.
A critical methodological problem in computational validation is data leakage: if the same drug-disease associations appear in both training and test sets, performance metrics are inflated and do not reflect generalization to truly novel predictions. Proper evaluation requires time-split validation, where the model trains on associations known before a specific date and tests on associations that were established afterward. Only a handful of published repurposing models have been evaluated under strict time-split conditions.
Cross-indication validation offers another layer of computational evidence: if a computational repurposing platform predicts drug A treats disease X, and independently predicts drug B (which shares a mechanism with drug A) also treats disease X, the mechanistic coherence of the predictions strengthens the overall signal. This type of mechanistic triangulation can be formalized through gene set enrichment analysis (GSEA) applied to the predicted target sets of the candidate compounds, checking for consistent enrichment in disease-relevant pathways across multiple predicted hits.
In Vitro Validation: Disease-Relevant Cell Models
Standard high-throughput cell viability or target engagement assays confirm that a repurposed compound produces a measurable cellular effect at pharmacologically relevant concentrations. The critical phrase is ‘pharmacologically relevant’: many repurposing candidates show activity in cell assays at concentrations that are 50 to 100 times the achievable plasma Cmax in humans. Activity at supratherapeutic concentrations does not constitute evidence for clinical utility.
Disease-relevant cell models present a second selection pressure. Cancer repurposing studies frequently use immortalized cell lines that bear limited resemblance to primary patient tumors. For CNS indications, the barrier between a simple neuronal monoculture and a physiologically relevant model of the blood-brain barrier is substantial. Induced pluripotent stem cell (iPSC)-derived disease models, including organoids, have improved the translational fidelity of in vitro repurposing validation considerably, but they remain slower and more expensive than classical cell line screens and are not yet scalable to primary screening campaigns.
Target engagement confirmation in cells, through thermal shift assays, NanoBRET, or proximity ligation assays, is particularly important in repurposing contexts because the drug’s primary target in the original indication may not be the target responsible for the hypothesized effect in the new indication. Confirming that the compound actually engages the predicted target in a disease-relevant cellular context at therapeutically achievable concentrations is a non-negotiable validation step before advancing to animal models.
In Vivo Validation: Animal Model Considerations
Animal model translation is the stage where computational repurposing programs most frequently fail to produce clinical signals. The failure modes are well-characterized: poor target conservation between species (particularly relevant for immunology indications), animal models that do not recapitulate the human disease mechanism, and pharmacokinetic differences that make it difficult to achieve the target plasma exposure required for efficacy in humans.
For repurposing programs advancing into animal efficacy studies, one advantage over novel compounds is the availability of prior human PK data. The team can calculate the free drug concentration at the target exposure required for efficacy in the animal model and ask directly whether that exposure is achievable in humans at a clinically tolerable dose. This target-attainment analysis, borrowed from anti-infective pharmacokinetics/pharmacodynamics (PK/PD) frameworks, dramatically sharpens the go/no-go decision at the in vivo stage and has reduced late-stage attrition in repurposing programs at academic centers that apply it rigorously.
Real-World Evidence: Electronic Health Record Mining
Electronic health records represent a massive, if imperfect, observational dataset for testing repurposing hypotheses in humans before committing to a prospective trial. Retrospective EHR analysis can ask whether patients who received drug A for indication X show lower incidence or delayed progression of disease Y compared to matched controls who received drug A’s comparator. These analyses require careful confounding control because patients receiving any given drug are not randomly assigned, and numerous confounders related to disease severity, comorbidity burden, and prescribing practice can produce spurious associations.
The FDA Sentinel System, which aggregates claims and EHR data on more than 500 million patients, has been used for pharmacovigilance and is increasingly being accessed for repurposing signal detection under the BEST (Biologics Effectiveness and Safety) Initiative. Several academic medical centers have developed federated EHR analysis pipelines that allow repurposing hypotheses to be tested across multiple institutional datasets without sharing patient-level data, addressing both privacy concerns and statistical power limitations.
Mendelian randomization (MR) offers a causal inference approach to EHR-based repurposing validation. MR uses genetic variants as instrumental variables to estimate the causal effect of modulating a drug’s target on disease risk, using GWAS summary statistics rather than clinical records. If the genetic variant that mimics a drug’s mechanism of action is associated with lower disease incidence, that is causal evidence for the repurposing hypothesis that cannot be produced by confounding. Several large-scale MR-based repurposing analyses, including the Drug Target MR platform from the MRC Integrative Epidemiology Unit at Bristol, have generated clinically actionable repurposing signals that have been incorporated into clinical trial design.
