1. Why Drug Naming Is an IP Function, Not a Marketing Task
The name on the bottle is not chosen by a copywriter the week before launch. It is a layered legal, regulatory, linguistic, scientific, and commercial artifact that a company begins constructing years before a drug reaches Phase III. Most large pharma organizations spend between $1 million and $3 million on the naming process for a single global brand, not counting the cost of regulatory submissions, legal clearances, and post-launch brand maintenance. The EMA rejects approximately 55% of proposed brand names on first submission. The FDA uses a computational similarity tool, the Phonetic and Orthographic Computer Analysis (POCA) system, to filter out names that score above a 75% phonetic or orthographic similarity threshold against the universe of approved drug names. Getting a name wrong means lost time at launch, forced post-market name changes, medication error liability, and the destruction of brand equity that may have taken years to build.
The core argument of this guide is simple: drug naming is an IP function. It sits at the intersection of trademark law, patent strategy, regulatory affairs, pharmacovigilance, and competitive intelligence. The pharmaceutical companies that treat it as such, starting the process in Phase I or early Phase II, integrating it with lifecycle management planning, and using naming data as a source of competitive insight, consistently outperform those that bolt it on at the end.
This guide covers every layer of that process at the depth required by IP teams and portfolio managers: the science of INN stems, the psychology of brand creation, the mechanics of POCA and NRG review, the LASA case record, the biologic suffix battle, the anatomy of an evergreening patent estate, and how to use naming data as a forward-looking competitive intelligence tool.
2. The Naming Trinity: Chemical, Generic, and Brand Names
Every approved drug carries three distinct names simultaneously. Each occupies a different legal, regulatory, and commercial space. Conflating them is a common error among analysts and even within some corporate strategy teams. The distinctions matter practically because each name type generates different kinds of IP, expires on different timelines, and exposes a company to different risks.
2.1 The Chemical Name: IUPAC Structure as Legal Foundation
The International Union of Pure and Applied Chemistry (IUPAC) name is the molecular description of the active pharmaceutical ingredient (API). It encodes atom count, bond structure, stereochemistry, and functional groups into a systematic nomenclature. Acetaminophen’s IUPAC name is N-(4-hydroxyphenyl)acetamide. Diazepam’s is 7-chloro-1,3-dihydro-1-methyl-5-phenyl-2H-1,4-benzodiazepin-2-one.
The chemical name is the anchor of the composition-of-matter patent. Patent claims drafted around a novel molecular entity use the IUPAC name or its CAS registry number as the precise legal definition of what is being protected. This precision matters enormously in Paragraph IV challenge litigation, where a generic manufacturer arguing non-infringement will scrutinize every atom in the claim structure. Small deviations in stereochemistry (R vs. S enantiomers, for instance) can define the boundary between a valid composition-of-matter patent and a design-around opportunity. The chemical name is not used in clinical practice, but it is the bedrock document that patent attorneys, medicinal chemists, and regulatory scientists rely on throughout the drug’s legal life.
IP Valuation Note. The composition-of-matter patent, indexed to the IUPAC structure, is the most valuable patent in any pharmaceutical portfolio because it blocks all competing products containing that molecular entity, regardless of formulation, indication, or route of administration. When analysts value a pharma asset, the presence or absence of an unexpired composition-of-matter patent is the single largest variable in the net present value (NPV) calculation. A drug with five years of remaining composition-of-matter exclusivity is a fundamentally different asset from one protected only by formulation or method-of-use patents, even if the revenue profile looks identical today.
2.2 The Generic (Nonproprietary) Name: Public Property, Permanent Identifier
The generic, or nonproprietary, name is the official designation of the active ingredient assigned through the USAN/INN process. Unlike the brand name, it is not owned. It cannot be trademarked. It is free for any manufacturer to use once the relevant patents expire, and it is mandatory for all manufacturers to use it consistently.
Sildenafil is the generic name for Pfizer’s Viagra. Atorvastatin is the generic name for Pfizer’s Lipitor. Semaglutide is the generic name for Novo Nordisk’s Ozempic and Wegovy. The generic name persists through the entire product lifecycle, from early Phase I trial registration through the post-patent generic market. It is the identifier used in regulatory submissions (NDA, BLA, ANDA), pharmacopoeia monographs, and scientific literature worldwide.
The generic name is the engine of the global generic drug market, which accounts for roughly 90% of all prescriptions dispensed in the United States by volume, though far less by value. Without a standardized generic identifier, bioequivalence assessments, Orange Book listings, and Abbreviated New Drug Application (ANDA) filings would be practically impossible to administer. The generic name is what makes the generic entry pathway function. Generic applicants file their ANDA against a Reference Listed Drug (RLD), identified by its generic name and the innovator’s NDA number, and are required to demonstrate pharmaceutical equivalence to that specific chemical entity.
2.3 The Brand (Proprietary) Name: Perpetual Trademark, Commercial Engine
The brand name, also called the proprietary or trade name, is a trademarked commercial identifier owned exclusively by the innovator company. Unlike a patent, a trademark does not expire as long as the owner continues to use it and renew it. Pfizer still owns Viagra. Bristol-Myers Squibb still owns Pravachol. AstraZeneca still owns Prilosec (the renamed version of the original Losec). These are perpetual IP assets.
The brand name is the primary vehicle for marketing investment. Pharmaceutical companies in the United States routinely spend more on direct-to-consumer advertising and physician detailing for their branded products than they spent on the clinical development that produced them. The commercial logic is straightforward: the brand name captures the premium price, builds prescriber loyalty, and, if strong enough, sustains market share even after generic entry. Lipitor retained substantial branded market share for years after atorvastatin generics became available, driven by the name recognition and physician habit that decades of marketing had built.
IP Valuation Note. Brand name equity is a real, measurable balance sheet asset in some jurisdictions, though it is often undervalued or inconsistently accounted for in pharma M&A. When a major brand is acquired, intangible asset appraisers use income approaches (the royalty relief method, discounted cash flow of branded sales premium over generic pricing) to assign a discrete value to the trademark separate from the underlying molecule and its patents. For blockbuster drugs approaching the patent cliff, the brand trademark’s residual value, reflecting the premium that a significant subset of prescribers and patients will continue to pay, can constitute 10-25% of the total IP valuation.
Key Takeaways: Section 2
The chemical name anchors composition-of-matter patent claims, the most protective and highest-value IP in any drug portfolio. The generic name is the permanent public identifier and the foundation of the ANDA/generic entry mechanism. The brand name is a perpetual trademark that generates revenue long after patent expiry, provided the marketing investment is maintained. Analysts who fail to distinguish the IP rights and expiry timelines of each name type will consistently misprice pharmaceutical assets.
3. The Science of Generic Nomenclature: USAN, INN, and the Stem System
3.1 The Architects: WHO INN Programme and the USAN Council
The World Health Organization’s International Nonproprietary Name (INN) Programme has operated since 1953. Its mandate is narrow but critical: assign one unique, universally recognized name to every pharmaceutical substance. The INN list now contains over 7,000 names, growing by roughly 120 to 150 new entries per year. Each INN is public property, free to use by any party globally, which is the legal foundation of biosimilar and generic markets alike.
In the United States, the United States Adopted Names (USAN) Council executes the domestic equivalent. The USAN Council is a formal partnership among the American Medical Association (AMA), the United States Pharmacopeial Convention (USP), and the American Pharmacists Association (APhA), with an FDA liaison. A USAN is a legal prerequisite for marketing approval in the US. There are no material exceptions.
The two bodies operate in close coordination. Their shared objective is to make the USAN and the INN identical in virtually all cases. The acetaminophen/paracetamol divergence (USAN versus INN, respectively) is a pre-harmonization historical artifact. Since the modern collaboration framework was established, global name divergences are rare and quickly flagged. This harmonization is not just a patient safety convenience; it is a commercial accelerator. Multinational clinical trials, which now routinely enroll patients across 30 to 60 countries simultaneously, require a single compound identifier. The existence of one harmonized generic name eliminates a class of protocol confusion errors and simplifies regulatory submissions to each country’s health authority. The net effect is a measurable reduction in clinical cycle time.
3.2 The INN Stem System: A Structured Code for Competitive Intelligence
The INN stem system is the core analytical tool that distinguishes generic nomenclature from arbitrary labeling. Every INN contains a stem, typically a suffix or occasionally a prefix, that identifies the drug’s pharmacological class or chemical family. The stem is, in the words of Michael Quinlan, Pfizer’s former naming director, the drug’s ‘family name.’ The unique prefix attached to that stem distinguishes one compound from its class siblings.
The strategic importance of this system extends well beyond patient safety. A new INN proposal, submitted publicly to the WHO during the pINN review period, is one of the earliest public disclosures of a competitor’s compound and its mechanism of action. A company monitoring WHO INN publications that sees three new ‘-tinib’ proposals (tyrosine kinase inhibitors, predominantly used in oncology) from a single sponsor over 18 months now has specific, actionable early-warning intelligence: that company is building an oncology kinase inhibitor pipeline and has at least three distinct compounds in development. This data precedes any clinical trial registration, any conference presentation, and any press release by potentially two to three years.
The table below covers the most commercially significant INN stems across small molecules and biologics, with their precise mechanistic definitions.
