A deep-dive pillar for pharma IP teams, generic market strategists, portfolio managers, and institutional investors tracking ANDA timelines and biosimilar entry risk.
1. What REMS Actually Is, and What It Has Become
1.1 Statutory Foundation
Risk Evaluation and Mitigation Strategies are mandatory post-market safety programs authorized under Section 505-1 of the Federal Food, Drug, and Cosmetic Act, codified through the Food and Drug Administration Amendments Act of 2007. The FDAAA gave the FDA explicit authority to require REMS either before a drug reaches the market or at any point after approval when new safety signals emerge.
Before 2007, the FDA used Risk Minimization Action Plans (RiskMAPs) as the predecessor framework. RiskMAPs were less standardized and lacked the binding enforcement structure that ETASU-based REMS now carry. The 2007 formalization created clearer legal obligations, but it also created the architecture that brand manufacturers now exploit.
REMS programs fall into a rough taxonomy by component complexity. The simplest require only Medication Guides, FDA-approved patient-facing documents distributed at dispensing. A step up, some REMS include Communication Plans, direct manufacturer-to-prescriber outreach designed to flag specific adverse event profiles. At the most restrictive tier sit Elements to Assure Safe Use, which can mandate prescriber certification, pharmacy certification, restricted dispensing locations, laboratory test prerequisites before dispensing, patient enrollment in registries, and ongoing monitoring documentation. As of January 2025, 94.5% of all active REMS programs incorporate ETASU.
Each REMS also requires a timetable for assessment reports, giving the FDA a mechanism to determine whether the program is meeting its stated risk mitigation goals and whether modifications are warranted. This assessment requirement is simultaneously the regulatory tool most amenable to genuine safety refinement and the procedural mechanism most easily weaponized to slow down shared-system negotiations with generic entrants.
1.2 The Dual-Use Problem
The FDA’s Dr. John Jenkins, former Director of the Office of New Drugs, said publicly that brand companies are ‘abusing the system’ and that REMS has become ‘an evergreening system for avoiding generic competition.’ That statement did not come from a generic industry lobbying group. It came from inside the agency charged with administering these programs.
This contradiction is the central analytical challenge: REMS simultaneously protect patients from drugs that carry risks severe enough to keep them off the market otherwise, and provide brand manufacturers with a legal architecture that can indefinitely delay the competition that drives prices toward marginal cost. The two functions are not separable by design. They are separable only by intent, and intent is precisely what generic manufacturers, the FTC, and now federal courts have to prove.
The FDA’s 2023 guidance updates, which aimed to reduce stakeholder burden through standardized REMS language and consolidated assessment reporting, signal that the agency recognizes the administrative weight REMS have accumulated. But guidance updates do not resolve the core incentive problem: every month of delayed generic entry is worth hundreds of millions of dollars to brand manufacturers for top-tier products.
Key Takeaways: Section 1
The statutory authority for REMS is legitimate and the underlying safety purpose is real. The problem is not the statute. The problem is that the same ETASU architecture that restricts patient access for safety reasons also restricts generic and biosimilar manufacturers from obtaining reference drug samples, running bioequivalence studies, and progressing through ANDA review. Brand companies discovered this dual function and built a litigation and negotiation playbook around it. The rest of this document maps that playbook in operational detail.
2. The Anti-Competitive Playbook: Five Tactics, One Goal
Brand companies do not need to win every anti-competitive move. They need only delay generic market entry long enough to recover revenue that a patent cliff would otherwise eliminate. Here are the five primary REMS-based tactics, in order of operational prevalence.
2.1 Sample Denial Under REMS Pretext
Generic bioequivalence testing under the Abbreviated New Drug Application pathway requires physical samples of the Reference Listed Drug. The brand manufacturer controls those samples. For products with REMS, particularly those with restricted distribution, brand companies have refused to sell samples to generic developers, citing safety program requirements as justification for the refusal.
The claimed rationale is that the restricted distribution system means samples cannot be diverted to parties not certified under the REMS. In practice, the FDA’s own guidance made clear that bioequivalence testing can be conducted under safety protocols acceptable to the agency, and that a brand manufacturer’s provision of samples for that purpose does not constitute a REMS violation. The FDA issued draft guidance in 2014 outlining how generic developers could obtain an FDA opinion letter to this effect. Brand manufacturers disputed these letters’ enforceability in court, and the dispute dragged on until CREATES.
The economic logic of sample denial is straightforward. A brand manufacturer that prevents a generic from ever submitting an ANDA prevents the FDA from ever approving that generic. No approval, no market entry, no price compression. The 180-day generic exclusivity window granted to first-filers under Hatch-Waxman never triggers. The entire competitive dynamic downstream of ANDA approval is frozen.
2.2 Shared REMS Negotiation as Delay Instrument
Under FDA guidance, when a brand drug has a REMS with ETASU, any generic version must have the same or comparable ETASU. The FDA’s strong preference, codified in guidance, is a single, shared system (SSS REMS) that covers both brand and generic products under one administrative infrastructure. This reduces burden on prescribers and pharmacies, who otherwise must enroll in multiple programs for clinically equivalent drugs.
Brand manufacturers recognized that the SSS REMS preference created an exploitable chokepoint. If the FDA will not approve a generic with separate REMS unless the brand agrees to a shared system or the FDA grants a formal waiver, and if the brand can control the pace and terms of shared system negotiations, the brand can delay generic market entry indefinitely without ever formally refusing to cooperate.
Negotiation impasses arise over cost-sharing formulas, governance voting structures, confidentiality provisions, product liability indemnification, and antitrust concerns around competitor coordination. None of these are trivial. But the GAO, the FTC, and multiple generic manufacturers have documented that brand companies selectively drag out discussions on commercially resolvable terms for years, using procedural complexity as a strategic asset rather than a genuine obstacle.
2.3 Proprietary Restricted Distribution Outside Formal REMS
The Association for Accessible Medicines’ 2018 Matrix Global Advisors report found that $10.3 billion of the $13.4 billion in annual lost savings came not from formal FDA-mandated REMS, but from proprietary restricted distribution systems that brand companies created unilaterally, outside the REMS framework. These programs mimic REMS architecture, restrict access to specialty pharmacies, and impose enrollment requirements on prescribers, but they have no FDA mandate and no regulatory purpose beyond competitive exclusion.
This tactic is analytically distinct from REMS abuse because it does not even require the invocation of a safety rationale. The brand simply contracts with a limited set of specialty distributors, excludes open-market wholesalers, and asserts that sample sales for generic testing violate distribution agreements. Courts have had to parse whether these agreements constitute antitrust violations under Section 1 of the Sherman Act, a more complex evidentiary path than the CREATES Act’s direct statutory claim.
2.4 REMS Patent Claims to Block Waiver Grants
CREATES Act provisions allow the FDA to grant waivers of the single, shared system requirement in circumstances where an aspect of the ETASU is protected by an unexpired patent or trade secret. Brand companies have begun filing patents on REMS program components, including database structures, patient registry software, enrollment certification workflows, and algorithm-based monitoring systems. If a REMS component is patented, the generic manufacturer must certify that it sought but could not obtain a license before the FDA will grant a waiver.
