The complete analytical guide for pharma IP teams, portfolio managers, and generic entrants: from claim construction and Markush deconstruction to prosecution history estoppel, post-Amgen enablement attacks, and building a freedom-to-operate program that functions as a market entry engine.
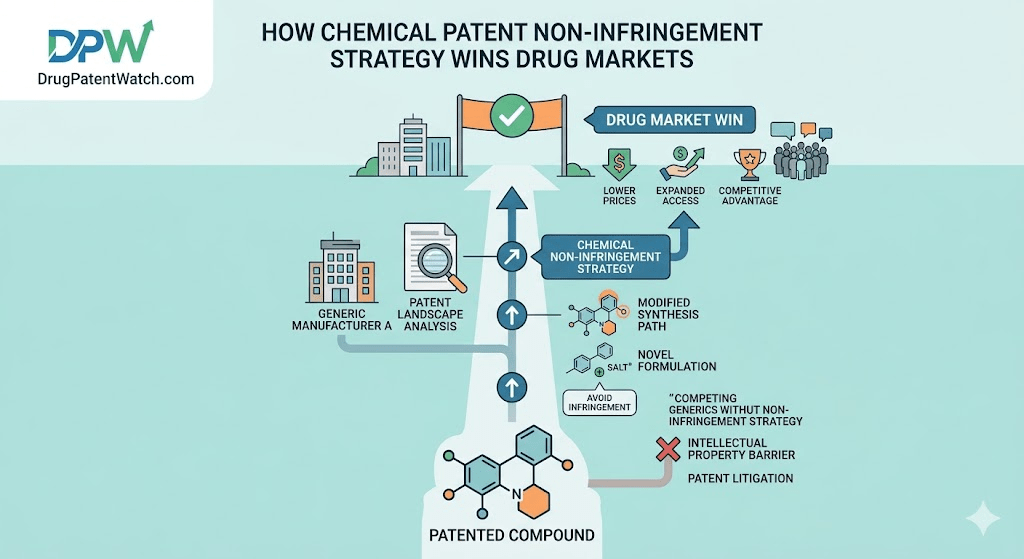
Why Non-Infringement Is a Commercial Weapon, Not a Legal Defense
A pharmaceutical patent is the only mechanism that allows a company to recover a drug development investment averaging $2.6 billion before a single generic enters the market. That single number explains why patent litigation in this industry is less a legal dispute and more an extension of corporate strategy. For every Lipitor, Humira, or Keytruda, the patents protecting the franchise are worth more than most entire companies. The conventional view treats a non-infringement position as reactive: something you build after the lawsuit arrives. That framing is wrong, and it costs companies real money. A well-developed non-infringement argument, assembled proactively and tied to commercial development timelines, does several things that a purely defensive posture cannot. It deters litigation before a complaint is filed. It forces favorable settlement terms, including earlier licensed market entry. It shifts negotiating leverage in your favor before a single deposition is taken. The primary audience for your non-infringement analysis is rarely a judge. It is opposing counsel and the CFO sitting across from them, both doing the same math: does it make sense to spend $3 million over two-plus years of litigation when the defendant has a credible, documented argument that the patent doesn’t reach their product? In roughly 40% of patent cases, the answer is no, and the case settles.
What the Invalidation Data Actually Tells Generic Entrants
That 26% invalidation rate for litigated pharmaceutical patents looks modest until you disaggregate it. The 2021 analysis of Orange Book patents issued between 2000 and 2018 found that 94% of invalidated pharmaceutical patents were not primary API patents but follow-on patents covering formulations, dissolution profiles, salt forms, or new uses. Primary API patents, those covering the active pharmaceutical ingredient itself, almost never fall to an invalidity challenge. The strategic implication is direct. Against a primary API patent, non-infringement is your primary path, not invalidity. The patentee has invested in that patent’s strength for a decade. It has been through post-grant scrutiny, and the specification is usually well-supported. Attempting to knock it out on obviousness or written description is, statistically, an uphill fight. Non-infringement, by contrast, sidesteps the patent’s strength entirely, arguing that regardless of how valid the patent is, your compound simply does not fall within its claims. Against a follow-on patent, the calculus reverses. High invalidation rates mean that an Inter Partes Review (IPR) petition at the PTAB or an enablement challenge in district court is a viable primary attack, not just a backup. A dual-track approach combining non-infringement with invalidity, what trial lawyers call the pincer movement, creates the most pressure and the best settlement leverage.