3.3 The INN/USAN Application Process: Mechanics, Timeline, and Intelligence Value
A pharmaceutical company submits its INN/USAN application when a compound enters Phase I or early Phase II clinical trials. This timing is not incidental. It reflects both regulatory necessity (scientific publications and conference presentations require an official non-proprietary name) and strategic preference (avoiding internal compound codes in public disclosures).
The application itself is a structured negotiation. The sponsoring company submits a set of candidate names, each constructed with the appropriate stem for the compound’s pharmacological class and a proposed unique prefix. The prefix must satisfy four substantive criteria. First, it must be phonetically and orthographically distinct from all existing generic and brand names. Second, it must use only letters that are common to most Roman-alphabet languages: the letters Y, H, K, J, and W are excluded from INN prefixes because they carry confusing or difficult phonetic values in major pharmaceutical markets including Germany, France, Spain, and Japan. Third, it must not contain promotional or laudatory elements. Fourth, it must not imply a specific therapeutic indication, because a drug approved for indication A that later demonstrates efficacy for indication B will carry the same generic name regardless.
Once the USAN Council and the WHO INN Expert Group reach agreement, the proposed INN (pINN) is published for a four-month public comment period. Any party, including a competitor, can file a substantive objection during this window. If no valid objection is sustained, the recommended INN (rINN) is finalized. From that point, the name enters all WHO publications, global pharmacopoeias, and national adopted name lists.
The intelligence value of monitoring this process cannot be overstated. The pINN publication is the first public confirmation of a competitor’s molecular investment in a specific class. It is not speculative; the company has committed sufficient resources to file an INN application, which typically occurs only after positive Phase I safety data. For analysts tracking a therapeutic area, a USAN/INN monitoring program costs a fraction of what a commercial intelligence subscription service charges, yet it consistently delivers some of the earliest actionable signals in the industry.
Key Takeaways: Section 3
The INN stem system is both a public safety infrastructure and a competitive intelligence feed. Monitoring WHO pINN publications provides class-level pipeline signals 18 to 36 months before clinical trial database registration becomes informative. The USAN application process is a formal commitment of R&D resources and should be treated as such by competitor analysts. Name harmonization between USAN and INN reduces multinational trial complexity and is a meaningful but underappreciated accelerator of clinical cycle time.
4. The Art of Brand Naming: Psychology, Agencies, and the Viability Funnel
4.1 The Commercial Logic of Brand Naming
A brand name is a trademark, and a trademark is a monopoly right of indefinite duration. Pfizer’s composition-of-matter patent on sildenafil expired in the United States in 2020. The brand name Viagra did not expire. It is Pfizer’s property indefinitely, provided they continue using it commercially and renewing the registration. This distinction defines the long-term commercial calculus of brand naming investment.
Pharmaceutical companies spend heavily on brand names because those names carry value that outlasts the underlying IP exclusivity. When the first generic atorvastatin entered the U.S. market in November 2011, Lipitor’s price dropped precipitously within months. But Pfizer retained a branded market segment, physicians and patients willing to pay the premium, specifically because the Lipitor name had 14 years of marketing behind it. Brand equity functions as a partial substitute for patent exclusivity at the margin.
The brand naming process is distinct from generic naming in almost every respect. Generic naming is rule-bound, standardized, and supervised by scientific bodies. Brand naming is creative, legally intensive, psychologically sophisticated, and subject to rejection by regulatory agencies that have no interest in your company’s commercial objectives. The regulatory bodies treat the brand name as a potential public health hazard, not a marketing asset, which shapes the entire dynamic.
4.2 The Psychology of Pharmaceutical Brand Names
Effective pharmaceutical brand names operate on several psychological registers simultaneously. They need to be memorable for prescribers who see dozens of names daily, reassuring or empowering for patients managing chronic or serious conditions, phonetically distinct enough to survive verbal communication in noisy clinical environments, and emotionally resonant without crossing into promotional claim territory.
Naming theorists identify several structural archetypes that dominate successful pharmaceutical brand naming. Evocative names create an emotional or conceptual association without making a direct efficacy claim. Lyrica (pregabalin), for nerve pain management, draws on ‘lyric’ and ‘lyrical’, connoting harmony and relief, qualities that resonate with a patient population managing persistent pain. Paxil (paroxetine) draws on ‘pax’, the Latin word for peace, targeting the anxiety relief that defines the drug’s primary value to patients. Celebrex (celecoxib) references ‘celebration’, evoking the freedom of movement that arthritis management aims to restore.
Mechanism-adjacent names take a different approach, targeting prescriber recognition over patient resonance. Lipitor (atorvastatin) compresses ‘lipid inhibitor’ into five syllables, giving a cardiovascular physician an immediate pharmacological anchor. This approach works particularly well in hospital formulary decisions where the prescriber is the primary audience and clinical credibility matters more than patient marketing.
‘Empty vessel’ names create a blank phonetic slate that the company can define entirely through its own narrative. Xeljanz (tofacitinib) has no inherent meaning. Its phonetic profile, the hard ‘X’, the sharp ‘j’, the short ‘anz’ ending, suggests precision and efficacy without making any specific claim. The company builds the entire brand story from zero, which is expensive but provides complete messaging control.
What all successful brand names share, regardless of archetype, is phonetic memorability: a distinctive sound pattern that a busy physician can reliably recall and reproduce verbally to a pharmacist. This requirement creates a specific phonetic engineering problem: the name must be distinctive enough to be memorable, but not so similar to any existing name that it triggers POCA flags or LASA errors.
4.3 The Naming Agencies: Brand Institute, Brandsymbol, and Addison Whitney
Most large pharmaceutical companies do not run brand naming in-house. They engage specialized agencies with the regulatory knowledge, linguistic resources, and trademark search infrastructure to manage the process professionally. The three largest dedicated pharmaceutical naming firms are Brand Institute (Miami), Brandsymbol (Philadelphia), and Addison Whitney, now a subsidiary of Syneos Health.
These agencies differ in philosophy and methodology. Brand Institute emphasizes its proprietary safety databases and claims to have named more than half of all prescription drugs currently on the U.S. market. Brandsymbol runs an integrated model that combines naming with brand identity and packaging. Addison Whitney takes a human-centered strategy approach, evident in their documented methodology for the Ozempic/Wegovy dual-brand development for Novo Nordisk.
A full-service brand naming engagement typically runs 12 to 18 months from initial brief to final regulatory submission. The process begins with a strategic positioning session: the agency and client define the drug’s core value proposition, its target prescriber and patient profiles, its competitive differentiation, and the emotional register the name should occupy. From that brief, the agency’s creative teams generate an initial pool of candidate names. At serious agencies, this pool numbers in the hundreds, sometimes exceeding a thousand names for a major product. Many agencies will not use pre-existing name banks, preferring to start from a clean creative slate for each engagement to ensure the name is genuinely tailored to the product and its market moment.
4.4 The Viability Funnel: From 1,000 Candidates to One Global Brand
The funnel analogy used by Pfizer’s Michael Quinlan is accurate: the process is a series of increasingly demanding screens, each eliminating a large proportion of candidates, until a handful of names survive to face the final regulatory review.
The first screen is trademark clearance. Trademark paralegals run each candidate against global trademark databases covering not just pharmaceutical categories but all commercial categories in major markets. A name that conflicts with a registered trademark in an unrelated field can still be rejected, particularly if that trademark has strong recognition. This screen typically eliminates 60 to 70% of initial candidates, often more.
The second screen is linguistic and cultural review. Agencies maintain networks of in-country, native-speaking linguistic consultants in major pharmaceutical markets. Each surviving candidate is evaluated for negative connotations, offensive meanings, difficult pronunciations, or confusing associations in German, French, Spanish, Italian, Portuguese, Japanese, Mandarin, and Arabic, among others. A name that sounds aspirational in English may carry a clinical or embarrassing meaning in another language. This review eliminates a further substantial portion of surviving candidates.
The third screen is market research. Focus groups and quantitative surveys test surviving candidates with target prescribers and patients. The research measures unaided recall, pronunciation accuracy, spelling accuracy, perceived appropriateness for the indication, and emotional response. Names that score below threshold on recall or generate consistently negative reactions are dropped.
The fourth screen is safety review, which the regulatory section below covers in detail. This involves POCA-style computational analysis, simulated handwritten prescription tests, verbal order simulations, and computer-entry tests to identify any LASA risk against the existing drug name database.
What emerges from this funnel, typically, is a shortlist of three to eight names that the company prepares to submit for regulatory review. Even at this late stage, the rejection rate at the FDA and EMA is substantial enough that companies routinely prepare multiple regulatory submissions simultaneously or in rapid sequence. A single submission strategy for a global launch is a high-risk approach that most major pharma companies abandoned years ago.
The increasing crowding of the pharmaceutical lexicon is a structural headwind that raises the cost and time of brand naming over time. Every approved drug adds two new names (generic and brand) to the universe of existing names, expanding the pool against which every new candidate must be checked for similarity. The letters X, Q, Z, and J have become systematically overrepresented in new pharmaceutical brand names precisely because these unusual letters create phonetic and orthographic distinctiveness in a crowded field. This is not aesthetic preference; it is a rational response to market saturation.