This tactic grafts patent law onto regulatory procedure. A brand company that patents its REMS infrastructure converts what should be an administrative negotiation into an IP licensing dispute, adding legal surface area and delay. The strategy is particularly effective against small or mid-sized generic entrants that lack resources for parallel ANDA prosecution, REMS negotiation, and patent licensing or invalidation proceedings.
2.5 Procedural REMS Assessment Manipulation
REMS programs require periodic assessment reports. Brand companies have used the assessment process to propose expansions or modifications to REMS requirements at moments when generic market entry is imminent. A proposed REMS modification triggers a new FDA review period, and if that modification affects the ETASU structure, the FDA may require generic applicants to demonstrate that their proposed REMS meets the new standard. This delays approval.
This tactic is the hardest to prove and the hardest to counter because REMS assessment is a legitimate regulatory obligation. Distinguishing a good-faith safety-driven REMS modification from a strategically timed one requires contemporaneous documentation of the brand manufacturer’s competitive intelligence activities, which is rarely available to generic challengers without formal discovery.
Key Takeaways: Section 2
Brand manufacturers do not use one tactic. They use layered combinations. A company facing multiple ANDAs and biosimilar applicants can simultaneously deny samples to applicants in early development stages, stall shared REMS negotiations with applicants who have already obtained samples, patent REMS infrastructure to block waiver-based workarounds, and propose REMS modifications when an applicant nears approval. Each layer adds months or years of delay, and the cumulative effect is a monopoly extension that operates entirely within the legal grey zones the CREATES Act was written to close.
3. IP Valuation Under REMS Siege: Celgene, Actelion, and the Cost of the Moat
Understanding the commercial stakes of REMS manipulation requires quantifying how much IP value these tactics protect and how that value flows into corporate transactions. Two case studies anchor this analysis.
3.1 Celgene: Revlimid, Thalomid, and the REMS-as-Patent-Extension Structure
Celgene built the most documented case study in REMS weaponization. Revlimid (lenalidomide) and Thalomid (thalidomide) both carried REMS with restricted distribution due to severe teratogenicity. Both products generated combined revenues exceeding $10 billion annually at peak. The REMS programs were not incidental to those revenues; they were structural.
Mylan’s April 2014 antitrust lawsuit against Celgene alleged that Celgene refused to sell Mylan bioequivalence testing samples of both Revlimid and Thalomid, even after the FDA had determined that Mylan’s proposed safety protocols were acceptable for testing purposes. The complaint documented a deliberate scheme to prevent sample access despite repeated requests. Celgene’s position was that its distribution agreements with its specialty distributor, and its REMS obligations, precluded sample sales to potential generic competitors.
From an IP valuation standpoint, Celgene’s REMS strategy extended the effective exclusivity of Revlimid well beyond its compound patent expiration. Lenalidomide compound patents began expiring in 2019, but Celgene negotiated volume-limited generic entry agreements with several manufacturers, phased in over years, as part of patent litigation settlements. The REMS infrastructure reinforced this settlement architecture by creating an additional barrier that generic manufacturers would need to clear independently of the patent position. Bristol Myers Squibb acquired Celgene in 2019 for $74 billion, with Revlimid’s long-dated exclusivity through settlement agreements treated as a core valuation driver. The REMS component of that exclusivity was not separately disclosed in transaction documents but was embedded in the revenue ramp projections that justified the acquisition premium.
The analytical lesson for IP teams: in a transaction context, REMS-based exclusivity must be modeled as a separate risk-adjusted exclusivity layer on top of patent exclusivity, not as a redundant safety program. When that layer collapses, the revenue curve collapses faster than the patent expiry schedule suggests.
3.2 Actelion: Tracleer, Zavesca, and Preemptive Litigation as Moat Defense
Actelion took a different approach. Rather than passively refusing sample requests, Actelion in 2011 preemptively sued the generic applicants Apotex and Roxane seeking a declaratory judgment that it had no legal duty to supply samples of Tracleer (bosentan) and Zavesca (miglustat). Both products carried REMS with restricted distribution systems linked to embryofetal toxicity and severe neurological risks.
Tracleer was Actelion’s primary revenue driver, generating over $1 billion annually in the U.S. before generic entry pressure. The company’s strategy of filing first, before any generic had completed bioequivalence testing, was designed to establish judicial precedent protecting sample denial as a legal right. The FTC filed an amicus brief arguing that this position, if upheld, would allow monopolists to use REMS programs to prevent any generic from ever entering the market.
The case settled, which meant the strategic goal of establishing pro-brand precedent failed. But the delay achieved was real. Actelion was acquired by Johnson & Johnson in 2017 for $30 billion, with Tracleer and its successor Opsumit central to the valuation. J&J’s due diligence would have modeled the REMS-protected exclusivity window as a component of the enterprise value of the pulmonary arterial hypertension franchise.
IP teams at acquirers and targets must now run a specific REMS due diligence module: catalog the ETASU components, assess which are patent-protected, identify ANDA filers and biosimilar applicants in queue, assess the status of any shared REMS negotiations, and model the revenue impact if the FDA grants a waiver allowing separate generic REMS on a compressed timeline.
3.3 REMS IP Valuation Framework
A REMS program’s value as an exclusivity asset depends on four variables. First, the revenue at stake, which is the peak net U.S. sales of the brand product multiplied by the generic erosion curve for that therapeutic class. Second, the REMS defensibility score, which is a function of whether REMS components are patent-protected, how complex the ETASU infrastructure is, and whether the brand has documented a history of good-faith shared REMS negotiations. Third, the ANDA queue depth, meaning how many applicants are in queue, at what stage of ANDA review, and whether any have already cleared bioequivalence testing. Fourth, CREATES Act exposure, which is the probability that a court would compel sample sales or find the brand liable for willful refusal.
Analysts who reduce REMS to a line item in a compliance cost model are mispricing the asset. For high-revenue ETASU drugs with multiple ANDA filers, REMS-related exclusivity can extend effective market protection by 24 to 48 months beyond patent expiration. At a blended gross margin of 80% for specialty products, that extension on a $2 billion revenue drug is worth $2.4 to $4.8 billion in gross profit. It is not a compliance detail. It is the most expensive regulatory question in the transaction.
Key Takeaways: Section 3
REMS-based exclusivity has been priced into major pharma M&A transactions without being explicitly labeled. Acquiring teams at BMS (Celgene) and J&J (Actelion) paid strategic premiums partly based on revenue projections that assumed delayed generic entry. Post-CREATES, that assumption requires a haircut. The REMS moat is narrower and less defensible than it was in 2018, and any DCF model for a REMS-restricted specialty product that does not account for CREATES Act litigation risk is incomplete.