KEY TAKEAWAYS
1. AUPR is a more informative validation metric than AUROC when known drug-disease associations are sparse, which is the standard condition in repurposing datasets.
2. Time-split validation is the minimal acceptable standard for evaluating computational repurposing model performance; retrospective-only evaluation on datasets that overlap training data inflates reported metrics.
3. Target engagement confirmation in disease-relevant cells at pharmacologically achievable concentrations is required before in vivo studies; cellular activity at supratherapeutic concentrations is not actionable evidence.
4. Target-attainment PK/PD analysis using prior human PK data can be applied at the in vivo stage to sharpen go/no-go decisions before committing to clinical development.
5. Mendelian randomization provides the closest approximation to causal evidence for a repurposing hypothesis in human population data and is increasingly integrated into trial design rationale.
PART IV: Case Studies with IP Valuation Analysis
Baricitinib: From JAK-STAT Rheumatology to COVID-19 Emergency Authorization
Baricitinib (Olumiant), developed by Eli Lilly and Incyte, is a selective JAK1/JAK2 inhibitor approved in 2018 by the FDA for moderate-to-severe rheumatoid arthritis refractory to one or more DMARDs. Its initial indication carried a full safety data package including long-term extension study data covering cardiovascular risk, malignancy, and serious infection. That established safety package was the primary asset enabling the COVID-19 repurposing program to move at pandemic speed.
The computational lead for baricitinib in COVID-19 came from BenevolentAI in early 2020. The platform identified baricitinib’s putative activity against AAK1 (AP2-associated protein kinase 1), a regulator of clathrin-mediated endocytosis that SARS-CoV-2 exploits for host cell entry, as a secondary mechanism beyond JAK-STAT immunomodulation. This dual-mechanism hypothesis differentiated baricitinib from other JAK inhibitors and contributed to Lilly’s decision to pursue emergency authorization aggressively. The ACTT-2 trial, conducted under NIAID sponsorship, demonstrated a shorter time to recovery compared to remdesivir in hospitalized COVID-19 patients, and the COV-BARRIER trial subsequently showed a 13% relative reduction in 28-day mortality in patients receiving systemic corticosteroids.
Baricitinib IP Valuation: Core Assets
The baricitinib IP estate includes composition-of-matter patents held jointly by Lilly and Incyte, with US expiration dates in the 2028-2029 range. Method-of-use patents covering the RA indication extend into the early 2030s. The COVID-19 emergency use authorization (EUA), and subsequent approval under the tradename Olumiant for COVID-19, did not generate new patent protection, but the regulatory exclusivity associated with the COVID-19 indication added commercial shelf life. The FDA’s grant of non-patent five-year NCE exclusivity (new chemical entity) expires earlier than the composition-of-matter patents, leaving patent protection as the primary exclusivity pillar.
The JAK inhibitor class faces ongoing post-market safety scrutiny. The FDA’s 2021 class-wide boxed warning update covering cardiovascular risk and malignancy, driven by data from tofacitinib’s ORAL Surveillance trial, affected all approved JAK inhibitors including baricitinib. From an IP valuation perspective, the safety warning has not materially undermined baricitinib’s patent value, but it has reshaped the commercial trajectory by limiting first-line use in RA. The COVID-19 indication added peak sales that were not modeled in original valuation analyses and accelerated revenue toward the period of strongest patent protection.
Sildenafil: The Original Repurposing IP Case Study
Sildenafil’s transition from angina candidate to erectile dysfunction treatment and, subsequently, to pulmonary arterial hypertension (PAH) therapy illustrates the full range of repurposing IP strategies across a single molecule’s lifecycle. Pfizer’s original composition-of-matter patents on sildenafil citrate covered the molecule broadly. The erectile dysfunction indication was protected by a separate use patent (US 5,346,905) claiming the treatment of male erectile dysfunction with phosphodiesterase inhibitors. This patent was aggressively enforced by Pfizer against generic manufacturers through the mid-2000s.
The PAH repurposing program, commercialized as Revatio, added a distinct regulatory and IP layer. Pfizer received a new NDA approval in 2005 for 20 mg sildenafil three times daily in PAH, a dose and regimen distinct from the 25-100 mg doses used in erectile dysfunction. This formulation and dosing patent position, combined with orphan drug designation for PAH (which is classified as a rare disease affecting fewer than 200,000 patients in the US), generated seven years of orphan exclusivity that was distinct from, and layered on top of, the existing Viagra patent position.