Key Takeaways: Section 4
Brand names are perpetual trademark assets with measurable residual value after patent expiry. The naming process is a 12 to 18 month, multi-stage funnel that eliminates most candidates before any regulatory submission. Specialized naming agencies provide regulatory knowledge, global linguistic infrastructure, and trademark search capability that no in-house team can cost-effectively replicate. The saturation of the pharmaceutical lexicon is a long-term structural driver of naming cost and complexity that will intensify as the number of approved drugs grows.
5. The Regulatory Gauntlet: FDA, EMA, and the Global Approval Matrix
5.1 The FDA’s Dual Review Structure: DMEPA and OPDP
The FDA reviews proposed brand names through two distinct divisions with separate mandates that frequently pull in opposite directions. Understanding how each division evaluates a name is essential for anticipating rejection risk.
The Division of Medication Error Prevention and Analysis (DMEPA) focuses exclusively on patient safety. Its job is to determine whether a proposed name, in its written, handwritten, and spoken forms, is likely to be confused with any existing drug name in a way that could cause a medication error. DMEPA’s methodology is systematic and multi-layered.
The first layer is computational. DMEPA runs every proposed name through the POCA system, which calculates phonetic similarity scores and orthographic similarity scores against all approved drug names in its database. A combined score above 75% similarity is a significant red flag and, in practice, a near-certain rejection trigger. Names that clear the POCA threshold move to the second layer: human simulation testing. DMEPA staff and external consultants conduct handwritten prescription simulations, where clinician volunteers write prescriptions for the candidate name and reviewers assess whether those handwritten scripts could be read as other drug names. They also conduct verbal order simulations, playing back recorded speech of the candidate name and assessing whether listeners might transcribe a different existing drug name. Computer-entry testing, where participants type the candidate name into simulated pharmacy and EHR systems, identifies disambiguation problems in dropdown menus, a common source of real-world dispensing errors.
The Office of Prescription Drug Promotion (OPDP) reviews the same name from a completely different angle. OPDP’s concern is not safety but promotional integrity. Under the Federal Food, Drug, and Cosmetic Act, a brand name that misbrands the drug, by overstating its efficacy, minimizing its risks, implying broader indications than approved, or suggesting superiority over competitors, is grounds for rejection. Names that contain the word ‘fast’, ‘safe’, ‘pure’, ‘max’, ‘cure’, or any other claim-like element will be flagged. Names that incorporate the drug’s indication (e.g., ‘OncoCure’ for a cancer drug) raise concern about indication broadening and promotional messaging.
The structural tension between DMEPA and OPDP is real and practical. A name distinctive enough to pass DMEPA’s safety screens (unusual letters, unique phoneme combinations) may strike OPDP as improperly suggestive or promotional. A name evocative enough to build brand equity may be too similar to an existing name for DMEPA to pass. The sweet spot is narrow, which is why companies invest so heavily in pre-submission screening.
The FDA’s review timeline allows for a tentative acceptability determination after Phase II completion, though this is not a binding approval. Final review and approval occurs in parallel with, or just prior to, the NDA/BLA review. The tentative acceptability determination has strategic value: it allows companies to begin building marketing materials and training sales forces around a specific name before the drug is approved. But it is conditional, and the ‘reservation dilemma’ described below means even tentative acceptability does not fully insulate a name from last-minute disruption.
5.2 The Reservation Dilemma: An Unresolved Structural Risk
The FDA’s brand name review is confidential. When a company submits a name for tentative review, neither the submission nor the tentative outcome is publicly disclosed. This confidentiality is commercially rational; no company wants to reveal its brand identity years before launch. But it creates a systemic problem: two companies can simultaneously submit confusingly similar names for drugs in different therapeutic areas, both receive tentative acceptability, and neither knows about the other. When the first drug receives full marketing approval, its name enters the public database. If the second company’s name then fails the expanded POCA screen against the newly approved name, the second company faces a forced name change at launch, with all the marketing, supply chain, regulatory, and commercial disruption that entails.
The FDA explored a formal name reservation system in a 2014 Federal Register notice but never implemented it, citing the complexity of managing confidential commercial information across competing applications. The dilemma remains unresolved as of 2026. This is not a theoretical risk. Post-market name change events (Brintellix to Trintellix, Kapidex to Dexilant) often trace back to exactly this dynamic: a name cleared screening against the existing database but conflicted with a name that was approved during the review period. The practical implication for any global launch team is to develop and maintain a portfolio of at least four to six regulatory-ready name candidates simultaneously, not a single preferred name with contingency options.
5.3 The EMA’s Name Review Group: The Strictest Gatekeeper in the World
The European Medicines Agency’s Name Review Group (NRG) is, by most industry accounts, the most demanding brand name review body globally. The 55% rejection rate on first submission is not an anomaly; it reflects the NRG’s genuinely comprehensive and multi-lingual review methodology.
The NRG draws reviewers from all EU member states, covering more than 20 languages and their associated phonetic systems. A name that is phonetically distinct in English may collapse into near-identity with an existing name in Polish, Romanian, or Finnish. The NRG’s multi-language simultaneous review catches conflicts that single-country review processes routinely miss.
The EMA’s prohibition on INN stems in brand names is an absolute rule with no practical exceptions. A brand name for an ACE inhibitor may not contain ‘-pril’. A brand name for a monoclonal antibody may not contain ‘-mab’. A brand name for a statin may not contain ‘-statin’. This rule forces a clean separation between the scientific and commercial naming spaces, preventing brand names from embedding pharmacological class information that could confuse prescribers about a drug’s mechanism or misrepresent its properties.
Additional EMA-specific grounds for rejection include: use of single letters or numbers (confused with dosage designations), use of company names or abbreviations (creates perception of endorsement or exclusivity beyond the specific product), and names that are identical or confusingly similar to a product that has been withdrawn from the EU market within the last five years, regardless of the reason for withdrawal.
The EMA permits up to two name submissions per review request and accepts applications up to 18 months before the planned regulatory submission. Approved names receive a three-year validity window before requiring re-confirmation. This three-year timeline aligns reasonably well with a typical Phase III program but can cause complications for drugs with extended development programs, orphan disease designations, or accelerated approval pathways.
5.4 Comparative Regulatory Matrix: FDA vs. EMA Brand Name Review
Criterion
FDA
EMA
Primary Review Bodies
DMEPA (safety) + OPDP (promotion)
Name Review Group (NRG)
Computational Safety Tool
POCA system (phonetic + orthographic)
Multi-language expert panel from all member states
INN Stem Rule
Strongly discouraged, not absolute prohibition
Absolute prohibition; stems cannot appear in brand names
Number of Submissions per Request
Typically one; second required if first rejected
Up to two per request
Rejection Rate
Not publicly disclosed; estimated 20-30%
Approximately 55% on first submission
Tentative Review Availability
Yes, post-Phase II
Yes, up to 18 months pre-submission
Approval Validity
Valid until marketing approval or regulatory decision changes
Three years from acceptance
Language Scope
English-primary with some translation consideration
All EU official languages evaluated simultaneously
Name Reservation System
Not available
Not available
Key Takeaways: Section 5
FDA review operates through two independent divisions (DMEPA and OPDP) with different mandates that create conflicting pressures on name selection. A POCA similarity score above 75% is effectively disqualifying. The EMA’s NRG is the stricter gatekeeper by most industry measures, with a 55% first-submission rejection rate and an absolute prohibition on INN stems in brand names. The absence of a name reservation system at either agency is a structural risk that no amount of pre-submission screening fully eliminates; the only rational response is a portfolio of multiple viable candidate names.
6. LASA Errors: Quantifying the Human and Commercial Cost of Confusion
6.1 The Scale of the Problem
Look-Alike, Sound-Alike (LASA) drug name confusion is not a fringe concern. The Institute for Safe Medication Practices (ISMP) reports that name confusion accounts for approximately 25% of all medication errors reported to national tracking systems. In the United States, medication errors cause at least one patient death per day and injure an estimated 1.3 million people annually. LASA confusion is a principal contributing factor in a significant proportion of those events.
The error occurs at multiple points in the medication use pathway. A physician writes an illegible prescription. A pharmacist mishears a verbal order. A technician selects the wrong item from an alphabetically sorted dropdown menu in a pharmacy management system. A nurse pulls the wrong unit-dose packet from an automated dispensing cabinet. Each of these failure modes is directly addressable through name design choices: distinct phoneme patterns reduce verbal order errors; distinct orthographic profiles reduce handwritten prescription errors; distinct first letters reduce dropdown-menu selection errors; distinct packaging and labeling reduce cabinet-pull errors.
LASA risk escalates dramatically when the drugs involved are high-alert medications. Opioids, anticoagulants, insulin formulations, chemotherapy agents, and cardiovascular drugs with narrow therapeutic windows are the categories where LASA mix-ups are most likely to cause rapid, severe, or fatal harm. The Reminyl/Amaryl confusion resulted in fatal hypoglycemia in at least two documented Alzheimer’s patients. The Losec/Lasix confusion resulted in at least one patient death and triggered a mandatory brand name change. These are not edge cases; they are the expected outcome of insufficiently screened names placed into the hands of busy practitioners.