4. The Hatch-Waxman Bargain Being Broken
4.1 What Hatch-Waxman Actually Agreed To
The Drug Price Competition and Patent Term Restoration Act of 1984 was a compromise. Innovator companies received patent term restoration, up to five years, to compensate for time lost during the FDA approval process. In exchange, the ANDA pathway allowed generic manufacturers to reference innovator clinical data and to demonstrate bioequivalence rather than conduct full independent clinical trials, dramatically lowering the cost of generic development.
The ANDA pathway’s bioequivalence standard is the foundation of the entire agreement. If a generic cannot run bioequivalence studies, it cannot file an ANDA. If it cannot file an ANDA, the Hatch-Waxman framework produces no competitive result. Brand companies that deny samples for bioequivalence testing are not fighting within the Hatch-Waxman framework. They are dismantling it.
4.2 The Paragraph IV Certification Mechanism
Hatch-Waxman created the Paragraph IV certification pathway for ANDAs that challenge Orange Book-listed patents. A Paragraph IV filer certifies that the listed patent is invalid, unenforceable, or will not be infringed by the proposed generic. Filing a Paragraph IV certification triggers a 30-month stay of ANDA approval if the brand company sues for patent infringement within 45 days.
This mechanism is the primary legal battleground for most small-molecule generic competition. It is well-documented, heavily litigated, and, despite its complexity, reasonably well-functioning as a competitive pathway. REMS abuse operates upstream of the Paragraph IV system. If a generic manufacturer cannot obtain samples to run bioequivalence studies, it cannot even file the ANDA that would trigger a Paragraph IV certification. The REMS barrier prevents competition from entering the patent litigation arena at all.
4.3 The Orange Book Patent Listing Problem as Complement to REMS Abuse
The FTC’s September 2023 policy statement on improper Orange Book patent listings is directly relevant here. The FTC has cited a pattern of brand manufacturers listing patents in the Orange Book that cover drug delivery devices, packaging components, and REMS software tools, rather than the drug substance or formulation. When these listings trigger 30-month stays upon a generic’s ANDA filing, they extend market exclusivity without a valid pharmaceutical IP basis.
The combination of improper Orange Book listings and REMS sample denial creates a two-layer barrier: generic companies that somehow obtain samples face extended 30-month stays based on device or REMS-related patent listings, and those stays give brand companies additional time to litigate or negotiate settlements that delay competitive market entry further.
Key Takeaways: Section 4
The Hatch-Waxman Act’s generic pathway depends on bioequivalence testing. Bioequivalence testing depends on sample access. Sample denial collapses the entire framework. REMS abuse is not a sophisticated legal strategy exploiting regulatory complexity at the margins. It is a direct attack on the foundational precondition of generic competition. The CREATES Act addresses this specifically, but the complementary tactics of improper Orange Book listings and proprietary restricted distribution require parallel enforcement action from the FTC and the FDA’s Office of Pharmaceutical Quality.
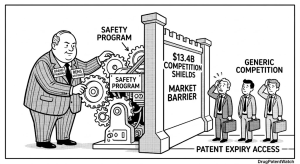
5. Economic Damage: The $13.4B Annual Tax on Generic Entry
5.1 The Core Numbers and Their Composition
Matrix Global Advisors’ July 2014 study, commissioned by the Association for Accessible Medicines, quantified $5.4 billion in annual lost savings to the U.S. health system from REMS and REMS-like access blocks, based on analysis of 40 drugs across eight generic manufacturers. By September 2018, the same analytical framework applied to an updated dataset produced a figure of $13.4 billion, a 250% increase in four years.
The composition of that $13.4 billion is analytically important. Only $3.1 billion was attributable to formal FDA-mandated REMS programs. The larger portion, $10.3 billion, came from proprietary restricted distribution systems that brand companies created outside the REMS framework. This split tells IP and policy teams that the CREATES Act, which specifically addresses REMS-related access barriers, addresses roughly 23% of the documented economic harm. The remaining 77% requires a different legal and regulatory response, likely grounded in antitrust enforcement of distribution agreements and the FTC’s authority over unfair methods of competition.
Breaking the $13.4 billion down by payer reveals a policy map. The federal government loses $5.2 billion annually, a figure that spans Medicare Part D, Medicaid, and federal employee health programs. Private insurers lose $2.4 billion. State and local governments and other payers collectively lose over $500 million. Patients pay an additional $1.8 billion out of pocket in costs they would not face if generic alternatives were available. The remaining portion is distributed across other payers including PBMs taking margin hits and employer self-insured plans absorbing higher claims.
5.2 The Biosimilar Multiplier
Matrix Global Advisors estimated that delayed biosimilar market entry costs approximately $140 million for every $1 billion in biologics sales. Given that the U.S. biologics market now exceeds $200 billion in annual sales, the potential lost savings from biosimilar access barriers are an order of magnitude larger than the numbers cited in the 2018 REMS-specific study. REMS programs apply to biologics as well as small molecules. The Biologics Price Competition and Innovation Act created the biosimilar pathway, but biosimilar manufacturers face REMS-related barriers analogous to those that ANDA filers face, with the additional complexity that biosimilar interchangeability designations require additional clinical data that brand companies can use as a second layer of access resistance.
5.3 Downstream Cost Cascades
The AJMC analysis of opioid REMS economics estimated that drug diversion and misuse impose $72.5 billion annually on payers, with Medicare and Medicaid bearing two-thirds of that burden. This figure is typically cited to justify REMS programs. What is underanalyzed is the counterfactual: delayed generic entry in opioid analgesics maintains higher prices for brand opioids, reduces price-competitive incentives in the opioid market, and concentrates distribution through restricted specialty channels that are harder to monitor for diversion than standard retail pharmacy networks.
REMS programs intended to reduce diversion can, by maintaining restricted distribution as a market structure, actually reduce the network visibility that community pharmacists and state prescription monitoring programs use to identify diversion patterns. The policy is more complicated than either the brand safety argument or the generic access argument captures.
Key Takeaways: Section 5
The $13.4 billion annual figure is the right order of magnitude for the economic damage, but it likely understates the biosimilar-specific losses and does not fully account for second-order costs including reduced prescriber willingness to initiate high-REMS-burden therapies, administrative cost pass-throughs to payers, and the concentration of distribution in specialty pharmacy networks that may reduce diversion surveillance effectiveness.
6. Patient Access as Collateral Damage
6.1 The Price-Access Transmission Mechanism
Generic drugs price at 75% to 85% below brand prices on average, with price compression accelerating as more generic manufacturers enter the market. A brand drug with one generic entrant typically sees a price reduction of 20% to 30%. By the sixth generic entrant, the price may be 80% to 90% below brand. Every month of delayed generic entry is a month in which patients and payers pay the difference.
A 2022 survey found that 15% of U.S. adults reported foregoing prescription drugs due to cost. That figure is not an abstraction. For REMS-restricted specialty drugs, which tend to be high-cost therapies for serious conditions, the affordability gap between brand and generic pricing can mean the difference between adherence and discontinuation.