Literal Infringement vs. the Doctrine of Equivalents: How Courts Draw the Boundary
Every pharmaceutical patent infringement analysis starts from the same duality. You face two distinct but connected threats: literal infringement and the doctrine of equivalents. Ignoring either one is a strategy failure.
What Literal Infringement Requires, and Where It Breaks Down
Literal infringement requires that an accused product contain each and every limitation recited in a patent claim. The test is binary. If your compound is missing even one required structural element, no literal infringement exists for that claim. A claim to a composition comprising active ingredient X, disintegrant Y, and polymer coating Z cannot be literally infringed by a product using a different coating, regardless of how similar the products behave in practice. This is why the claim chart, a line-by-line comparison of each claim limitation against your product’s actual composition, is the foundational document of any non-infringement defense. It is also the document that settles cases: a well-built claim chart that shows a clean, documented missing element often ends the conversation before trial.
How the Doctrine of Equivalents Works Against Generic Entrants
The doctrine of equivalents (DoE) exists because courts recognized that sophisticated parties could make trivial, insubstantial changes to a patented compound and capture its full commercial value while technically evading the literal claim. The doctrine allows a patentee to argue that a substitute element in your product is legally equivalent to the missing claimed element. Two tests govern equivalence analysis. The function-way-result (FWR) test asks whether the substitute performs substantially the same function, in substantially the same way, to achieve substantially the same result. The insubstantial differences test asks whether a person of ordinary skill in the art would view the change as trivial. Both tests operate element-by-element: the Supreme Court in Warner-Jenkinson Co. v. Hilton Davis Chemical Co. made clear that you cannot argue the overall product is equivalent; equivalence must be proven for every individual claim element not literally present. Your defense against a DoE argument has three legal pillars, covered in depth below: prosecution history estoppel, ensnarement, and the vitiation rule.
Claim Construction: The Single Most Consequential Event in Pharmaceutical Patent Litigation
Patent cases are won and lost at the Markman hearing, not at trial. The Supreme Court’s ruling in Markman v. Westview Instruments, Inc. held that claim construction is a question of law for the judge alone. Before anyone asks whether your product infringes, the court decides what the patent claims actually mean. A single term, construed broadly or narrowly, can expand or contract the patent’s reach enough to determine the outcome before a fact-finder ever hears the merits.
The Intrinsic Evidence Hierarchy: Claims, Specification, and Prosecution History
Courts follow a strict hierarchy of evidence for claim construction. Intrinsic evidence, the public record of the patent itself, controls. Extrinsic evidence, which includes expert testimony, dictionaries, and scientific literature, can inform but cannot override a meaning that is clear from the patent’s own record. Within intrinsic evidence, the claims themselves come first: words in a claim are given their ordinary and customary meaning as understood by a person of ordinary skill in the art (POSA) at the time of the invention. The specification comes next, functioning as a dictionary for the claims. It can define terms explicitly, and the Federal Circuit has repeatedly held that the specification is the best guide to claim scope. The prosecution history is the third element, the complete record of all office actions, rejections, applicant responses, and amendments filed during the patent’s journey through the USPTO. This record is a public document and is, for sophisticated defendants, the most tactically valuable piece of intrinsic evidence available.
How to Use Prosecution History Disclaimer to Lock Patentees Into Narrow Scope
Prosecution history disclaimer occurs when a patent applicant, to get a claim allowed over prior art, makes an argument that distinguishes the invention using specific language or a specific definition. Courts hold applicants to those distinctions during litigation. The patentee cannot “recapture” the broader scope they surrendered to secure the patent. The tactical approach is to excavate the file wrapper looking for any argument that narrows the claim’s scope in a direction favorable to you. If an applicant argued during prosecution that “heating” in their process claim means only conventional thermal conduction, not microwave heating, and you use microwave heating, that argument becomes the foundation of a non-infringement position at the Markman stage. The investigation should extend beyond the single asserted patent. Prosecution histories from parent applications, divisionals, and continuations are all intrinsic evidence for construing the claims of later-issued family members. An admission made a decade ago to obtain a parent patent can limit the scope of a new continuation asserted against you today. This is a frequently overlooked but extremely productive line of research.