Sildenafil IP Valuation: Layered Exclusivity Architecture
The Revatio program demonstrates how repurposing can create a second exclusivity tower over the same molecule. The Viagra exclusivity tower expired in 2012 in the US, admitting generic sildenafil for erectile dysfunction. The Revatio tower, protected by orphan exclusivity and PAH-specific dosing patents, had a different expiration structure. Generic manufacturers filing ANDAs for Revatio faced the orphan exclusivity barrier in addition to the listed patents, requiring them to certify under Paragraph IV against both types of protection. Pfizer settled several of these Paragraph IV challenges with reverse payment agreements that delayed generic Revatio entry until 2017.
The commercial value generated by the PAH repurposing program was substantial: Revatio reached annual revenues of approximately $400 million at peak, creating a meaningful second revenue stream from a molecule whose primary exclusivity had substantially expired. For investors and IP teams, the Revatio case shows that orphan drug designation applied to a repurposed compound can generate six-figure-per-patient pricing power and multi-year exclusivity independent of patent status.
Zidovudine: Antiretroviral to Oncology Repurposing
Zidovudine (AZT), approved in 1987 as the first antiretroviral treatment for HIV/AIDS, has been computationally identified as a potential candidate across multiple oncology indications based on mechanistic analysis of its reverse transcriptase inhibitory activity and its capacity to inhibit transposable element (TE) activity in cancer cells. The mechanistic rationale connects LINE-1 and other endogenous retroviral element (ERE) activity in cancer cells to immune evasion and genome instability, suggesting that nucleoside reverse transcriptase inhibitors (NRTIs) like AZT might suppress TE-driven immunosuppression in tumor microenvironments.
The computation-to-clinic pathway for AZT in oncology has been slower than for baricitinib in COVID-19, in part because AZT’s composition-of-matter patents expired decades ago and there is no IP holder with a strong commercial incentive to fund a prospective randomized trial. Academic-initiated repurposing programs for off-patent drugs face this structural incentive problem consistently. The NRTIC trial program, an NCI-funded initiative examining NRTI repurposing in various cancers, represents the public sector’s attempt to fill this gap, but trial timelines are extended and evidence generation is slow.
Zidovudine IP Analysis: The Off-Patent Repurposing Problem
AZT’s patent landscape illustrates the most challenging IP environment for computational repurposing: a molecule with no surviving composition-of-matter protection, multiple generic manufacturers, and no obvious commercial sponsor for new indication development. The theoretical IP play for AZT in oncology would require either a novel combination patent (AZT plus a specific checkpoint inhibitor, for instance) or a patient selection patent claiming a genetically or molecularly defined subpopulation. These secondary patent strategies are viable in principle but require sufficiently narrow and non-obvious claims to survive obviousness challenges.
For institutional investors, off-patent drug repurposing creates a different valuation model than on-patent repurposing. Revenue capture depends on regulatory exclusivity (particularly orphan designation if the target population qualifies), branded formulation patents, and first-mover pricing power in specific hospital formulary settings. The probability of achieving durable exclusivity is lower than in on-patent repurposing, but the development cost is also substantially lower, and grant funding from NIH, BARDA, or Wellcome Trust is more accessible for academic sponsors.
Baricitinib/COVID-19 to Broad AI Repurposing: The REMEDi4ALL Framework
The REMEDi4ALL consortium, an EU-funded Horizon Europe initiative, represents the most systematic attempt to date to catalog, evaluate, and standardize computational repurposing tools. The consortium published a structured evaluation of the in-silico repurposing landscape in Nature Reviews Drug Discovery, identifying 15 open-source, openly accessible tools rated highly by a panel of domain experts. The selection criteria covered scientific validity of the underlying methodology, software maintenance quality, documentation standard, and breadth of applicable disease areas.
Three case studies anchored the REMEDi4ALL evaluation: SARS-CoV-2 repurposing (applying all 15 tools to the COVID-19 context), pancreatic cancer (a disease with poor molecular characterization and low standard-of-care treatment efficacy), and multiple sulfatase deficiency (MSD), an ultra-rare lysosomal storage disorder caused by SUMF1 mutations. The choice of MSD as a case study was deliberate: it represents the subset of rare diseases where computational repurposing has the clearest utility advantage over traditional drug development because no commercial development program would otherwise be economically viable.