6.2 Recent REMS Modifications That Demonstrate the Problem
Several recent FDA actions on specific REMS programs illustrate both the access problem and the possibility of regulatory correction.
The Clozapine REMS, approved in September 2015, required strict monitoring of Absolute Neutrophil Count levels due to the risk of severe neutropenia. The monitoring protocol required weekly blood draws initially, with the frequency gradually decreasing based on stability. In March 2025, the FDA removed the ANC reporting requirement from the Clozapine REMS, concluding the program was no longer necessary to ensure benefits outweighed risks. For a drug treating severe treatment-resistant schizophrenia, the administrative burden of weekly blood draws was a documented barrier to initiation. Psychiatrists were reluctant to prescribe it; patients were reluctant to comply. The REMS removal will expand access to a drug with significant clinical utility.
In April 2025, the FDA eliminated REMS requirements for embryofetal toxicity risk for endothelin receptor antagonists including Ambrisentan and Macitentan-containing products, citing two decades of human pregnancy data that did not show a consistent pattern of congenital malformations. The ERA REMS had required patient enrollment, pharmacy certification, and periodic pregnancy testing, creating administrative friction for every prescription. The FDA’s determination that standard labeling language was sufficient to communicate the teratogenicity risk was based on evidence that had accumulated over years of post-market surveillance.
In June 2025, Bristol Myers Squibb announced FDA approval to remove REMS programs from its cell therapy products including Breyanzi and Abecma and to streamline patient monitoring requirements. The FDA accepted the medical oncology community’s argument that experience with cytokine release syndrome and neurologic toxicity management had matured to the point where center-based REMS administration was no longer necessary, and that proximity requirements to certified treatment centers were a disproportionate access barrier for patients in geographically underserved areas.
The mifepristone situation is different in kind. As of June 2025, multiple state Attorneys General filed a citizen petition urging the FDA to remove the mifepristone REMS. Their petition argued that the existing requirements, including prescriber certification, pharmacy certification, and patient agreement forms, are medically unnecessary given the drug’s established safety profile over more than 20 years of use, and that the REMS disproportionately restricts access in rural and medically underserved areas. The FDA had already removed in-person dispensing requirements in 2021, formalized in 2023, allowing mail-order pharmacy dispensing, but the remaining requirements still reduce pharmacy participation and prescriber willingness to offer the treatment.
6.3 Healthcare Provider Burden as a Market Structure Variable
Administrative burden on prescribers and pharmacies is not only a patient access issue. It is a market structure variable. When a REMS program requires physicians to complete certification training, enroll in a registry, submit periodic attestations, and document laboratory results before dispensing, the marginal cost of prescribing increases. Physicians facing that cost will preferentially prescribe less-restricted alternatives when clinical equivalence is plausible. For indications where REMS-restricted drugs are first-line, physician burden becomes a de facto utilization barrier that suppresses market volume below the clinically appropriate level.
For generic manufacturers, this has a compound effect. If the brand REMS suppresses total market volume, and if shared REMS negotiations require the generic to assume proportional administrative cost for a smaller-than-expected market, the generic’s projected return on ANDA investment is lower than the nominal market size suggests. This is a secondary deterrent effect that REMS abuse creates even for generic manufacturers who successfully clear sample access and ANDA approval.
Key Takeaways: Section 6
The FDA’s recent REMS removals for Clozapine, the ERA class, and cell therapies confirm the agency’s capability and willingness to recalibrate REMS requirements as post-market data accumulates. These decisions create a template for continued REMS reassessment across the broader portfolio of active programs. For pharma IP and regulatory teams, the risk is not only that a REMS program is challenged by generic entrants but also that the FDA independently determines it is no longer warranted, eliminating the exclusivity layer without litigation. That regulatory obsolescence risk belongs in every REMS-dependent IP valuation model.
7. The CREATES Act: Mechanics, Limits, and Enforcement Gaps
7.1 Legislative Architecture
The Creating and Restoring Equal Access to Equivalent Samples Act, enacted in December 2019 as Section 610 of the Further Consolidated Appropriations Act of 2020, had been introduced in various forms since 2016. Its delay in passage was itself a product of brand industry lobbying pressure, which for three years successfully blocked a bill that had bipartisan support in both chambers.
CREATES establishes a private right of action allowing generic and biosimilar manufacturers to sue brand companies that refuse to sell product samples in sufficient quantities for bioequivalence testing. The sale must be on ‘commercially reasonable, market-based terms.’ Courts can order sample sales, award attorneys’ fees and litigation costs to prevailing generic manufacturers, and impose monetary penalties on brand companies to deter repeat refusals.
7.2 The Covered Product Authorization Mechanism
For drugs with REMS containing ETASU, CREATES adds a prerequisite before a generic manufacturer can file suit. The generic developer must first obtain a Covered Product Authorization from the FDA. A CPA confirms that the proposed testing protocols are comparable to the existing REMS safety requirements, providing the FDA’s official determination that the generic can safely handle the samples before a court orders the brand to provide them.
The CPA mechanism is designed to prevent brand manufacturers from claiming that sample provision creates a genuine safety risk. Once the FDA has issued a CPA, the safety argument is foreclosed. What remains is a commercial dispute about price and quantity, which is exactly what CREATES was designed to resolve through the ‘commercially reasonable, market-based terms’ standard.
7.3 The Separate REMS Option
CREATES grants the FDA expanded discretion to approve separate, comparable REMS for generic manufacturers rather than requiring a single, shared system. Specifically, the FDA can grant a waiver of the SSS requirement when an aspect of the brand’s ETASU is claimed by an unexpired patent or trade secret and the generic applicant certifies that it sought but could not obtain a license. This removes the brand’s leverage to block generic entry simply by refusing to grant a REMS software license or by claiming proprietary rights over enrollment database architecture.
7.4 The Indemnification Provision
CREATES includes a provision giving brand manufacturers an affirmative defense and specific indemnification protection from liability arising from a generic manufacturer’s handling of samples provided under the Act. This directly addresses one of the brand industry’s stated justifications for sample denial: the claim that if a generic manufacturer mishandles a dangerous REMS-restricted drug during testing, the brand could face liability for the resulting harm. The indemnification provision eliminates that exposure, removing the legal basis for the safety-based refusal argument.
7.5 Remaining Enforcement Gaps
CREATES does not address proprietary restricted distribution systems created outside the REMS framework. That $10.3 billion segment of the documented economic harm remains dependent on antitrust enforcement under Sections 1 and 2 of the Sherman Act, which requires proving competitive harm through a rule-of-reason analysis rather than relying on a specific statutory entitlement. The evidentiary burden is substantially higher, and the litigation timeline is longer.
The Act also leaves ‘sufficient quantities’ undefined beyond specifying that the amount must allow the generic manufacturer to conduct the necessary testing and fulfill regulatory requirements. Brand companies can comply narrowly with CREATES by providing minimal quantities, forcing multiple rounds of requests and litigation to establish the actual amount required for a full bioequivalence study. The first CREATES Act case, Teva v. Amicus Therapeutics, resolved when Amicus agreed to provide adequate quantities after suit was filed, but the definition of ‘sufficient quantities’ was not tested by the court.