Prosecution History Estoppel: The Most Powerful Defense Against the Doctrine of Equivalents
Prosecution history estoppel (PHE) is the dominant legal tool for defeating DoE claims and the first line of attack any sophisticated defendant should develop. The framework comes from the Supreme Court’s ruling in Festo Corp. v. Shoketsu Kinzoku Kogyo Kabushiki Co., which created a strong presumptive bar against equivalents whenever a patentee narrowed a claim during prosecution for reasons related to patentability.
How the Festo Presumption Works in Chemical Patent Cases
The analysis is three steps. First, you show that the patentee amended a claim to narrow its scope, and that the amendment was made for a patentability reason, most commonly to overcome a prior art rejection under 35 U.S.C. § 102 (novelty) or § 103 (obviousness). Second, the law presumes the patentee surrendered the entire territory between the original broader claim and the final narrower claim. Third, the burden shifts to the patentee to rebut that presumption by proving one of three narrow exceptions: the equivalent was unforeseeable at the time of the amendment, the rationale for the amendment bears only a tangential relationship to the equivalent at issue, or there was some other reason the patentee could not have described the equivalent in the amended claim. Those exceptions are deliberately difficult to satisfy. “Unforeseeable” is a high standard in chemistry, where structural analogs are common and their synthesis is routinely practiced. “Tangential” requires that the connection between the amendment and the accused equivalent be genuinely peripheral, not just indirect. The practical effect is that a confirmed narrowing amendment for patentability creates a near-impenetrable barrier to DoE arguments in the surrendered territory.
Applying PHE to Chemical Substituent Amendments: A Worked Example
Suppose a patent application originally claimed “a compound comprising an alkyl substituent at position 3.” An examiner rejects the claim as obvious over a prior art compound with an ethyl group at that position. To overcome the rejection, the applicant amends the claim to “a compound comprising a methyl substituent at position 3.” Your product uses a propyl group. PHE analysis proceeds directly. The amendment from “alkyl” to “methyl” was a narrowing amendment made for a patentability reason. The presumption of surrender covers all alkyl groups that were eliminated when the scope narrowed, including propyl. The patentee must now prove that a propyl group at position 3 was completely unforeseeable at the time of the amendment, a nearly impossible argument given the well-understood chemistry of alkyl substituent series. The DoE claim fails.
Ensnarement and Vitiation: Two Additional Defenses Against Doctrine of Equivalents Claims
Ensnarement: When the Patentee’s Equivalents Would Swallow the Prior Art
The ensnarement doctrine prevents a patentee from using the doctrine of equivalents to claim subject matter that was already in the public domain at the time of the invention. If the patentee’s asserted range of equivalents would cover prior art, the DoE simply cannot reach that far. Courts apply a hypothetical claim analysis. They construct a hypothetical claim broad enough to literally cover the accused product and ask whether that hypothetical claim would have been patentable. If the prior art would have anticipated or rendered the hypothetical claim obvious, the DoE cannot be used to achieve that scope. The doctrine correctly recognizes that the public cannot be taxed twice: once through the original patent grant, and again through a DoE expansion that consumes what already belongs to the public domain. In practice, this means your invalidity search and your DoE defense use overlapping evidence. Prior art references that push against the hypothetical claim’s patentability also destroy the patentee’s equivalents theory. Building your invalidity case and your ensnarement defense simultaneously from the same prior art body is both efficient and strategically coherent.