The consortium’s analysis revealed significant variability in predicted candidates across tools applied to the same disease, reflecting differences in underlying data sources, algorithmic assumptions, and target scope. This variability is informative for research teams: computational repurposing predictions that appear consistently across multiple independent tools using different methodological approaches carry substantially higher confidence than predictions generated by a single platform.
KEY TAKEAWAYS
1. Baricitinib’s COVID-19 repurposing succeeded partly because its established safety data package eliminated Phase I requirements, allowing direct entry into Phase III under NIAID sponsorship at pandemic speed.
2. The Revatio (sildenafil PAH) case establishes the template for layered repurposing exclusivity: orphan drug designation plus dosing-regimen patents can create a second exclusivity tower over an off-patent molecule.
3. Zidovudine’s oncology repurposing exposes the structural incentive failure in off-patent drug repositioning: no IP holder, no commercial sponsor, and slow academic trial timelines.
4. Consistent prediction of the same drug-indication pair across multiple independent computational platforms, using different methodologies, is a stronger signal than a high-confidence prediction from a single platform.
5. The REMEDi4ALL framework’s multi-tool evaluation approach should be adopted as a standard practice in any R&D organization designing a systematic computational repurposing program.
Investment Strategy: Evaluating Repurposing Programs by IP Architecture
SIGNAL
WHAT TO WATCH
ANALYST IMPLICATION
On-patent repurposing (composition-of-matter still active)
Remaining patent term, new method-of-use filings, sNDA timeline to indication expansion
Long runway; strongest exclusivity position; evaluate whether new indication fits within existing safety label
Off-patent + orphan drug overlay
Orphan designation status, qualifying rare disease prevalence (<200K US patients), competing designations
7-year US exclusivity is durable if designation holds; evaluate litigation risk and ultra-rare disease market size
AI platform repurposing programs (pre-clinical)
Platform track record of prospective clinical hits, pharma partnership terms, proprietary data assets
Assign probability of success adjustment; single-asset AI repurposing companies carry higher risk than multi-program platforms
Low direct investment opportunity but can identify signals early; watch for licensing or spin-out following Phase II readout
505(b)(2) repurposing NDA strategy
Reference listed drug, listed Orange Book patents, Paragraph IV exposure, three-year exclusivity for new clinical investigations
Three-year exclusivity from clinical investigations provides limited but real protection; assess Paragraph IV risk before investing around NDA filing
PART V: Regulatory Pathways for Repurposed Drugs
The 505(b)(2) NDA: The Primary Regulatory Tool
Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act allows an NDA applicant to rely, at least in part, on published literature or FDA’s findings of safety and effectiveness for an existing approved drug. This pathway was designed precisely for products that modify or build on previously approved drug substances, including repurposed drugs seeking new indications, new formulations, new dosing regimens, or new patient populations.
A 505(b)(2) application for a new indication of an approved drug can reference the approved drug’s existing safety data for the common toxicology profile while generating new clinical efficacy data for the target indication. The regulatory efficiency gain is meaningful: a Phase I safety study may be waived or reduced to a bridging pharmacokinetic study if the population’s characteristics do not suggest meaningfully different drug exposure. The remaining development burden is typically a Phase II proof-of-concept study followed by at least one adequately powered Phase III trial, depending on the indication’s regulatory requirements and the strength of existing mechanistic evidence.
The exclusivity protections available under 505(b)(2) for a new indication are three years of exclusivity for new clinical investigations if those studies were essential to approval, layered over any existing patent protection for the drug. This three-year exclusivity is narrower than the five-year NCE exclusivity available to new molecular entities and does not block ANDA filers from referencing the original drug’s approval, only from using the new clinical data submitted in the 505(b)(2) application.
Orphan Drug Designation: The Repurposing Accelerator for Rare Indications
ODD is one of the most powerful exclusivity tools available to repurposing programs targeting diseases with a US prevalence below 200,000 patients. Beyond the seven-year market exclusivity, ODD confers a 25% tax credit on qualified clinical trial expenses (reduced from 50% by the Tax Cuts and Jobs Act of 2017 and subject to ongoing IRA-related adjustments in 2026), waiver of the PDUFA application fee (approximately $3.4 million for FY2026), and eligibility for the FDA’s Rare Pediatric Disease Priority Review Voucher program if applicable pediatric criteria are met.
The ODD market exclusivity is product-specific and indication-specific, not compound-specific in a broad sense. Multiple sponsors can hold ODD for the same drug in different rare diseases, and a second company can obtain ODD for the same drug in the same disease if it demonstrates clinical superiority or that the first product is not available in sufficient quantity. The clinical superiority standard is defined as greater effectiveness, greater safety, or a major contribution to patient care. For repurposing programs, the clinical superiority pathway is relevant when an improved formulation or dosing regimen for an already-designated compound is the primary differentiation strategy.