The 14-day window in which a brand manufacturer can avoid liability by making a qualifying offer creates a procedural safety valve, but it also creates a negotiating game around what constitutes a ‘qualifying offer.’ If the brand’s initial offer is commercially unreasonable but technically responsive to the request, the generic must decide whether to accept a suboptimal offer or return to litigation.
Key Takeaways: Section 7
CREATES is the most significant legislative intervention in the REMS abuse problem since the FDAAA created REMS. Its private right of action is functional, its CPA mechanism is structurally sound, and the indemnification provision eliminates the primary non-frivolous safety argument for sample denial. The gaps, specifically the exclusion of non-REMS restricted distribution systems and the undefined ‘sufficient quantities’ standard, require either judicial clarification through future CREATES Act litigation or Congressional amendment. IP teams advising generic manufacturers should treat CREATES as a necessary but not sufficient tool, requiring parallel antitrust strategy for non-REMS distribution barriers.
8. Litigation Record: Pre- and Post-CREATES Case Analysis
8.1 Pre-CREATES: The Antitrust Problem
Before CREATES, generic manufacturers seeking sample access had one primary legal tool: federal antitrust claims. The Sherman Act Section 2 refusal-to-deal doctrine, as shaped by Aspen Skiing and its progeny, requires plaintiffs to show that the monopolist terminated a voluntary course of dealing with competitors and that the conduct makes sense only as a strategy to maintain monopoly power rather than serve legitimate business purposes. Proving that specific REMS-related sample denials met this standard was difficult and fact-intensive.
Lannett Co. v. Celgene Corp., filed in 2008, challenged Celgene’s refusal to sell Thalomid samples and settled without establishing judicial precedent. Actelion Pharmaceuticals Ltd.’s preemptive declaratory judgment action against Apotex and Roxane in 2011, seeking confirmation that Actelion had no duty to supply Tracleer and Zavesca samples, triggered FTC intervention by amicus brief before also settling without precedential outcome. Mylan Pharmaceuticals Inc. v. Celgene Corp., filed in April 2014 over Revlimid and Thalomid samples, was the most extensively litigated pre-CREATES case. A federal judge allowed the suit to proceed, which itself was a notable development, but the case remained in early litigation when CREATES passed in 2019.
The pattern across these cases is consistent: suits filed, extensive pretrial litigation over the scope of the antitrust claims, then settlement. The settlements resolved the individual cases but produced no published opinions establishing when REMS sample denial constitutes an antitrust violation. Every new generic manufacturer facing the same situation started from zero.
8.2 Post-CREATES: Teva v. Amicus Therapeutics
The first CREATES Act lawsuit was filed by Teva Pharmaceuticals against Amicus Therapeutics in the Eastern District of Pennsylvania in July 2021. Teva sought samples of Galafold (migalastat), a Fabry disease treatment, for its ANDA bioequivalence program. The dispute began in August 2020 when Teva requested two wallet packs from Amicus. Amicus delivered them in April 2021, more than seven months later and well beyond the 31-day statutory deadline. When Teva sent a second request for 25 wallet packs to support analytical and bioequivalence studies, Amicus offered two additional packs.
Teva filed under CREATES, alleging refusal to provide sufficient quantities. Amicus then agreed to provide the requested quantities, and Teva dismissed the suit. The case never proceeded to substantive briefing on the ‘sufficient quantities’ definition.
The quick resolution is both evidence of CREATES’ deterrent effect and a limitation on its precedential value. Brand companies facing CREATES suits now know that holding out through pretrial litigation carries the risk of court-ordered sample sales, attorneys’ fees, and monetary penalties. Many will settle early, as Amicus did. But without published opinions defining ‘sufficient quantities,’ ‘commercially reasonable market-based terms,’ and the scope of ‘ancillary materials’ that generic manufacturers can request alongside drug samples, each new CREATES case starts fresh on those definitional questions.
8.3 FTC Enforcement as Parallel Track
The FTC’s June 2025 listening session on anticompetitive conduct by pharmaceutical companies impeding generic or biosimilar competition signals renewed enforcement attention to the broader category of access barriers. The agency’s 2023 policy statement on improper Orange Book patent listings is being implemented through challenges to specific patents, and the FTC has explicitly framed both improper Orange Book listings and REMS misuse as unfair methods of competition under FTC Act Section 5.
The agency’s combination of litigation activity, amicus brief filing, enforcement guidance publication, and Congressional testimony creates a regulatory and legal environment in which brand companies maintaining REMS-based access barriers face escalating scrutiny from multiple directions simultaneously.
Key Takeaways: Section 8
The litigation record shows that the private right of action in CREATES functions as designed, at least in generating early settlements that produce sample access. What it has not yet produced is a body of judicial interpretation defining the key statutory terms. That interpretive gap will be filled by future cases, and the companies that bring those cases, whether they are generic manufacturers pushing for broad definitions of ‘sufficient quantities’ or brand manufacturers defending narrow ones, will shape the competitive landscape for the next decade.
For investors modeling generic market entry timing for ETASU drugs, the current CREATES litigation environment implies that sample denial as a delay tactic carries a shorter expected delay period than it did in the antitrust era. Where pre-CREATES antitrust cases could drag for years without forcing sample access, CREATES cases appear to resolve in months when the brand company calculates that litigation costs exceed the economic value of delay. For a product generating $500 million annually in U.S. sales, the brand’s calculus changes significantly once CREATES penalties are on the table. Model the CREATES settlement probability as high and the expected delay reduction as 12 to 24 months compared to pre-CREATES baselines.
9. FDA’s Evolving Role as Market Structure Regulator
9.1 From Safety Mandator to Competition Facilitator
The FDA’s statutory mandate does not include market competition. Its authorities under the FD&C Act and FDAAA are grounded in public health, drug safety, and efficacy. But the agency’s actions on REMS over the past decade reveal an institution that increasingly understands the market structure consequences of its regulatory decisions and that has incorporated competition-adjacent considerations into its operational approach to REMS administration.
Dr. Janet Woodcock, as CDER Director, characterized REMS abuse as a ‘growing major problem’ that ‘delayed the availability of generics.’ Dr. John Jenkins used the phrase ‘evergreening system.’ These are not the words of regulators focused purely on safety outcomes. They reflect an institutional awareness that REMS administration has market structure implications that the FDA cannot ignore without becoming complicit in the harms it is publicly documenting.
9.2 The ‘Name and Shame’ Initiative
Beginning in May 2018, the FDA began publishing lists of brand companies using REMS or proprietary restricted distribution programs to deny generic manufacturer sample access. These lists are a transparency mechanism with no direct legal consequences. Their purpose is reputational pressure and public accountability.