The Vitiation Rule: When Equivalence Would Erase a Claim Element
The vitiation rule prevents the doctrine of equivalents from being applied so broadly that it eliminates, or “vitiates,” a claim limitation by rendering it meaningless. The principle comes from Warner-Jenkinson‘s all-elements rule and operates as a check against overreach. The rule is most powerful when the accused feature is the chemical or functional opposite of what is claimed. A patent claiming an “anhydrous” formulation cannot reach a formulation in which water is the primary solvent: a finding of equivalence would effectively delete the word “anhydrous” from the claim. A patent claiming an “acidic excipient” cannot reach an alkaline excipient for the same reason. In each case, the difference is categorical, not merely quantitative, and the claim language gives defendants a textual anchor for the vitiation argument.
Defense
Core Principle
Key Evidence
Strategic Advantage
Controlling Case
Prosecution History Estoppel
Patentee cannot recapture surrendered scope via DoE
Prosecution file showing narrowing amendment for patentability
Creates strong legal presumption; burden shifts to patentee
Festo Corp. v. Shoketsu
Ensnarement
DoE cannot extend over prior art
Prior art closer to accused product than to patented invention
Blocks scope expansion into public domain; overlaps with invalidity evidence
Wilson Sporting Goods v. David Geoffrey
Vitiation Rule
DoE cannot erase a claim limitation
Categorical difference in accused product (e.g., opposite chemical property)
Textually grounded; powerful against polar-opposite substitutions
Warner-Jenkinson Co. v. Hilton Davis
Prosecution Disclaimer
Patentee bound by definitions given during prosecution
Arguments in prosecution history narrowing a claim term’s meaning
Limits claim scope at Markman stage before infringement analysis begins
Omega Eng’g v. Raytek Corp.
Deconstructing Markush Claims: How Closed Groups Create Open Opportunities
Pharmaceutical compound patents rely heavily on Markush claims, a drafting format created after the USPTO upheld Eugene Markush’s right to claim “a compound where substituent R1 is selected from the group consisting of…” The phrase “consisting of” is legally significant: it creates a closed group. Nothing outside the enumerated members is included.
Why the Closed Group Is Your Literal Non-Infringement Path
If your compound’s substituent falls outside the list, you do not literally infringe. The analysis is that direct. A claim covering methyl, ethyl, and isopropyl at position R1 cannot literally capture a cyclopropyl or n-butyl group, regardless of how similar the compounds’ properties are. The patentee chose to enumerate specific members, and that choice has legal consequences. This is not a minor tactical point. A large share of pharmaceutical compound claims use Markush format at one or more substituent positions. Every closed group in a claim is a potential exit door for a structurally differentiated compound.
Using Prosecution History Estoppel Against Markush Claims
The Federal Circuit’s analysis in Amgen v. Amneal is the definitive guidance here. Amgen’s cinacalcet formulation patent recited specific binders and disintegrants in Markush format, a format adopted during prosecution to overcome prior art. The court found that the amendment to enumerate only those specific excipients created prosecution history estoppel, barring Amgen from arguing that Amneal’s use of pregelatinized starch, not listed in the closed group, was an equivalent. The patentee’s deliberate choice to narrow the claim to a specific list during prosecution is, itself, a surrender of everything outside that list. The practical research task is to find out why the Markush group was written as it was. If the prosecution file shows it was narrowed from broader language to overcome a prior art rejection, the estoppel argument is straightforward. If the group was always in its current form from the original application, the estoppel argument is weaker, though vitiation may still apply to the most structurally distinct alternatives.
Attacking Markush Group Validity: The Structural Similarity and Common Use Requirements
A secondary offensive strategy is to challenge whether the Markush group itself is proper. The USPTO requires that a valid Markush group have members sharing both a single structural similarity and a common use within the context of the claimed invention. A group that mixes members from chemically unrelated classes without a rational basis for their common function may be invalid as an improper Markush group, opening an additional invalidity route that runs parallel to the non-infringement argument.
Enantiomer Patents and the Patentability-Infringement Paradox
Chiral drugs account for a substantial portion of the top-selling pharmaceutical compounds. When a racemate is known prior art and a patentee isolates a single enantiomer and patents it, the basis for patentability is the “unexpected properties” of that specific stereoisomer: superior potency, reduced toxicity, better pharmacokinetics, or improved solubility. That patentability argument becomes a trap in infringement proceedings.