6.2 Documented Name Change Events: The Commercial Anatomy of a LASA Failure
The following case record covers the five most commercially significant brand name changes driven by LASA risk in the U.S. market over the past 25 years. Each represents a different failure mode and a different lesson.
Losec to Prilosec (omeprazole, AstraZeneca)
Losec was the original global brand name for omeprazole when it launched in the United States in 1989. Within months, dispensing errors involving the potent loop diuretic Lasix (furosemide) began appearing in error reporting systems. Both names were visually similar in cursive script. Both were available in 20 mg doses. Both were dispensed in busy outpatient pharmacies where speed and volume created conditions for misreading. At least one patient death was attributed to the confusion. AstraZeneca changed the U.S. brand name to Prilosec, a name with a distinct phonetic and orthographic profile. The rest of the world retained Losec.
The commercial cost included immediate marketing investment to rebuild brand recognition under a new name, all package labeling reprinted and restocked, and re-education of the prescriber base. The change came early enough in the product lifecycle that Prilosec went on to become one of the bestselling drugs in history, but the lesson is clear: a name cleared at launch can still fail once the real-world medication use environment exposes risks that simulated screening did not catch.
Reminyl to Razadyne (galantamine HBr, Janssen)
Reminyl, approved for Alzheimer’s disease management, was repeatedly confused with Amaryl, a sulfonylurea used in type 2 diabetes. Both drugs were available as 4 mg tablets. Both names share the ‘Rem/Am’ opening syllable phonetically and the ‘-il’ ending visually. In the clinical environment serving both populations (elderly patients with dementia and comorbid metabolic disease), the names were functionally indistinguishable to fatigued or distracted pharmacy staff. Two documented cases resulted in Alzheimer’s patients receiving Amaryl, causing fatal hypoglycemia. Janssen changed the brand name to Razadyne in 2005.
This case illustrates the demographic overlap problem: a LASA risk becomes most dangerous when both drugs are plausibly prescribed to the same patient population. Safety screening programs should weight LASA risk scores by the degree of prescriber and patient population overlap between the candidate drug and its closest phonetic or orthographic neighbors.
Brintellix to Trintellix (vortioxetine, Lundbeck/Takeda)
The FDA received 50 confirmed reports of confusion between Brintellix (an antidepressant) and Brilinta (ticagrelor, a P2Y12 platelet inhibitor). Both names begin with ‘Bri-‘, are five to six syllables long, and appear similar in both print and handwriting. In 12 confirmed cases, the wrong drug was dispensed. No patients were reported to have taken the wrong medication in those 12 cases, but the FDA judged the continuing risk unacceptable. The manufacturer changed the name to Trintellix in 2016. The name change forced a full relabeling exercise, reprinting of all patient education materials, retraining of pharmacy staff, and a fresh round of marketing investment to rebuild prescriber and patient recognition.
The commercial cost estimate for a LASA-driven name change, when accounting for marketing reset, relabeling, inventory write-down, and prescriber re-education, typically ranges from $50 million to $150 million for a drug with meaningful market penetration. For a drug in growth phase, the disruption to prescribing momentum can exceed the direct rebranding cost.
Kapidex to Dexilant (dexlansoprazole, Takeda)
Kapidex was approved in 2009 for gastroesophageal reflux disease management. It immediately generated confusion with two existing drugs: Casodex (bicalutamide, a prostate cancer drug) and Kadian (morphine sulfate extended-release). The potential for harm from either of those mix-ups was acute. Takeda initiated a voluntary name change to Dexilant in 2010 before any patient harm was reported. This proactive response allowed the company to frame the change as quality improvement rather than a reactive safety response, preserving some degree of market confidence.
Celebra to Celebrex (celecoxib, Pfizer)
This case is the only one on record where a LASA risk was caught before launch and corrected pre-approval. A pharmacy professor flagged the similarity between the proposed name Celebra and the already-approved antidepressant Celexa (citalopram, Forest Laboratories). The FDA agreed the names were too similar and required a change. Pfizer modified the name to Celebrex, adding a single letter that created sufficient orthographic distinction. Celebrex went on to become one of the top-selling drugs of its era. The pre-launch catch cost nothing beyond a minor marketing adjustment; a post-launch catch would have cost tens of millions and disrupted the blockbuster trajectory the drug eventually achieved.
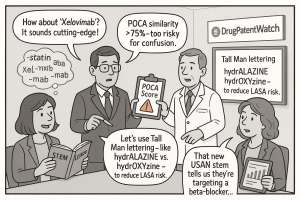
6.3 Mitigation Infrastructure: Tall Man Lettering, BCMA, and CDS Systems
Tall Man Lettering (TML) is the most widely deployed pharmacological LASA mitigation tool in the United States. Endorsed by both the FDA and ISMP, TML applies deliberate mixed case to visually distinguish the dissimilar portions of two look-alike names. Hydralazine and hydroxyzine become hydrALAZINE and hydrOXYzine. Prednisolone and prednisone become predniSOLONE and predniSONE. Quinine and quinidine become quiNINE and quiniDINE. The visual disruption breaks the cognitive pattern-matching tendency that causes a reader to gloss over a familiar-looking word without fully processing it.
Barcode Medication Administration (BCMA) systems require a nurse to scan both the patient’s wristband and the medication barcode before administration. When functioning correctly, BCMA systems can catch wrong-patient and wrong-drug errors at the bedside, including those driven by LASA name confusion that made it through the dispensing process. Hospital BCMA compliance rates in the U.S. have improved steadily since the 2004 FDA rule requiring barcodes on all unit-dose medications, but implementation gaps remain in small community hospitals and long-term care settings.
Clinical Decision Support (CDS) alerts within EHR platforms flag LASA-prone prescriptions at the point of order entry. When a physician types the first four letters of a drug name, a well-designed CDS system can surface a disambiguation alert if any nearby entry in the dropdown menu is a high-alert medication with a similar name. The challenge is alert fatigue: when CDS systems fire too frequently, clinicians habituate to overriding the alerts without reading them, which eliminates the protective value. Effective CDS design reserves LASA alerts for high-risk drug pairs, not every phonetic neighbor in the database.
Key Takeaways: Section 6
LASA errors account for roughly 25% of all reported medication errors. The commercial cost of a post-launch LASA-driven name change ranges from $50 million to $150 million and disrupts prescribing momentum beyond the direct rebranding cost. The case record shows that demographic overlap between a candidate drug’s patient population and a LASA neighbor’s patient population amplifies the risk. Pre-launch screening must include simulation studies under realistic clinical conditions, not only computational similarity scores. The Celebra/Celebrex case demonstrates that early catches are cheap; late catches are catastrophically expensive.
7. Technology Roadmap: Biologic Naming, Biosimilar Suffixes, and the Interchangeability Fight
7.1 The FDA’s Four-Letter Suffix Policy for Biologics
In 2017, the FDA finalized a naming policy requiring that all biologics licensed under Section 351 of the Public Health Service Act carry a unique four-letter, randomly assigned, hyphenated suffix appended to the INN. The reference biologic infliximab (Remicade, Janssen) became infliximab-dyyb for the Celltrion biosimilar (Inflectra) and infliximab-abda for the Samsung Bioepis biosimilar (Renflexis). Adalimumab (Humira, AbbVie) became adalimumab-atto for Amgen’s Amjevita and adalimumab-adbm for Boehringer Ingelheim’s Cyltezo.
The FDA’s stated rationale for this policy is pharmacovigilance. When a patient experiences an adverse event after receiving a biologic, the prescribing physician and the FDA’s reporting system need to identify precisely which product was administered, because different biologic products with the same INN may have subtle manufacturing differences that affect immunogenicity, efficacy, and safety. A four-letter distinguishing suffix makes that specific product identification possible in adverse event reports, claims databases, and electronic health records.
The policy is genuinely contested in ways that go beyond procedural disagreement. The suffix system affects market dynamics. When a physician who has always prescribed ‘adalimumab’ (Humira) must now choose between adalimumab-atto, adalimumab-adbm, adalimumab-afzb, adalimumab-bwwd, and eight other biosimilar suffixes, the cognitive complexity of the decision may itself bias prescribing toward the reference biologic, whose name (Humira) carries decades of brand recognition while its INN form (adalimumab) is the cleanest string in the list. This is not a hypothetical: prescribers in surveys consistently report that the INN suffix system increases their perceived uncertainty about biosimilar products.
IP Valuation Note: Adalimumab (Humira, AbbVie)
Humira (adalimumab) had peak global annual sales of approximately $21.2 billion in 2022, making it one of the highest-revenue pharmaceutical products ever. AbbVie’s patent estate around adalimumab comprised over 100 U.S. patents at its peak, covering the composition of matter, manufacturing processes, formulation, and methods of use for multiple indications. AbbVie used this patent thicket to delay U.S. biosimilar entry until January 2023, despite the original composition-of-matter patent expiring in December 2016. The brand name Humira was a constant commercial anchor throughout the biosimilar entry period: AbbVie’s ‘Humira Citrate-Free’ high-concentration formulation launched in 2017 under the same brand name, giving physicians a product differentiation narrative that emphasized patient comfort over cost. This is a textbook example of brand name as a lifecycle management tool operating in parallel with the patent thicket strategy.