Accelerated Approval and Breakthrough Therapy Designation in Repurposing
Breakthrough Therapy Designation (BTD) is available to repurposing programs targeting serious conditions where preliminary clinical evidence indicates substantial improvement over available therapy on at least one clinically significant endpoint. Repurposed drugs with established safety profiles and early efficacy signals from investigator-initiated trials can be strong BTD candidates because the FDA’s rolling review and intensive guidance engagement begins early in clinical development, and the safety evidence base reduces the primary development risk to efficacy confirmation.
Accelerated Approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit is available in oncology and other serious indications. Several repurposing programs have entered Accelerated Approval pathways on the basis of early Phase II tumor response data, with confirmatory trials required post-approval. The regulatory risk in this pathway is the confirmatory trial requirement: if the confirmatory Phase III trial fails, approval can be withdrawn, as FDA demonstrated with multiple oncology Accelerated Approvals in the 2020-2023 period.
KEY TAKEAWAYS
1. 505(b)(2) NDA provides three-year exclusivity for new clinical investigations and allows partial reliance on existing safety data; it is the primary regulatory pathway for most indication-expansion repurposing programs.
2. Orphan Drug Designation generates seven-year US market exclusivity independent of patent status, plus meaningful tax and fee benefits; it is applicable to any rare-disease repurposing target regardless of the drug’s original indication.
3. Breakthrough Therapy Designation is accessible to repurposing programs with preliminary clinical evidence of substantial improvement and can compress Phase II-to-NDA timelines through intensive FDA guidance.
4. Accelerated Approval based on surrogate endpoints in oncology repurposing carries post-approval confirmation risk; the FDA’s willingness to withdraw approvals on failed confirmatory trials should be modeled in program risk assessments.
PART VI: Challenges, Failure Modes, and Mitigation Strategies
Data Quality and the Training Distribution Problem
The predictive validity of any computational repurposing platform is constrained by the quality of the underlying data. The human drug-target interaction network is sparse and biased: well-studied protein families like kinases, GPCRs, and nuclear receptors are represented by orders of magnitude more interaction data than poorly studied families. A computational model trained primarily on kinase interaction data will perform well on kinase-based repurposing predictions but will systematically underperform on targets from underexplored biology. This training distribution problem is not solvable through algorithmic improvements alone; it requires broader and more chemically diverse experimental interaction data.
The public drug-target interaction databases also contain a substantial proportion of low-quality or incorrectly annotated records. ChEMBL, despite curation efforts, contains duplicate entries, incorrect target assignments from automated literature extraction, and assay protocol variations that make cross-assay comparison unreliable without normalization. Research groups that fail to apply rigorous data cleaning and activity cliff analysis before training models on ChEMBL may produce repurposing models with inflated apparent performance on retrospective benchmarks.
The Translation Gap: Why Computational Hits Fail Clinically
The rate at which computational repurposing predictions survive to clinical confirmation is not well characterized in the literature, partly because failed predictions are rarely published. Conservative estimates from groups that track prospective predictions suggest that fewer than 10 percent of computationally generated repurposing hypotheses produce a positive clinical signal at Phase II. This rate is not catastrophically below the general Phase II success rate (~50-60% for drugs with strong preclinical rationale) when one considers that many computational predictions are not subjected to rigorous preclinical prioritization before advancing to clinical testing.
The most common specific failure mode is inadequate target exposure in the relevant tissue compartment at a clinically tolerable dose. A drug may bind a target with high affinity in a biochemical assay and produce the expected pharmacological effect in a cell line, but if free drug concentration in the target tissue at therapeutic plasma exposure is insufficient to engage the target at clinically relevant occupancy, the mechanism will not translate to efficacy. This exposure-response relationship is sometimes characterized in the original indication’s clinical data package, but the target tissue is often different in the repurposed indication, making tissue distribution data in the new disease context a prerequisite for confident clinical translation.
Intellectual Property and Evergreening Critique
Drug repurposing has attracted criticism as a vehicle for pharmaceutical evergreening: the practice of filing incremental patents on known drugs to extend effective market exclusivity beyond the period justified by original R&D investment. The criticism is most pointed when method-of-use patents on new indications generate exclusivity over drugs that have been off-patent for years and are available generically at low cost, but where the new indication-specific label creates a branded market that commands premium pricing.