Literal Non-Infringement of Single-Enantiomer Patents
A compound consisting of the pure (R)-enantiomer does not literally infringe a claim to the pure (S)-enantiomer. These are different chemical entities. A racemic mixture equally does not infringe a claim to either pure enantiomer. The stereochemical specificity in the claim language gives defendants a clean literal non-infringement argument when the structural identity of the accused compound is confirmed analytically.
How to Trap the Patentee Using Their Own Prosecution Arguments
The interesting fight comes at the DoE stage, where the patentee will argue that the other enantiomer, or the racemate, is an insubstantial equivalent. This is where the patentee’s own prosecution record becomes their liability. To overcome an obviousness rejection for a single enantiomer when the racemate is known prior art, the patentee must argue that their enantiomer has unexpected, superior, and unpredictable properties compared to the racemate or the other enantiomer. They need that argument to be strong and credible to get the patent allowed. Every statement they made about the enantiomer’s unique biological activity, its unexpected selectivity, its surprising metabolic stability, those statements are now admissions that the difference between the two mirror-image molecules is inventive, substantial, and entirely non-trivial. Presenting this to the court is direct: the patentee cannot argue to the USPTO that the difference between the two enantiomers is inventive and profound, and then argue to the court that the very same difference is insubstantial and should be ignored under the doctrine of equivalents. The tension between these two positions, which can be established entirely from the patent’s intrinsic record, is often decisive.
Post-Amgen v. Sanofi: How the Supreme Court Put Broad Genus Claims in the Crosshairs
The Supreme Court’s unanimous 2023 decision in Amgen Inc. v. Sanofi is the most important patent ruling for pharmaceutical IP strategy in years. The case involved Amgen’s patents for Repatha (evolocumab), a PCSK9 inhibitor. The patents claimed not the specific antibodies Amgen had developed but the entire functional genus: all antibodies that bind PCSK9 at certain residues and block it from binding LDL receptors.
Why Amgen Lost and What It Means for Generic and Biosimilar Challengers
The Court invalidated the claims for lack of enablement under 35 U.S.C. § 112(a). Despite Amgen’s two-step method for identifying qualifying antibodies, the Court found this amounted to a research program requiring extensive trial-and-error experimentation. The patent did not teach skilled scientists how to make and use the full scope of the functionally-defined genus; it taught them how to go looking for new members of that genus. That is not enablement. The practical implication is that any broad, functionally-defined genus claim, whether for antibodies or small molecules, is now a credible target for an enablement challenge. The wider the gap between the number of compounds the patentee actually reduced to practice and the number of compounds covered by the functional definition of the claim, the stronger the enablement attack.
The Enablement-Obviousness Tension: A Strategic Trap for Patentees
The most sophisticated use of Amgen‘s holding is the strategic tension it creates between enablement and obviousness defenses. When a patentee argues against your enablement challenge, they will contend that the chemistry in their field is predictable enough that their limited examples adequately enable the full scope of the genus. They must make this argument to survive your § 112 attack. But that argument cuts against them in your obviousness case. If the patentee claims the field is so predictable that a handful of examples enables a genus of thousands of compounds, then a skilled chemist would have had a reasonable expectation of success in making any member of that predictable genus, including your compound, starting from the prior art. Predictability in chemistry simultaneously supports enablement and destroys non-obviousness. The patentee who argues predictability to defeat your § 112 challenge is handing you a loaded gun for your § 103 argument.
Litigation Intelligence
The PTAB’s institution rate for IPR petitions challenging pharmaceutical patents has declined since its peak in 2016, but enablement and written description grounds in district court have become more potent post-Amgen. For broad genus claims covering functional antibody classes or structurally diverse small-molecule families, a § 112 challenge in district court now carries higher probability of success than it did before 2023.
Challengers should assess whether a given genus patent’s specification supports the full claim scope across the entire functional or structural range claimed, not just the specific exemplified compounds.