7.2 Biosimilar Interchangeability and Its Naming Implications
The FDA’s interchangeability designation under the Biologics Price Competition and Innovation Act (BPCIA) grants a biosimilar the status of a substitutable product, meaning a pharmacist can dispense the interchangeable biosimilar in place of the reference biologic without prescriber intervention, identical to the generic substitution process for small molecules in most U.S. states. Interchangeability requires additional clinical switching studies demonstrating that alternating between the reference biologic and the biosimilar does not produce greater risk than staying on either product alone.
Interchangeability has direct naming implications. An interchangeable biosimilar retains its four-letter suffix under the current FDA policy, which creates an inconsistency: a product legally substitutable for the reference biologic still carries a distinct suffix that visually and administratively differentiates it. Industry groups representing biosimilar manufacturers have argued that interchangeable products should either carry the same nonproprietary name as the reference biologic or display the suffix only in the background on labeling, not prominently. The FDA has not moved to this position. AbbVie and other reference biologic manufacturers have opposed name convergence, arguing the suffix system protects the integrity of pharmacovigilance regardless of interchangeability status.
7.3 EMA Biosimilar Naming: The Alternative Model
The EMA does not require the four-letter suffix system. In the EU, a biosimilar carries exactly the same INN as the reference biologic. Infliximab is infliximab, whether it is Remicade or Inflectra or Remsima. Product differentiation in the EU is achieved through the brand name and the Marketing Authorization Holder (MAH) identification, not through INN modification. The EMA’s position is that robust pharmacovigilance is achievable through brand name tracking, batch number identification, and MAH records without artificially distinguishing the nonproprietary names of products with demonstrated biosimilarity.
The regulatory divergence between FDA and EMA on this question is one of the most significant unresolved naming policy issues in global pharmaceutical regulation as of 2026. A company launching a biosimilar globally must navigate two systems with different INN policies, which creates asymmetric labeling, different prescribing conventions, and complex market access challenges. For the reference biologic manufacturer, the FDA suffix system provides a structural market advantage in the U.S. that does not exist in the EU.
7.4 mRNA and Gene Therapy Naming: The Emerging Frontier
The COVID-19 pandemic accelerated the development and approval of mRNA-based therapeutics. The INN system responded with the ‘-meran’ stem for mRNA products. BNT162b2, Pfizer/BioNTech’s mRNA COVID-19 vaccine, received the INN tozinameran. mRNA-1273, Moderna’s vaccine, received the INN elasomeran. This stem designation creates a searchable, monitorable class identifier for the emerging mRNA platform, which now extends beyond vaccines to mRNA-based cancer immunotherapies, mRNA lipid nanoparticle therapies for rare genetic diseases, and mRNA-based protein replacement approaches.
Gene therapy naming is still less codified. The FDA’s Center for Biologics Evaluation and Research (CBER) handles most gene therapy INN applications, and the WHO has issued guidance on naming for gene therapy products, but a fully standardized stem system equivalent to the ‘-mab’ paradigm for monoclonal antibodies has not yet emerged. Analysts tracking gene therapy pipelines should monitor WHO INN proposals and the CBER advisory committee meeting calendar for emerging nomenclature conventions that will signal how the class is being clinically stratified.
Key Takeaways: Section 7
The FDA’s four-letter biologic suffix policy is a genuine structural advantage for reference biologic manufacturers in biosimilar competition. The EMA’s non-suffix approach demonstrates that robust pharmacovigilance is achievable without INN modification, and the regulatory divergence between the two systems adds complexity and cost to global biosimilar launches. Interchangeability designation does not currently change the naming framework in the U.S., despite being the closest functional equivalent to generic substitution for biologics. mRNA and gene therapy naming conventions are in active development and represent a monitoring priority for analysts in those therapeutic areas.
8. Evergreening Tactics: How Naming Fits the Lifecycle Management Playbook
8.1 What Evergreening Is and Why Naming Matters
Evergreening is the commercial practice of extending effective market exclusivity beyond the expiry of a drug’s original composition-of-matter patent through a sequence of follow-on IP filings. The tactics include new formulation patents, new salt form patents, new method-of-use patents for additional indications, new dosage strength patents, new route-of-administration patents, pediatric use patents enabled by FDA Pediatric Research Equity Act (PREA) and Best Pharmaceuticals for Children Act (BPCA) exclusivities, and new manufacturing process patents. Each layer of the patent stack can be listed in the FDA’s Orange Book if it meets the statutory criteria, triggering a 30-month stay of any ANDA approval upon a Paragraph IV certification by a generic filer.
The brand name intersects with this strategy at multiple points. When a company launches a new formulation under the existing brand name (e.g., an extended-release or subcutaneous version), the brand equity built around the original product transfers to the next-generation product without rebuilding from zero. The brand name is the continuity signal that allows a physician already familiar with and comfortable prescribing Effexor (venlafaxine immediate-release) to write a prescription for Effexor XR (venlafaxine extended-release) without a detailed clinical education campaign. This naming continuity is commercially critical to the financial success of lifecycle management investments.
8.2 The Evergreening Technology Roadmap: A Stage-Gated IP Estate
A complete pharmaceutical evergreening strategy, when executed systematically, follows a predictable stage-gated progression. The sequence is not arbitrary; each stage targets a different legal and clinical vulnerability in the asset’s lifecycle, and the naming strategy aligns with each stage.
Stage 1: Composition-of-Matter Protection (Years 0-20 from filing, typically 12-15 years effective exclusivity after accounting for development time). The original NCE patent protects the molecule itself. At launch, the brand name is introduced and associated with the composition-of-matter estate. This is where brand equity construction begins.
Stage 2: Formulation and Salt Form Patents (Typically filed 3-7 years after original NDA, expiring 5-10 years after composition-of-matter). Common tactics include conversion from an acidic to a basic salt (e.g., from hydrochloride to mesylate for improved stability), new polymorphic crystal forms with superior bioavailability, microencapsulation or nanoparticle reformulation for modified release, and new delivery systems (transdermal patch from oral tablet; long-acting injectable from daily oral). Each new formulation is typically listed in the Orange Book under the existing brand name, or under a new formulation-specific brand name suffix (e.g., the ‘-XR’, ‘-LA’, ‘-CD’ designations that systematically accompany extended-release versions).
Stage 3: New Indication Patents and Supplemental NDAs (Filed when Phase II data supports expansion, typically 5-10 years after original approval). Method-of-use patents protecting new indications extend the regulatory exclusivity window if the new indication receives priority review or orphan disease designation. The brand name’s therapeutic scope expands with each new indication approval, and companies often invest in distinct marketing campaigns for each new indication under the umbrella brand, reinforcing name recognition while expanding the prescriber base.
Stage 4: Pediatric Exclusivity (Filed when BPCA or PREA studies are complete, granting 6 months additional exclusivity beyond all existing patents and exclusivities). The FDA’s pediatric exclusivity grant is unique in that it extends all Orange Book-listed patents by six months, not just the one that prompted the pediatric study. For a drug with multiple layered patents, this six-month extension can be worth hundreds of millions of dollars in revenue. The brand name remains constant; only the label changes to reflect the pediatric indication.
Stage 5: Next-Generation Molecule and New Brand (Filed when the follow-on compound has distinct clinical advantages, typically 7-12 years into the original drug’s commercial life). The clearest example is Prilosec (omeprazole) to Nexium (esomeprazole), where AstraZeneca launched the S-enantiomer of omeprazole under an entirely new brand name with a composition-of-matter patent, effectively restarting the exclusivity clock. The new brand name signals a distinct product, requires a fresh set of marketing investment, but inherits the therapeutic category recognition that the original brand built.
8.3 Orange Book Patent Listing and Paragraph IV Dynamics
The Orange Book (FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations) lists patents that cover an approved drug product. Any patent listed in the Orange Book for the Reference Listed Drug (RLD) is subject to Paragraph IV certification by a generic filer, who must notify the innovator and certify that the listed patent is invalid, unenforceable, or will not be infringed by the proposed generic product. If the innovator sues within 45 days, a 30-month stay of ANDA approval is automatically triggered, giving the innovator additional time to litigate.
From the naming perspective, the Orange Book listing strategy has a direct commercial connection to the brand name. Formulation patents listed in the Orange Book under the same NDA as the brand protect the specific product that physicians and patients know by name. When a generic manufacturer files a Paragraph IV against a formulation patent, the resulting litigation determines whether the generic can market a product under the same INN as the brand at a price point that undercuts the brand. If the innovator loses the Paragraph IV challenge and the generic enters the market, the brand name’s commercial value becomes dependent entirely on the remaining brand loyalty among prescribers and patients who are aware that a generic equivalent is available.
Investment Strategy Note. For portfolio managers evaluating a pharma asset with an approaching composition-of-matter patent cliff, the Orange Book patent listing for that drug is the most detailed public document available for constructing a patent expiry timeline and assessing generic entry risk. Count the number of Orange Book-listed patents, identify their expiry dates, and map each patent against its claim type (formulation, method of use, process). If the entire Orange Book estate expires within two years of the composition-of-matter patent, the brand faces rapid and deep generic penetration. If formulation patents extend 5-7 years beyond the composition-of-matter expiry, the brand may retain premium pricing for a meaningful transitional period, particularly if the new formulation has a clear clinical or compliance advantage. Services like DrugPatentWatch automate this Orange Book patent mapping and track Paragraph IV filings in real time, making the monitoring tractable for even small IP teams.