The legal boundary between legitimate new-indication development and impermissible evergreening is not always clear. Courts and the USPTO evaluate method-of-use patent claims on conventional patentability grounds (novelty, non-obviousness, enablement) without reference to the underlying drug’s age or price. From an IP strategy perspective, the question of whether a repurposing patent will survive validity challenge is distinct from the policy question of whether it should. R&D teams and their IP counsel need to address both: the former through prior art searches and claim drafting, the latter through proactive payer engagement and access program design that can pre-empt reimbursement challenges.
Regulatory Complexity and the Label Problem
A repurposed drug approved for a new indication faces a specific regulatory challenge: physicians prescribing the drug for the new indication must use the labeled dose and formulation, which may differ from what was approved in the original indication. If the optimal dose for the new indication is lower, a generic manufacturer can undercut the branded repurposing program by recommending off-label dose reduction from the cheaper original formulation, capturing prescriptions without having conducted the clinical trials. This label arbitrage problem is particularly relevant in oncology, where weight-based dosing and dose modifications are routine.
KEY TAKEAWAYS
1. The training distribution bias toward well-studied protein families (kinases, GPCRs) means that computational repurposing platforms systematically underperform on understudied biology; platforms with proprietary broad-biology interaction data have a structural advantage.
2. Fewer than 10% of prospective computational repurposing predictions generate a positive Phase II clinical signal; rigorous preclinical target-attainment analysis before advancing to clinical testing would improve this rate.
3. Tissue-level drug exposure at therapeutic plasma concentrations is the most common specific mechanistic explanation for clinical failure after a positive computational and in vitro result.
4. Evergreening critiques of repurposing IP strategies have regulatory and legal dimensions that are separable; R&D and IP teams should address both through claim design and proactive payer engagement.
5. Label arbitrage by generic manufacturers can undermine repurposing program economics even when method-of-use patents are valid; formulation patents and REMS programs provide partial but imperfect protection.
PART VII: Future Directions and Emerging Opportunities
Precision Repurposing: Patient Selection as IP Strategy
The convergence of computational repurposing with biomarker-driven precision medicine is reshaping both the science and the IP strategy of the field. Rather than seeking broad new indications for existing drugs, the leading edge of repurposing research now focuses on identifying molecularly defined patient subgroups in whom a known drug will generate a signal that is not detectable in an unselected population. This approach, called precision repurposing, produces companion diagnostic requirements, biomarker patents, and patient selection patents that create layered IP protection distinct from the drug’s original patent estate.
The regulatory and commercial framework for precision repurposing benefits from the FDA’s co-development guidance for therapeutics and companion diagnostics. A repurposing NDA filed with a required companion diagnostic generates a more defensible exclusivity position than a repurposing NDA with an optional biomarker, because the required companion diagnostic is listed in the drug’s label and creates a barrier to off-label generic substitution. The companion diagnostic itself may also be separately patentable, adding an additional IP layer.
Quantum Computing: Horizon Technology for Binding Prediction
Quantum computing’s potential contribution to drug repurposing lies primarily in quantum chemistry simulation of drug-target binding at full electronic-structure resolution. Classical force-field-based molecular dynamics, the current standard for characterizing protein-ligand interactions, uses empirical energy functions that sacrifice accuracy for computational tractability. Quantum mechanical (QM) methods provide more accurate descriptions of electronic polarization, charge transfer, and dispersion interactions that are important for predicting binding affinity, but the computational cost of QM calculations currently limits their application to small molecular fragments or short simulation timescales.
Variational quantum eigensolvers (VQEs) and quantum phase estimation algorithms running on error-corrected quantum hardware could, in principle, perform full QM binding free energy calculations for drug-protein complexes at biologically relevant sizes within practically useful timescales. Current quantum hardware, still in the noisy intermediate-scale quantum (NISQ) era, cannot yet demonstrate quantum advantage on practically relevant drug-binding problems. Horizon estimates from quantum hardware developers place commercially useful fault-tolerant quantum computation in the early-to-mid 2030s, making quantum binding prediction a planning horizon for R&D strategy rather than a current operational tool.
Single-Cell and Spatial Genomics: Resolving the Tissue Problem
Single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics are addressing one of computational repurposing’s most persistent weaknesses: the inability to specify which cell type within a tissue is actually the relevant site of drug action for a given indication. Bulk transcriptomic approaches average gene expression across heterogeneous cell populations within a tissue, obscuring cell-type-specific drug effects that are clinically meaningful. ScRNA-seq resolves this ambiguity by profiling gene expression in individual cells.