The practical effect is limited but not zero. Brand companies with institutional investor bases care about FDA public lists linking them to anti-competitive conduct. Hospitals, PBMs, and government formulary managers have incorporated FDA transparency data into procurement and contracting decisions. The naming mechanism is a soft regulatory tool, but it creates information that plaintiffs’ attorneys, the FTC, and Congressional staff use in enforcement and legislative proceedings.
9.3 Shared REMS Facilitation: What FDA Does and Does Not Do
FDA guidance on shared system REMS development describes the agency’s facilitation role in detail. The FDA convenes kick-off meetings between brand and generic applicants, sets milestone timelines for shared system development, and issues correspondence documenting where negotiations stand. What the FDA does not do is arbitrate commercial disputes over cost-sharing formulas, governance voting rights, or liability allocation between competing manufacturers.
This limitation is the structural gap that brand companies exploit in shared REMS negotiations. The FDA can document that negotiations have stalled but cannot compel a brand company to accept the generic manufacturer’s proposed cost-sharing arrangement. CREATES’ provision allowing the FDA to grant waivers and approve separate generic REMS reduces the brand’s negotiating leverage by creating an exit for the generic, but the waiver process itself takes time and requires FDA resources.
9.4 Recent REMS Removal Decisions as Regulatory Signal
The FDA’s 2025 decisions to remove REMS requirements for Clozapine, ERA products, and Bristol Myers Squibb’s cell therapies, and to prospectively narrow REMS requirements for other categories, are regulatory signals that the agency is actively reassessing the proportionality of existing programs. The standard it is applying, whether the REMS is ‘no longer necessary to ensure that the benefits of a drug outweigh its risks,’ is evidence-based and post-market surveillance driven.
For brand companies treating existing REMS as permanent exclusivity infrastructure, these decisions are a warning. A REMS that the FDA determines is no longer necessary will be eliminated regardless of the brand company’s commercial interests. The FDA’s authority to modify or remove REMS unilaterally means that REMS-dependent IP valuation carries a regulatory obsolescence risk that is independent of litigation risk and patent expiry.
Key Takeaways: Section 9
The FDA is not a competition regulator, but its operational decisions on REMS administration function as market structure interventions. The combination of transparency lists, shared REMS facilitation, waiver authority under CREATES, and active REMS reassessment creates a regulatory environment in which REMS-based exclusivity is less durable than it was five years ago. Teams building revenue models for REMS-restricted brand products should add a regulatory obsolescence scenario to standard patent cliff modeling.
10. Evergreening Technology Roadmap: REMS in the Full Life Cycle Management Stack
10.1 What Evergreening Actually Involves
Evergreening is the aggregate strategy through which brand pharmaceutical manufacturers extend the effective commercial exclusivity of a drug beyond the expiration of its primary compound patent. The full life cycle management stack includes multiple layers, and REMS manipulation is one component of a broader system.
The primary compound patent covers the active pharmaceutical ingredient itself, typically filed at or before the IND stage and expiring 20 years from the priority date, adjusted for patent term restoration under Hatch-Waxman. Compound patents are the first line of defense.
Formulation patents cover specific delivery mechanisms, extended-release matrices, prodrug structures, particle size specifications, and co-crystal forms. These patents often expire 5 to 10 years after the compound patent, extending exclusivity through secondary IP without requiring new clinical development beyond bioequivalence studies for the modified formulation.
Method-of-use patents cover specific indications, dosing regimens, patient populations defined by biomarker status, or combination therapies. These are the basis for authorized generic strategies and for Paragraph IV certifications that challenge indication-specific Orange Book listings.
Pediatric exclusivity adds 6 months of exclusivity upon completion of FDA-requested pediatric studies, independent of underlying patent position. Companies frequently sequence pediatric study completion to align with compound patent expiry, maximizing the additive exclusivity.
New chemical entity exclusivity provides 5 years of data exclusivity for drugs containing a previously unapproved active moiety, during which no ANDA can be submitted that references the NDA’s clinical data. Biologics carry 12 years of reference product exclusivity under the BPCIA, with an additional 4-year period during which no biosimilar application can be approved.
10.2 Where REMS Fits in the Stack
REMS manipulation sits at the regulatory exclusivity layer of the stack, operating after patent and data exclusivity but before market competition fully develops. Its function is not to generate new IP rights but to delay the operationalization of existing generic rights. A generic manufacturer that has won a Paragraph IV case, invalidated the relevant Orange Book patents, and obtained ANDA approval can still be blocked from commercial launch if it cannot obtain a shared REMS agreement and the FDA has not yet granted a waiver for a separate system.
This positioning makes REMS manipulation particularly dangerous as a competitive strategy because it operates at the moment of maximum commercial vulnerability for brand companies. Patent cliffs produce the steepest revenue declines in the first 6 to 18 months of generic competition. Any mechanism that delays that first generic launch by even 6 months is worth hundreds of millions of dollars for a large specialty product.
10.3 Patent Thickets and REMS Integration
The most sophisticated life cycle management programs integrate patent thicket construction with REMS administration. A patent thicket is a dense web of overlapping patents covering multiple aspects of the drug, its formulation, its delivery device, its manufacturing process, its metabolites, and its clinical applications. The goal is to force generic applicants to navigate multiple Paragraph IV certifications, each triggering a potential 30-month stay, before any single claim collapses.
REMS integration into the thicket occurs when brand companies file patents on REMS infrastructure components: patient registry database structures, enrollment algorithm logic, monitoring reporting systems, and prescriber certification portal technology. When a generic manufacturer seeks a CREATES Act waiver for a separate REMS, the presence of these patents requires the generic to certify that it sought and could not obtain a license, a process that adds delay. If the brand refuses to license, the generic must either design around the patented REMS infrastructure or wait for the patents to expire.
10.4 Post-CREATES Life Cycle Management Adaptation
Post-CREATES, sophisticated brand companies are adapting their life cycle management stacks. The adaptations include earlier and more aggressive patent filing on REMS infrastructure components, more rapid generic licensing agreements for secondary indications (preserving exclusivity for primary high-revenue indications), increased reliance on proprietary restricted distribution systems outside the formal REMS framework (which CREATES does not address), and more active engagement with the shared REMS process to avoid CREATES Act liability while using commercial negotiation to delay finalization.
The shift toward proprietary restricted distribution is particularly significant. A brand company that transitions from a formal REMS-based distribution restriction to a proprietary specialty pharmacy network before an ANDA is approved retains most of the competitive exclusivity of the REMS while removing itself from CREATES Act jurisdiction. The generic manufacturer facing this structure must pursue antitrust claims rather than CREATES claims, returning to the more difficult evidentiary path of the pre-CREATES era.
Key Takeaways: Section 10
REMS is one layer in a multi-layer exclusivity stack. Understanding how it interacts with patent thickets, Orange Book listings, pediatric exclusivity, and proprietary distribution systems is essential for accurate modeling of generic and biosimilar market entry timelines. Post-CREATES, brand companies are shifting the exclusivity burden toward layers that CREATES does not reach, particularly non-REMS proprietary distribution and REMS infrastructure patents. Generic manufacturers and their legal teams need comprehensive visibility into the full stack to prioritize challenges effectively.