Key Takeaways: Section 8
Evergreening is a multi-stage IP strategy that extends effective exclusivity past the original composition-of-matter patent. The brand name is the commercial continuity mechanism that transfers the equity of the original product to each successive formulation, reducing the marketing cost of lifecycle management. Orange Book patent listings are public documents that map the full exclusivity timeline for any approved drug; analysts who build NPV models without reading the Orange Book are working from an incomplete picture. Pediatric exclusivity grants six months of extension across all listed patents, not just the one that triggered the study, making PREA/BPCA studies a high-return investment for drugs with large patient populations.
9. Patent Intelligence: Reading Competitor R&D Through Naming Filings
9.1 The Patent as a Competitor Notebook
A pharmaceutical patent is a public legal document. In exchange for the right to exclude others from using the claimed invention, the inventor must disclose the invention in sufficient detail that a person skilled in the relevant art could practice it. This disclosure requirement is a legal obligation, and it creates a rich intelligence resource that many pharma strategy teams significantly underutilize.
The three sections of a patent most valuable for competitive intelligence are the claims, the detailed description, and the examples. The claims define the legal scope of protection and simultaneously reveal exactly what the competitor considers novel and non-obvious. The detailed description and examples reveal the technical challenges encountered during development, the formulation strategies tested, the excipient combinations evaluated, and the manufacturing processes considered. A skilled analyst reading a competitor’s formulation patent will often learn more about the product’s underlying vulnerabilities (stability problems, solubility challenges, polymorphic instability) than from any published clinical paper.
The patent type is itself a strategic signal. A composition-of-matter filing is a foundational claim to a new molecule. A formulation patent filed 5-7 years after the composition-of-matter patent is an evergreening signal. A method-of-use patent for a new indication is an expansion signal. A process patent for improved synthesis efficiency is a manufacturing competitiveness signal. A patent on a new salt form often signals that the original form had stability or bioavailability problems the company is now trying to engineer around.
9.2 Pipeline Reconnaissance: Connecting USAN/INN Filings to Patent Estates
The most powerful pipeline intelligence workflow combines two public data sources: USAN/INN filings (which reveal compound identity and mechanism) and patent families (which reveal technical development strategy). When a competitor files a new INN for a ‘-tinib’ compound, the analyst’s next step is to identify the corresponding patent family, map the priority date (the earliest filing date, which predates the publication by 18 months), assess the breadth of composition-of-matter claims, and identify any early formulation or method-of-use filings that signal where the company plans to take the drug commercially.
This connected analysis can place a competitor compound accurately on a development timeline with a level of confidence that far exceeds what CLINICALTRIALS.GOV data alone provides. Patent priority dates are earlier than trial registration dates. They reveal the approximate date of first successful synthesis or preclinical efficacy demonstration. Combined with INN filing dates (Phase I/II entry signals) and clinical trial registration (Phase II/III signals), the analyst can construct a compound-level development timeline with multiple independently verifiable data points.
Patent landscape analysis at the therapeutic area level identifies white space: therapeutic targets, combination strategies, or delivery technologies where patent density is low relative to clinical interest. Low patent density in an area of high clinical need is a signal of opportunity for a company assessing where to direct its own R&D investment or which assets to license in. This analysis requires systematic coverage of the entire patent literature in the relevant area, which is the core capability that patent intelligence platforms like DrugPatentWatch are built to provide.
9.3 Constructing a Competitor’s Lifecycle Strategy from Public Filings
A practical example illustrates the analytical method. A competitor’s original NDA for a twice-daily oral tablet of Compound X is approved. The composition-of-matter patent expires in Year 12 post-approval. By Year 7, the competitor has filed two new patent families: one covering a once-daily extended-release formulation and one covering a long-acting injectable formulation. The USAN Council publishes a new INN application for the once-daily version the following year. The EMA NRG receives a brand name submission two years after that.
At this point, the analyst knows the following without any proprietary information: the competitor plans to launch a next-generation once-daily formulation before the composition-of-matter cliff, probably 3-4 years before, with a new brand name designed to distinguish the next-generation product commercially. The injectable formulation is in earlier development but represents the long-term franchise anchor. The competitor’s entire lifecycle management strategy is visible in the public record.
The commercial response options for a company competing in the same therapeutic area are now specific and actionable. Clinical development of a head-to-head efficacy study comparing the competitor’s once-daily formulation to its twice-daily original might support a marketing campaign emphasizing that the competitor’s own improvement on their original product. Independent formulation development of a sustained-release or injectable product using the company’s own molecule can be accelerated to compete with the announced competitor roadmap. Licensing or M&A targeting companies with delivery technology patents that could pre-empt the competitor’s next-generation formulation strategy becomes a specific strategic priority.
Key Takeaways: Section 9
A pharmaceutical patent is a detailed technical disclosure of the inventor’s knowledge at the time of filing. The claims reveal scope; the description and examples reveal technical challenges and development choices. Connecting INN/USAN filings to patent families gives analysts a compound timeline with multiple independently verifiable reference points. Patent landscape analysis identifies white space and competitive crowding. Platform services that systematically monitor USAN publications, Orange Book listings, and Paragraph IV challenges provide the intelligence infrastructure for proactive competitive strategy.
10. Advanced Franchise Strategies: Dual Branding, Line Extensions, and White Space
10.1 Dual-Brand Strategy: Semaglutide as the Canonical Case
Novo Nordisk’s decision to commercialize semaglutide under two distinct brand names, Ozempic for type 2 diabetes management and Wegovy for chronic weight management, is the most sophisticated executed example of pharmaceutical dual-brand strategy in the GLP-1 market. The decision was neither cosmetic nor operationally driven. It was a deliberate market segmentation choice with distinct implications for pricing, reimbursement, physician targeting, patient communication, and regulatory positioning.
The rationale begins with reimbursement architecture. The type 2 diabetes drug market and the obesity drug market operate under different formulary structures, different patient access criteria, and fundamentally different payer attitudes. A single brand spanning both indications would have been forced into a single pricing structure that could not simultaneously optimize for both markets. Ozempic could be priced and positioned to compete with the established type 2 diabetes drug formulary. Wegovy could be priced and positioned for a market where a premium for weight management outcomes was commercially defensible, with a separate access and reimbursement negotiation track. This pricing segmentation alone likely adds billions of dollars in cumulative revenue compared to the single-brand alternative.
Addison Whitney, the naming agency, documented their methodology for both names. Ozempic was constructed to be strong and forward-looking: ‘O’ for the cyclical journey of disease management, the phonetic link to ‘sem’ from semaglutide, ’emp’ for empowerment, and ‘pic’ for reaching a peak. Wegovy was constructed as a collaborative partner: ‘we’ for the shared patient-physician journey and a subtle reference to weight, ‘go’ for action and forward movement, and ‘vy’ as a soft cadence that keeps the name approachable. Whether or not prescribers consciously decode these phonetic cues, the emotional register of each name aligns with the psychological state of its intended patient population in a way that single-brand approaches cannot achieve.
IP Valuation Note: Semaglutide (Ozempic/Wegovy, Novo Nordisk)
Novo Nordisk’s semaglutide patent estate includes composition-of-matter protection, formulation patents covering the specific injection device and concentration, and method-of-use patents for both diabetes and obesity indications. The combined peak annual revenue for Ozempic and Wegovy exceeded $20 billion in 2024, making semaglutide the highest-revenue pharmaceutical molecule globally at that time. The GLP-1 receptor agonist class is experiencing its first Paragraph IV challenge filings as of 2025 from generic manufacturers assessing the composition-of-matter patent vulnerability. For portfolio managers: the dual-brand structure means that even if one indication faces earlier generic entry than the other, brand equity is not concentrated in a single asset. The company can defend the higher-margin brand (likely Wegovy, given obesity’s premium pricing) while accepting earlier generic pressure in the higher-volume, lower-margin diabetes market.
10.2 Line Extension Naming: The ‘-XR’, ‘-CD’, and Reformulation Playbook
The most common form of lifecycle management brand naming is the reformulation suffix. Extended-release versions of drugs are routinely launched under names like Wellbutrin XL, Effexor XR, Glucophage XR, Concerta, and Adderall XR, where the core brand name carries its established equity and the suffix signals the new formulation attribute. This naming approach is commercially efficient: it transfers brand recognition without requiring a full new brand-building campaign while simultaneously creating a new Orange Book-listable patent estate.
The regulatory risk in this approach is that the suffix must not constitute a promotional claim. ‘XL’ (extra-long) and ‘XR’ (extended-release) are widely accepted as formulation descriptors. Names like ‘Ultra’, ‘Max’, or ‘Pro’ appended to a brand name have historically drawn OPDP scrutiny. The European approach is more restrictive: the EMA generally permits only pharmacopeially standardized terminology in product name variations and discourages commercial suffix additions that imply superiority.
10.3 White Space Analysis: Identifying Naming Opportunity Across Therapeutic Categories
White space in the pharmaceutical naming context has two dimensions: clinical and linguistic. Clinical white space is a therapeutic target or indication with high unmet need and limited approved drug competition. Linguistic white space is a phonetic and orthographic zone in the existing drug name landscape where a new brand name can be placed without triggering POCA flags or NRG rejection.