The Human Cell Atlas, now comprising single-cell profiles from over 100 distinct tissue types and over 50 million cells, is the reference atlas for mapping drug perturbation effects to specific cell types. Repurposing platforms that integrate HCA-derived cell-type-specific expression profiles with CMap/LINCS perturbation data can make cell-type-resolved repurposing predictions that are substantially more mechanistically precise than bulk transcriptomic approaches. This level of resolution also creates the potential for identifying subtype-specific biomarkers that can anchor companion diagnostic development.
Federated Learning and Privacy-Preserving Repurposing
Multi-institutional EHR data generates far more statistical power for repurposing signal detection than any single institution’s records, but patient data privacy regulations (HIPAA in the US, GDPR in Europe) restrict the movement of identifiable health data across institutional boundaries. Federated learning, in which machine learning models train on locally held data at each institution and only model parameter updates (never raw data) are shared across the network, offers a technically sound approach to this problem.
The PCORnet (Patient-Centered Outcomes Research Network) infrastructure, which connects EHR data from over 80 million patients across US health systems in a federated architecture, is increasingly being accessed for repurposing research. The FDA’s Sentinel System and the European Health Data Space, which will go into full operation under the European Health Data Space regulation passed in 2024, are the major regulatory-grade federated data infrastructures that could support prospective repurposing signal detection at population scale.
KEY TAKEAWAYS
1. Precision repurposing targeting molecularly defined patient subgroups creates layered IP through companion diagnostic co-development, biomarker patents, and patient selection patents, producing a stronger exclusivity position than broad indication claims.
2. The Human Cell Atlas provides single-cell resolution reference data that substantially improves the mechanistic specificity of repurposing predictions over bulk transcriptomic approaches.
3. Federated learning across PCORnet and EU Health Data Space infrastructure will enable EHR-powered repurposing signal detection at population scale without requiring patient data transfer.
4. Quantum computing for drug-binding simulation represents a planning horizon technology for early-to-mid 2030s; current NISQ-era hardware does not yet demonstrate meaningful quantum advantage on practically relevant binding problems.
Investment Strategy: Sector-Level Positioning in Computational Repurposing
SIGNAL
WHAT TO WATCH
ANALYST IMPLICATION
AI platform companies with prospective clinical hits
ClinicalTrials.gov filings for computationally derived candidates; Phase II readout timelines
Probability-weight each active clinical program; platforms with more than two programs in Phase II carry lower binary risk
Layered IP creates better exclusivity than indication-only repurposing; evaluate diagnostic partner’s regulatory track record
Rare disease repurposing (ODD overlay)
FDA ODD grant notices; PDUFA date calendars; competing programs in the same indication
ODD-protected rare disease repurposing has pricing power that the underlying drug’s off-patent status would otherwise eliminate
Federated EHR data platform providers
Network growth (number of connected patient records); pharma collaboration revenue; regulatory data quality certifications
Infrastructure plays are less binary than single-drug repurposing bets; revenue scales with network effects and partnership volume
Academic spin-outs from repurposing consortia
IP assignment agreements from university technology transfer offices; NIH grant history; Phase I data in hand
Pre-clinical spin-outs carry full development risk; prior grant funding de-risks early-stage capital requirements
Master Key Takeaways
KEY TAKEAWAYS
1. Approximately 30% of newly marketed US drugs result from repurposing strategies; the primary development cost advantage is concentrated in programs where the drug’s mechanism maps directly to the new indication at the same or similar dose.
2. IP protection for repurposed drugs requires method-of-use, formulation, dosing regimen, and patient selection patents, all of which carry higher obviousness risk than composition-of-matter claims and require active maintenance against IPR challenges.
3. The 505(b)(2) pathway is the regulatory default for indication expansion; Orphan Drug Designation adds seven years of US market exclusivity independent of patent status and is applicable to any repurposed drug targeting a qualifying rare disease.
4. GNN architectures have displaced classical fingerprint-based ML for drug-target interaction prediction; biological foundation models (scGPT, scFoundation) now enable cell-type-specific perturbation prediction at single-cell resolution.
5. The network proximity hypothesis provides a scalable, coarse-grained repurposing filter; its highest validated use is as a multi-tool consensus screen, not as a primary evidence source for any single algorithm’s prediction.
6. Baricitinib’s COVID-19 program succeeded because JAK-STAT inhibitor safety data was already fully established, enabling direct Phase III entry at pandemic speed; the safety data package was the core asset, not the computational identification alone.