11. Biosimilar-Specific REMS Risk: Where the Next Battlefield Is
11.1 BPCIA and Biosimilar Interchangeability
The Biologics Price Competition and Innovation Act created the abbreviated biosimilar approval pathway, requiring biosimilar applicants to demonstrate that their product is ‘highly similar’ to the reference biologic and has no clinically meaningful differences in safety, purity, or potency. Biosimilar interchangeability, the designation allowing automatic substitution at the pharmacy level without a physician’s intervention, requires additional switching studies demonstrating that alternating between the reference biologic and the biosimilar does not produce greater immunogenicity or other risks than continuous use of either product.
For biosimilars of REMS-restricted biologics, the same ETASU requirement applies: the biosimilar must have the same or comparable REMS as the reference product. This creates a shared REMS negotiation dynamic identical to that for small-molecule generics, with the additional complexity that biologics manufacturing and characterization data may be proprietary to a degree that small-molecule bioequivalence testing is not.
11.2 Reference Standard Access for Biosimilars
CREATES applies to biosimilar applicants as well as ANDA filers. A biosimilar manufacturer that cannot obtain sufficient quantities of the reference biologic for comparative characterization, stability studies, and clinical lot release testing is blocked from completing its biosimilar application. For biologics with REMS restricted distribution, the same sample denial tactics apply.
The analytical challenge is greater for biosimilars because the quantity of reference product required for full biosimilar characterization and clinical development typically exceeds what a small-molecule bioequivalence study requires. A biosimilar development program may require multiple commercial-scale lots of reference product over several years. The ‘sufficient quantities’ question under CREATES is more complex for biosimilars than for small-molecule ANDA applicants.
11.3 Biosimilar Interchangeability as REMS Risk Modifier
A biosimilar with interchangeability designation can be substituted at the pharmacy without a new prescription, giving it a commercial adoption pathway analogous to automatic substitution for small-molecule generics under state generic substitution laws. Without interchangeability, biosimilar uptake depends on physician prescribing behavior, payer formulary design, and PBM contracting, all of which are more easily influenced by brand company marketing and rebate agreements.
Brand companies have responded to biosimilar interchangeability risk by seeking interchangeability for their own second-generation products, pricing their reference biologic competitively against biosimilars in formulary negotiations, and structuring rebate arrangements with PBMs that make formulary exclusion of biosimilars economically rational for payers even when biosimilar list prices are substantially lower.
REMS manipulation in the biosimilar context adds a delay layer that allows brand companies more time to implement these commercial countermeasures. Each month of delayed biosimilar launch is a month in which the brand can restructure contracting, shift patients to next-generation products with their own exclusivity, and build prescriber inertia against switching.
Key Takeaways: Section 11
The biosimilar REMS barrier is less documented than the small-molecule generic REMS barrier but structurally identical. Given that delayed biosimilar entry costs approximately $140 million per $1 billion of biologics sales, and given that the U.S. biologics market now exceeds $200 billion annually, the aggregate economic stakes dwarf the documented losses from small-molecule REMS abuse. Biosimilar manufacturers, payers, and policy teams should treat the CREATES Act as only the first layer of a biosimilar access strategy, with parallel attention to biosimilar interchangeability regulatory strategy, FTC engagement on distribution restrictions, and CMS formulary policy as complementary tools.
12. Investment Strategy for Analysts
12.1 REMS as an Underpriced Risk in Brand Pharma Valuations
Sell-side models for specialty pharmaceutical companies with REMS-restricted portfolios routinely underestimate the rate at which REMS-based exclusivity will erode. The standard analyst approach treats the patent expiry date as the primary revenue inflection point and models generic erosion curves from that date forward. This approach misses two scenarios that post-CREATES data suggests are more common than models assume.
The first scenario is accelerated generic entry. CREATES has shortened the expected timeline from patent expiry to first generic launch for ETASU drugs by reducing the leverage brand companies have in shared REMS negotiations and sample access disputes. A model that uses pre-CREATES historical generic launch timing for ETASU drugs is using stale base rates.
The second scenario is FDA-initiated REMS removal. The FDA’s 2025 decisions on Clozapine, ERAs, and cell therapies demonstrate that the agency will remove REMS programs when post-market data no longer supports the restriction. A REMS removal before patent expiry eliminates the regulatory exclusivity layer that brand companies rely on to cushion the patent cliff. Models that treat REMS as permanent infrastructure until patent expiry do not account for this scenario.
12.2 Screening Criteria for REMS-Sensitive Positions
Analysts evaluating positions in companies with REMS-restricted products should apply a screening checklist. First, identify all active REMS programs with ETASU in the portfolio and map the ANDA queue for each. Second, assess whether any ANDA filers have filed Paragraph IV certifications challenging Orange Book patents, and whether any have publicly disclosed bioequivalence study activity that implies sample access has been achieved. Third, determine whether the company has received any FDA requests regarding shared REMS negotiations or any CREATES Act demand letters. Fourth, review the company’s Orange Book listings for patents that the FTC or courts have flagged as potentially improper. Fifth, assess the FDA’s post-market surveillance data on the REMS program’s clinical outcomes to estimate the probability of FDA-initiated REMS modification.
12.3 Generic-Side Opportunity Mapping
For investors in generic and specialty generic companies, REMS-restricted drugs with large ANDA queues and near-term patent expirations represent potential high-margin first-to-market opportunities if the REMS barrier can be cleared. CREATES has reduced, though not eliminated, the time and cost of clearing that barrier.
The highest-return opportunities are typically ETASU drugs with a single active ANDA filer, where CREATES has produced sample access, where the brand has not filed REMS infrastructure patents, and where the FDA’s shared REMS waiver pathway is available. In these situations, the generic manufacturer faces a clear path from sample access through bioequivalence study completion to ANDA approval with a separate REMS, and first-to-market exclusivity in a previously uncontested generic space.
12.4 Biosimilar Entry Timing Models
For biologics with REMS, the combination of BPCIA 12-year reference product exclusivity, the biosimilar interchangeability standard, and REMS-based access barriers creates a multi-layer timing problem that standard DCF models do not adequately capture. A biologics-specific model should use a Monte Carlo simulation across four key variables: reference product exclusivity end date, biosimilar interchangeability designation probability and timing, REMS barrier resolution timeline under CREATES, and FDA-initiated REMS removal probability.
The output should be a probability-weighted biosimilar entry date distribution rather than a single-point estimate. Brand manufacturers can use this model to assess acquisition timing for biosimilar competitors. Generic and biosimilar manufacturers can use it to prioritize their pipeline investments.