Systematic white space analysis combines clinical database mapping (which indications have one or two approved agents vs. five or six), patent landscape review (which mechanistic approaches are patent-dense vs. sparse), and POCA pre-screening against the existing approved drug name database (which phonetic zones are still available for occupancy). Companies that invest in this analysis as part of pre-IND and Phase I strategy can select research programs where the probability of a successful launch, inclusive of regulatory naming approval, is materially higher than the industry average.
Key Takeaways: Section 10
Dual-brand strategies for multi-indication molecules unlock pricing, reimbursement, and marketing optimization that single-brand approaches cannot achieve. The semaglutide case demonstrates that the brand name is a commercial architecture decision, not an afterthought. Reformulation suffix naming is the most cost-efficient lifecycle management branding approach but carries regulatory constraints that vary significantly between the FDA and EMA. White space analysis that combines clinical, patent, and phonetic database mapping can identify naming opportunity zones where launch probability is systematically higher.
11. Case Studies: Viagra, Lipitor, Humira, Ozempic, and Wegovy
11.1 Viagra (Sildenafil, Pfizer): Creating a Market Category Through a Name
Sildenafil was synthesized at Pfizer’s Sandwich, Kent research facility in the early 1990s as a potential angina therapy. UK-92,480 was its internal compound code. It showed limited efficacy for angina but demonstrated consistent and reproducible effects on erectile function, reported by male trial participants across multiple study sites. Pfizer redirected the development program toward erectile dysfunction, a clinical area with virtually no approved pharmacological treatment at the time and a patient population estimated in the tens of millions in the United States alone.
The naming team’s problem was as much sociological as linguistic. Erectile dysfunction in the late 1990s was a subject most physicians and patients avoided discussing directly. A name that was clinical and technical would reinforce the stigma by treating the condition as a medical problem to be whispered about. A name that was accessible and aspirational could help normalize the conversation and accelerate physician adoption.
Viagra was crafted to convey vigor and vitality. The ‘Vi-‘ opening shares the phonetic signature with ‘vital’, ‘vigorous’, and ‘virile’. The ‘-agra’ suffix brings phonetic energy and, deliberately or not, evokes the power of Niagara Falls. Some naming theorists have noted the phonetic proximity to the Sanskrit ‘vyaghra’ (tiger), though Pfizer has not confirmed this as a deliberate source. What matters commercially is that the name projects strength and confidence without making any regulatory claim. It is a perfectly calibrated ’empty vessel’ name that Pfizer’s marketing team could fill with whatever narrative was most effective in each market.
When Viagra launched in March 1998, it achieved the fastest first-quarter commercial uptake in U.S. pharmaceutical history at that time, generating approximately $400 million in U.S. sales in its first full quarter. By the end of 1998, Pfizer had filled roughly 7 million prescriptions globally. The name was a central driver of that adoption speed: it was memorable, easy to request by name, and phonetically distinct enough that no pharmacist misheard it.
IP Valuation Note: Sildenafil/Viagra
Pfizer’s composition-of-matter patent on sildenafil expired in the United States in December 2017. The brand name Viagra remains active. Pfizer launched an authorized generic version of Viagra in December 2017, the same day generic entry was permitted, using the sildenafil INN. The brand Viagra continued at premium pricing for the brand-loyal segment. By 2020, the overall sildenafil market in the U.S. had shifted more than 90% to generic volume, but the Viagra brand continued to generate positive revenue from the premium segment, demonstrating the residual value of a strong pharmaceutical trademark even 20+ years after launch.
11.2 Lipitor (Atorvastatin, Pfizer): Winning a Crowded Market with a Mechanism-Adjacent Name
Atorvastatin entered the U.S. market in January 1997 as the fifth approved HMG-CoA reductase inhibitor. Merck’s Zocor (simvastatin) and Mevacor (lovastatin), Bristol-Myers Squibb’s Pravachol (pravastatin), and Lescol (fluvastatin, Sandoz) were already established. The competitive framing of the launch was not favorable: a ‘me-too’ drug entering a market where physicians had established prescribing habits and clinical comfort with existing agents.
Pfizer’s clinical strategy was to demonstrate superior LDL-lowering efficacy at all dose levels, which the clinical data supported. But the naming strategy also contributed to the drug’s commercial differentiation. Lipitor is a mechanism-adjacent name that immediately communicates pharmacological function to prescribers. ‘Lipid’ is the universal clinical term for the relevant biomarker category. ‘Inhibitor’ is the universal pharmacological verb for the enzyme-blocking mechanism. Compressed into ‘Lipitor’, these two terms create a name that gives a cardiologist or primary care physician an immediate pharmacological anchor without requiring a clinical education campaign to establish the drug’s scientific identity.
Lipitor became the bestselling pharmaceutical product in history by cumulative global revenues, exceeding $125 billion over its patent-protected life. Its composition-of-matter patent expired in the United States in November 2011. Generic atorvastatin entered the market the same day. Within 90 days of generic entry, the Lipitor brand’s market share by volume dropped below 10%. This rapid collapse, despite 14 years of marketing investment, illustrates the limits of brand equity when therapeutic equivalence is established and price differences are large. The brand equity value is real but bounded: it supports a premium-paying minority, not a majority, once equivalent generics are available and insurer substitution policies are in effect.
11.3 Humira (Adalimumab, AbbVie): The Brand That Survived a 100-Patent Thicket
Humira (adalimumab) is the defining example of brand naming combined with an aggressive patent thicket strategy. Abbott Laboratories (later AbbVie after the 2013 spin-off) built a patent estate of more than 100 U.S. patents around adalimumab covering its composition, formulation, manufacturing process, and methods of use across rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, plaque psoriasis, hidradenitis suppurativa, and uveitis. This multi-indication patent architecture meant that any generic or biosimilar manufacturer attempting to enter the adalimumab market faced not a single patent to challenge but a web of overlapping protections, most requiring separate Paragraph IV certification and litigation.
The Humira brand remained the commercial anchor throughout the biosimilar entry period beginning in 2023. AbbVie’s pre-entry strategic response to biosimilar competition included launching the Humira Citrate-Free formulation in 2017, a higher-concentration (100 mg/mL vs. 50 mg/mL) version that reduced injection volume and the burning sensation associated with citrate as a buffering agent. This new formulation, listed in the Orange Book under the Humira brand, was supported by a physician education campaign emphasizing the patient experience improvement, creating a clinical differentiation narrative between Humira and the incoming biosimilars. The brand name’s continuity from the original formulation to the citrate-free version was commercially essential to this strategy.
11.4 Ozempic and Wegovy (Semaglutide, Novo Nordisk): The Dual-Brand Master Class
This case is covered extensively in Section 10.1. The additional investment strategy dimension worth emphasizing is the regulatory sequencing. Ozempic (semaglutide 0.5 mg, 1 mg) received FDA approval for type 2 diabetes in December 2017. Wegovy (semaglutide 2.4 mg) received FDA approval for chronic weight management in June 2021. The four-year gap was not accidental: Novo Nordisk used the clinical data from the SUSTAIN trial program (diabetes) and the STEP trial program (obesity) to build separate regulatory dossiers, each with its own indication-specific clinical package. The brand names were developed and submitted to the FDA and EMA during the respective trial programs. The result is two independently defensible regulatory approvals, two separately negotiated reimbursement contracts with major U.S. payers, and two distinct brand identities with separate patient marketing campaigns.
12. Investment Strategy: How Analysts Should Read Naming Decisions
12.1 Naming as a Forward Indicator of Pipeline Value
Drug naming decisions generate public signals at multiple points in a drug’s development lifecycle. Analysts who systematically monitor these signals gain a timing advantage over peers who wait for clinical data readouts, press releases, or investor day presentations.
The first signal is the INN/USAN application. As discussed, this is typically concurrent with Phase I or early Phase II entry. The stem choice identifies the mechanism. The prefix choice, once published, allows correlation with the company’s patent estate to confirm the molecule. The combined signal confirms: (a) the compound exists and is in clinical development, (b) the company has committed enough resources to file regulatory name applications, and (c) the mechanism of action aligns with the patent claims filed under that compound’s priority date.
The second signal is the regulatory name submission. EMA NRG submissions are not publicly disclosed in real time, but NDA/BLA filing dates often become public through the FDA’s PDUFA date calendar. When a PDUFA date is announced, it confirms that an NDA/BLA has been submitted and that the brand naming process is either complete or in final review. A company that has not yet announced an NDA submission but has recently settled a brand name in a job posting, trademark filing, or conference presentation (where a drug’s brand name is sometimes previewed) is providing an early signal of imminent submission.
The third signal is trademark registration. U.S. trademark applications for pharmaceutical brand names are public filings searchable through the USPTO database. A company filing a trademark application for a name in the pharmaceutical category, while simultaneously having a drug in Phase III trials with a relevant mechanism, is giving advance public notice of its intended brand name. This signal can precede the NDA submission by 12-18 months.