The Written Description Requirement and Genus Claims in Small-Molecule Drug Patents
Written description under 35 U.S.C. § 112(a) requires that the patent specification demonstrate the inventor was in “possession” of the full scope of what is claimed as of the filing date. For genus claims in pharmaceutical patents, the specification must either disclose a representative number of species within the genus or identify common structural features that allow a skilled person to recognize genus members.
When a Broad Genus Claim Fails Written Description
A claim covering millions of structurally diverse compounds supported by two or three exemplified structures is highly vulnerable. The Federal Circuit in Juno Therapeutics, Inc. v. Kite Pharma, Inc. invalidated CAR-T cell therapy claims on written description grounds where the specification’s examples did not adequately represent the claimed genus of antigen-binding domains. The same principle applies to small-molecule claims: if the claimed genus is so broad that the disclosed examples represent only a tiny fraction of covered structures, and no common structural feature ties the genus together in a way that would allow a skilled chemist to identify members reliably, the written description requirement is not met. For defendants, the written description attack requires carefully mapping the claimed genus against the disclosed species, then demonstrating the gap. This is fundamentally a chemistry analysis: how many distinct structural classes fall within the claim? How many exemplified structures exist in the specification? Is there a unifying structural motif, or does the claim use purely functional language to define membership?
Building an Element-by-Element Claim Chart: The Forensic Foundation of Non-Infringement
The claim chart is the evidentiary backbone of every non-infringement defense. It is a structured, two-column comparison: one column deconstructs the patent claim element by element, the other describes the corresponding feature of the accused product, supported by analytical data. Courts rely on it. Opposing counsel negotiates over it. Settlement leverage depends on how clean and well-documented it is.
Starting with Independent Claims and the Logic of Dependent Claim Analysis
Every infringement analysis starts with independent claims. A dependent claim incorporates all the limitations of the claim from which it depends and adds more. If your product does not infringe an independent claim, it cannot infringe any dependent claim drawn from it: the math is inescapable. This means a single, well-established missing element in an independent claim protects you from the entire dependent claim family branching from it. The corollary is that you must analyze every independent claim in the patent. Infringement of a single independent claim is sufficient for the patentee to prevail. You need to establish non-infringement of each independent claim, or at minimum, have a credible argument for each.
Analytical Chemistry and Synthetic Reproduction as Evidentiary Tools
For chemical compound patents, the claim chart is built on data, not assertion. Structural confirmation of your API and its key intermediates, using NMR, X-ray crystallography, HPLC, and mass spectrometry, is the evidentiary foundation for every structural non-infringement argument. In Hatch-Waxman litigation, where the challenged product is often not yet on the market, it may be necessary to synthesize the proposed API or a critical intermediate specifically to generate the structural data needed to populate the claim chart. PharmaForensics and similar contract analytical chemistry firms specialize in exactly this work. The quality of this analytical data determines how persuasive the claim chart is. An expert’s statement that “my compound lacks element X” supported only by an expert declaration is weaker than the same statement backed by a crystal structure or a full NMR characterization. The evidentiary weight of the claim chart scales directly with the rigor of the underlying chemistry.
Why Obviousness in the Post-KSR Environment Favors Challengers
The Supreme Court’s 2007 ruling in KSR International Co. v. Teleflex Inc. dismantled the rigid teaching-suggestion-motivation (TSM) test that had governed obviousness analysis for decades. In its place, KSR installed a more flexible, common-sense approach: if there is a known problem, a finite number of identified solutions, and good reason to pursue them, the resulting compound is probably the product of ordinary skill, not inventive activity.
Structural Similarity and the Lead Compound Analysis
For pharmaceutical compound patents, obviousness analysis centers on lead compound identification. The challenger identifies a prior art compound with close structural similarity to the patented compound, establishes a motivation a skilled chemist would have had to modify the lead compound in the direction of the claimed structure, and argues there was a reasonable expectation of success that the modification would produce a compound with the desired properties. KSR‘s common-sense standard has made this analysis more accessible for challengers. Where the modification from lead compound to claimed compound involves predictable chemistry, standard medicinal chemistry optimization strategies, or well-understood structure-activity relationships within a compound class, the obviousness argument can be compelling even without a specific prior art “teaching” of the exact compound.