Key Takeaways: Section 12
REMS is a financial variable, not only a regulatory compliance variable. For brand companies, the post-CREATES erosion of REMS-based exclusivity requires haircuts to revenue projections that most current sell-side models do not apply. For generic and biosimilar companies, CREATES has opened opportunity windows that did not exist five years ago. For institutional investors in either category, the key analytical inputs are ANDA queue depth, CREATES exposure assessment, FTC enforcement risk, and FDA REMS reassessment probability, not just patent expiry dates.
13. Key Takeaways by Segment
For Pharma IP Teams
REMS programs with ETASU components should be audited annually for four characteristics: patent coverage of REMS infrastructure, current status of any shared REMS negotiations with generic applicants, receipt of any CREATES Act demand letters, and FDA assessment reports indicating whether the REMS is meeting its stated risk mitigation goals. Any REMS that fails on any of these dimensions represents a potential competitive vulnerability or enforcement exposure that requires active legal and regulatory management.
The CREATES Act has closed the sample denial loophole for formally mandated REMS programs but left the non-REMS proprietary restricted distribution gap open. Companies maintaining proprietary distribution systems should obtain antitrust counsel review of those systems before they become the subject of FTC scrutiny or private antitrust litigation, which is operating under an increasingly aggressive enforcement environment.
For Generic and Biosimilar Manufacturers
CREATES is your primary tool for sample access disputes involving REMS with ETASU. Use the Covered Product Authorization process with the FDA before filing suit. Document every sample request and every brand response in writing from the first contact. The procedural requirements for a valid CREATES Act request are specific: the request must be directed to a named corporate officer, identify a point of contact, and specify a mailing address. Failure to meet these requirements will give the brand a procedural defense.
For shared REMS negotiations, treat every communication as potential litigation evidence. Negotiate in writing. Document every delay and every commercially unreasonable position the brand takes on cost-sharing, governance, or confidentiality. If the brand is using REMS infrastructure patent claims to block a waiver, assess the patents and prepare a design-around or invalidity challenge in parallel with the CREATES claim.
For Portfolio Managers and Institutional Investors
Re-run your revenue models for every specialty drug position where REMS-based exclusivity is a significant component of the bull case. Apply a CREATES Act discount to expected generic entry timing for ETASU drugs. Add an FDA REMS removal scenario to your sensitivity analysis. Assess whether the brand companies in your portfolio are positioned in the aggressive sample-denial portion of the REMS spectrum or in the compliant shared-REMS portion, because the former carries FTC enforcement risk and CREATES litigation exposure that the latter does not.
For generic and biosimilar positions, CREATES-enabled first-mover advantages in previously REMS-blocked markets are real and underpriced. The highest-value opportunities are in ETASU drugs with limited ANDA competition, near-term patent expiry, and no REMS infrastructure patent coverage.
For R&D Leads
REMS design decisions made early in clinical development have competitive consequences at patent expiry. An REMS program built around proprietary digital infrastructure, patented database architecture, or novel monitoring algorithms creates exclusivity layers that persist beyond compound patent expiry. An REMS built around commercially standard technologies and designed for administrative simplicity does not. This is not an argument for or against either design approach; it is an observation that the REMS architecture decision is a long-term competitive strategy decision, not only a near-term regulatory compliance decision.
14. Recommendations for IP and Regulatory Teams
14.1 Operationalize CREATES Compliance Now
Every brand company with a REMS containing ETASU should have a documented sample request response protocol. The protocol should specify the legal basis for evaluating any request, the timeline for providing a response (the CREATES Act’s 14-day qualifying offer window is the operative deadline), and the commercial terms framework for a qualifying offer. Companies that have not developed this protocol are operating with CREATES liability exposure every time a generic manufacturer sends a sample request letter.
14.2 Map the Full Exclusivity Stack
IP teams should maintain a comprehensive exclusivity map for each REMS-restricted product that shows compound patent expiry dates, Orange Book patent listings with FTC challenge risk assessments, REMS infrastructure patent coverage and expiry, data exclusivity periods, pediatric exclusivity status, and the current ANDA or biosimilar application queue with each applicant’s documented stage of development. This map should be updated quarterly and shared with portfolio management, investor relations, and M&A diligence teams.
14.3 Engage the FDA on REMS Assessment Proactively
Companies with REMS programs that are approaching scheduled assessment submission dates should treat those assessments as an opportunity to right-size the REMS rather than a compliance exercise to be completed minimally. FDA-initiated REMS modifications that restrict access more than necessary create generic entry delays that generate CREATES litigation risk and FTC scrutiny. Proactively proposing proportionate REMS modifications that reflect current safety data is both better public health policy and better litigation risk management.
14.4 Antitrust-Proof Your Distribution System
Any proprietary restricted distribution program that was designed or timed to coincide with generic ANDA filings should be reviewed by antitrust counsel with Sherman Act Section 1 and Section 2 expertise. The FTC’s increasing focus on this area, combined with the agency’s expanded enforcement budget for pharmaceutical antitrust, means that distribution systems with thin competitive justifications face real enforcement risk. The economic benefit of delay must be weighed against the cost of antitrust exposure.
14.5 Model REMS Obsolescence Risk
The FDA’s 2025 REMS removals for Clozapine, ERA products, and cell therapies establish that the agency treats REMS reassessment as an active regulatory responsibility, not a passive post-approval monitoring obligation. For any REMS-restricted product where post-market safety data has accumulated substantially since the original REMS was designed, there is a non-trivial probability that the FDA will initiate a REMS modification or removal on its own motion. This probability should be quantified and incorporated into revenue projections as a discrete scenario, not as a footnote.
Conclusion
REMS programs serve a real and necessary public health function. The drugs subject to ETASU requirements carry serious risks, and those risks require active risk communication, prescriber training, patient monitoring, and sometimes restricted distribution. Nothing in the legal or policy critique of REMS abuse argues that these programs should not exist.
What the evidence, from the FDA’s own public statements, from Matrix Global Advisors’ economic analysis, from FTC amicus briefs and enforcement actions, and from federal antitrust litigation, establishes clearly is that REMS programs with ETASU have been used systematically and deliberately to extend commercial monopolies beyond the period Congress intended through the patent system and the Hatch-Waxman framework.
The CREATES Act is the most direct legislative response to date. Its private right of action has produced early settlements and increased compliance. Its CPA mechanism preserves FDA oversight of sample access safety. Its waiver authority reduces the leverage of shared REMS negotiations. But it leaves the non-REMS proprietary distribution problem unresolved, the ‘sufficient quantities’ standard undefined, and the biosimilar-specific REMS dynamic underaddressed.
For the $13.4 billion annual cost to become manageable, CREATES enforcement needs to mature through additional judicial interpretation, the FTC needs to maintain pressure on proprietary distribution systems under antitrust authority, the FDA needs to continue proactive REMS reassessment, and generic and biosimilar manufacturers need the competitive intelligence infrastructure to identify and pursue opportunities in previously REMS-blocked markets.
The barrier is real, the tools to break it exist, and the economic stakes justify sustained attention from every stakeholder in the pharmaceutical value chain.

















")