12.2 Name Change Events as Negative Signals
A post-market brand name change is an unambiguous negative signal. It indicates that the FDA has identified a medication error pattern severe enough to require commercial disruption. The direct cost (relabeling, restocking, re-education) is quantifiable but usually manageable. The indirect cost, the interruption to prescribing momentum and the erosion of physician confidence in the brand, is harder to model but typically larger. Analysts covering a drug that receives an FDA letter requesting a name change should model a 20-40% reduction in branded revenue growth for the following 12-18 months, depending on the drug’s market penetration at the time of the change and the availability of competitor alternatives.
12.3 Biosimilar Naming Policy as a Structural Indicator of Reference Biologic Defensibility
The current FDA biologic suffix policy is a specific factor in the financial modeling of biologic patent cliffs. Because the suffix system adds cognitive complexity to biosimilar prescribing, reference biologics with strong brand names retain a larger share of branded prescriptions in the face of biosimilar entry than small-molecule brands face in the face of generic entry. Humira’s brand retention rate in the first year of U.S. biosimilar entry was higher than the first-year small-molecule brand retention rate after generic entry for most major product categories. Analysts modeling biologic patent cliff NPV should apply a biosimilar penetration curve that is shallower in Year 1 and Year 2 than the generic penetration curve for comparable small molecules.
Key Takeaways: Section 12
INN/USAN filings, trademark registrations, and regulatory submission dates are public signals that collectively allow analysts to track pipeline assets and competitive positioning with 12-36 month lead times over conventional data sources. Post-market name changes are negative revenue signals with predictable short-term trajectory impacts. The FDA biosimilar suffix policy structurally supports slower biosimilar penetration curves than small-molecule generic curves, which is a meaningful input to biologic asset NPV models.
Investment Strategy Framework for Pharmaceutical Naming Events:
Naming Event
Signal Type
Analyst Action
New USAN/INN pINN publication
Early pipeline signal (Phase I/II entry)
Map to patent family; identify mechanism; assess competitive landscape
Trademark application filed (USPTO)
Pre-submission brand signal (12-18 months pre-NDA)
Confirm Phase III status; begin competitive positioning analysis
EMA NRG acceptance confirmed
Regulatory clearance signal
Add to launch probability model; adjust market entry timing
Model 20-40% branded revenue growth reduction for 12-18 months
Interchangeability designation granted
Biosimilar market acceleration signal
Steepen biosimilar penetration curve; revise reference biologic revenue model
Paragraph IV certification filed against Orange Book patents
Generic entry risk signal
Map remaining Orange Book patent estate; assess 30-month stay applicability
13. Frequently Asked Questions
Why do new drug names increasingly use X, Z, Q, and J?
The pharmaceutical lexicon now contains over 15,000 approved drug names globally. Every new name must be phonetically and orthographically distinct from all existing names to clear POCA screening and NRG review. Common letters in common phoneme combinations are largely exhausted in commercially viable configurations. Uncommon letters create natural distinctiveness because the human ear and eye are not habituated to them in the context of drug names. Xeljanz, Qsymia, Zofran, and Jakafi are not phonetically eccentric by accident. They occupy corners of the phonetic space that are genuinely less crowded, which improves their probability of clearing both the POCA screen and the EMA’s multi-language review.
Can a generic manufacturer use a brand name?
No. Generic manufacturers are legally prohibited from using the brand name without a license. They are required to use only the INN on their product labeling. The brand name remains the exclusive property of the trademark holder indefinitely, provided the trademark is maintained through continued commercial use and periodic renewal filings. In practice, pharmacists dispensing a generic atorvastatin tablet will typically affix a sticker noting it is substituted for Lipitor, or the pharmacy management system will note the substitution, but the generic product’s labeling will show only ‘atorvastatin’ and the generic manufacturer’s name.
What is the cost of developing a pharmaceutical brand name?
Full-service brand naming engagements with major pharmaceutical naming agencies typically cost between $500,000 and $3 million per drug, depending on the scope of services. This range covers strategic positioning, creative development, trademark screening (global, all categories), linguistic review (all major markets), focus group and survey research, LASA safety screening, and regulatory submission preparation. This cost does not include the subsequent investment in regulatory filing fees, legal defense of naming decisions, or the marketing investment required to build brand recognition once a name is approved.
How does a company handle a name that is accepted in the United States but rejected in Europe?
It is not unusual for a proposed name to receive tentative acceptability from the FDA but rejection from the EMA’s NRG, or vice versa. When this occurs, the company faces a choice: launch with different brand names in different jurisdictions, or find a new name that satisfies both regulatory bodies. Operating with different names across regions is commercially manageable (AstraZeneca operated with Losec in most of the world and Prilosec in the United States for years) but adds marketing complexity and cost. Most major companies now develop global naming candidates from the start, specifically designing names to satisfy both FDA and EMA requirements simultaneously, to avoid this bifurcation.
What happens to the brand name if a drug is approved and then withdrawn from the market?
The trademark remains the property of the owner after market withdrawal. However, the EMA’s NRG specifically considers recently withdrawn products in its LASA review; a new drug with a name similar to a recently withdrawn product may be rejected on safety grounds even though the withdrawn product is no longer on the market. The FDA applies a similar consideration. Additionally, a voluntary trademark abandonment occurs if the owner stops using the trademark commercially and fails to file renewal affidavits, at which point the name enters the public domain. In practice, companies typically maintain trademarks on withdrawn drugs for several years in case of re-approval or out-licensing.
How significant is the AI component in modern drug naming?
AI tools are now routinely used in the generation and initial screening phases of pharmaceutical brand naming. Large language models and phoneme-generation algorithms can produce thousands of candidate names in minutes against a defined strategic brief. These generated candidates are then fed into computational trademark and POCA pre-screening tools, reducing the human review workload significantly. However, AI has not replaced human judgment in the strategic positioning phase (what emotional register should this name occupy?) or the cultural nuance phase (how does this name land in rural Japan vs. urban Germany?). The creative-strategic briefing that anchors the entire process, and the final regulatory submission decision, remain human decisions. The most likely evolution over the next decade is AI assuming a larger role in the screening and triage phases while senior naming strategists focus on the positioning and regulatory navigation work that requires clinical, regulatory, and cultural knowledge that pure language models do not reliably generate.
14. Key Takeaways
The Naming Trinity The chemical name anchors composition-of-matter patent claims. The generic name is a permanent public identifier and the mechanism of generic market entry. The brand name is a perpetual trademark asset that generates revenue beyond patent expiry. Each carries distinct IP rights, different expiry timelines, and different commercial risk profiles.
Generic Nomenclature as Intelligence INN/USAN filings are public, occur 2-4 years before Phase III data, and contain stem assignments that identify competitor mechanism of action. Systematic monitoring of WHO pINN publications is one of the cheapest and most productive early-warning competitive intelligence tools available to a pharma strategy team.
Brand Naming Rejection Risk The EMA NRG rejects approximately 55% of proposed names on first submission. The FDA’s dual-review structure (DMEPA safety + OPDP promotion) creates conflicting pressures that narrow the viable candidate space. Any company relying on a single brand name candidate for a major global launch is accepting a risk that the industry’s empirical track record does not justify.
LASA Is a Commercial Catastrophe A LASA-driven post-market name change costs $50 to $150 million in direct costs and interrupts prescribing momentum for 12-18 months. The case record (Losec, Reminyl, Brintellix, Kapidex) demonstrates that this risk is not theoretical. Pre-launch simulation studies under realistic clinical conditions are the primary mitigation. Computational screening alone is insufficient.
Biologics and Biosimilar Naming Policy The FDA’s four-letter suffix policy is a structural advantage for reference biologic manufacturers during biosimilar entry, supporting slower penetration curves than small-molecule generic entry. The EMA’s non-suffix approach demonstrates that pharmacovigilance is achievable without INN modification. Analysts modeling biologic patent cliffs should calibrate biosimilar penetration assumptions accordingly.
Evergreening and Orange Book Architecture The brand name is the commercial continuity mechanism across the evergreening patent stack. Orange Book patent listings are public documents that map the full exclusivity timeline for any approved drug. Pediatric exclusivity grants six-month extension across all listed patents, making BPCA studies a high-return investment for large patient population drugs.
Patent Intelligence A pharmaceutical patent’s detailed description and examples reveal technical challenges, tested formulation alternatives, and development history that no clinical publication discloses. Connecting USAN filings to patent families gives analysts multi-point compound timelines. Paragraph IV challenge filings confirm which patents generic manufacturers consider legally vulnerable.
Dual-Brand and Franchise Strategy The semaglutide dual-brand case (Ozempic/Wegovy) demonstrates that a single molecule can support distinct commercial architectures in different indication markets, unlocking pricing, reimbursement, and marketing optimization that single-brand approaches cannot achieve. This strategy requires significant upfront investment in separate regulatory naming submissions and independent marketing infrastructure.
Investment Signals from Naming Events INN filings, trademark applications, regulatory naming acceptances, and post-market name change events are public signals at defined points in the drug development timeline. Building a systematic monitoring program around these signals gives analysts a 12-36 month timing advantage over teams that wait for clinical data and press releases.
For systematic patent monitoring, Orange Book tracking, and Paragraph IV filing alerts across the pharmaceutical pipeline, DrugPatentWatch provides the underlying data infrastructure referenced throughout this guide.