7. The Revatio/sildenafil case demonstrates that orphan drug designation applied to a repurposed molecule can reconstruct a full exclusivity tower over a compound whose primary patents have substantially expired.
8. Fewer than 10% of prospective computational repurposing predictions generate a positive Phase II clinical signal; rigorous target-attainment PK/PD analysis before clinical entry is the highest-yield intervention to improve this rate.
9. Time-split validation and multi-platform consensus screening are the minimum methodological standards for evaluating computational repurposing platform quality; single-algorithm retrospective performance metrics are insufficient.
10. Precision repurposing targeting molecularly defined patient subpopulations creates the strongest combined IP and commercial position, anchored by companion diagnostic co-development and layered biomarker patent protection.
Frequently Asked Questions
What is the legal difference between 505(b)(2) and a supplemental NDA for a new indication?
A supplemental NDA (sNDA) is filed by the holder of the original approved NDA. It adds a new indication, new dosing information, or new patient population to the existing approval without requiring a new reference drug. A 505(b)(2) NDA can be filed by any sponsor, including one without ownership of the original NDA, and relies on published literature or FDA’s prior findings for at least some of the safety and effectiveness data. For repurposing programs where the sponsor is not the original NDA holder, 505(b)(2) is the available pathway. Original NDA holders typically prefer the sNDA route because it keeps the new clinical data tightly controlled within their regulatory submission.
How does Paragraph IV certification apply to repurposed drugs?
When a generic manufacturer files an ANDA referencing an approved repurposed drug product and believes that a listed Orange Book patent is invalid, unenforceable, or will not be infringed by its proposed generic, it files a Paragraph IV certification and must provide notice to both the NDA holder and patent owner. If the brand sponsor files a patent infringement suit within 45 days, FDA automatically imposes a 30-month stay on ANDA approval. Repurposed drugs with method-of-use patents covering the specific new indication are the most common target for Paragraph IV certification in repurposing contexts, because the generic manufacturer’s proposed label can be ‘carved out’ to omit the patented indication and still proceed under a ‘skinny label’ strategy, which does not trigger the 45-day suit clock for those claims.
Which computational validation metric matters most for evaluating a repurposing platform?
For an organization evaluating a vendor’s or academic group’s repurposing platform, prospective clinical validation is the only metric that fully tests what the platform claims to do: predict which drug will work in which new indication before the clinical data exists. AUPR and AUROC on held-out retrospective datasets are necessary hygiene checks, not sufficient proof of platform validity. The practical question for evaluators is how many prospective predictions from this platform have been tested in a clinical context, what fraction of those tests generated a positive signal, and were those tests conducted in well-controlled investigator-initiated or sponsored trials rather than anecdotal off-label use.
Can AI-generated repurposing hypotheses be patented?
The patentability of AI-generated inventions is unsettled law in most jurisdictions. The USPTO’s February 2024 guidance on AI-assisted inventions holds that a patent can claim an AI-assisted discovery if a natural person made a significant contribution to the conception of the claimed invention. Under this standard, a repurposing hypothesis generated by an AI system and subsequently validated by human researchers who exercised scientific judgment in selecting, designing, and interpreting validation experiments likely meets the inventorship standard. The AI system itself cannot be named an inventor. From a claim-drafting perspective, the safest approach is to claim the validated biological finding (the drug-disease association and its mechanistic support) rather than the computational method that generated the hypothesis.
What is the minimum viable evidence package to support a repurposing IND?
The FDA does not prescribe a fixed evidence package for repurposing INDs beyond the requirements in 21 CFR 312.23, but the agency’s Division of Oncology Products and other divisions have issued informal guidance through pre-IND meeting feedback. For a repurposed drug with full clinical safety data from its original indication, the minimum viable package typically includes a mechanistic rationale document supported by published literature and in vitro or in vivo preclinical efficacy data in a disease-relevant model, a proposed clinical pharmacology section demonstrating that the planned clinical dose is achievable and was safe in the prior indication, and an initial clinical protocol with a clearly defined primary endpoint and patient population. The completeness of the package required in practice scales with how different the new indication is from the original: a JAK inhibitor repurposed for a second inflammatory condition requires less new preclinical work than a JAK inhibitor repurposed for a metabolic disease.
This analysis is produced for informational purposes only and does not constitute legal, financial, or regulatory advice. Patent landscapes and regulatory guidance are subject to change. Readers should consult qualified IP counsel and regulatory professionals before making strategic decisions based on information contained herein.