Obviousness of Single Enantiomers: When Racemate Prior Art Is Not Enough to Defeat a Patent
Courts have held that a pure enantiomer is not automatically obvious when the racemate is known prior art. The patentee must be given credit for discovering the enantiomer’s superior properties, which are not always predictable. However, if the methods for chiral resolution were well-established at the time, the motivation to separate enantiomers was clear from the literature, and the properties of the resolved enantiomers were at least somewhat predictable from the compound class, a strong obviousness case can be assembled. The analysis is heavily fact-dependent and turns on whether a POSA would have been motivated to separate the enantiomers with a reasonable expectation that one would have the desired properties.
Freedom-to-Operate Analysis as a Continuous Business Process, Not a One-Time Checkbox
An FTO analysis is not what happens when business development asks legal to “check the patents.” It is a dynamic, recurring process integrated into R&D decision-making from lead identification through commercial launch. Companies that treat FTO as a pre-launch clearance exercise routinely reach Phase 2 or Phase 3 development before discovering a blocking patent that could have been designed around at the lead optimization stage for a fraction of the cost.
When to Conduct FTO Analysis: Three Critical Inflection Points
The first FTO should run as soon as a lead compound and therapeutic target are identified. At this stage, the development program has maximum flexibility: the team can modify the compound’s structure, change the formulation approach, or explore a different synthetic route to avoid a blocking patent. The cost of a design-around at the lead optimization stage is measured in chemistry hours. The cost of the same design-around at Phase 2 is measured in tens or hundreds of millions of dollars, assuming it is even technically feasible. The second FTO update should come when the compound’s formulation and manufacturing process are locked, because those decisions create new patent exposure beyond the compound’s structure. Changes in particle size, polymorphic form, salt selection, excipient choices, or process parameters can bring a product within the scope of patents that were irrelevant to the bare API structure. The third, most comprehensive FTO clearance runs pre-launch, at the point when commercial manufacturing and distribution decisions are made. This opinion also serves as due diligence documentation for investors, partners, and acquirers evaluating the asset’s commercial marketability.
What a Risk-Stratified FTO Actually Produces
A rigorous FTO produces a risk-tiered patent landscape, not a binary green-light or red-flag. High-risk patents, those with claim language that likely reads on your product and whose prosecution history provides limited estoppel protection, require direct action: design-around, licensing negotiation, or a validity challenge. Medium-risk patents, those with colorable claim arguments but significant non-infringement or invalidity defenses, require monitoring and contingency planning. Low-risk patents, those clearly distinguishable from your product on claim language or prior art grounds, need only to be documented and revisited if they are asserted. The strategic outputs of this tiered analysis include design-around guidance for the chemistry team, a shortlist of patents for potential IPR challenge or licensing inquiry, and a formal opinion of counsel that protects against enhanced damages in willful infringement proceedings.
FTO opinion coverage across all asserted or assertable Orange Book patents for the lead asset
Prosecution history estoppel analysis for any follow-on patent claims the company is planning to use defensively
PTAB institution rate for any IPR petitions filed or threatened against the company’s key patents
Status of any pending Paragraph IV certification litigation and the 30-month stay expiration date
Revenue at risk from loss of exclusivity (LOE) events in the next 36 months, including formulation and method-of-use patents beyond the primary compound patent
Whether the company has a post-LOE lifecycle management strategy, including new formulations, new indications, or next-generation compounds in the pipeline
PTAB and Inter Partes Review: Using the Patent Trial and Appeal Board as a Parallel Weapon
An IPR petition at the PTAB is faster, cheaper, and uses a lower evidentiary standard than district court. The PTAB applies a “preponderance of the evidence” standard, compared to the “clear and convincing evidence” standard courts require for invalidity. IPRs typically conclude within 18 months of institution. Average cost through a full PTAB trial is a fraction of district court litigation. For challenging follow-on patents, whose invalidation rate exceeds that of primary API patents by a wide margin, an IPR petition is often the most efficient first move.
Coordinating IPR Filings with District Court Non-Infringement Defenses
The standard strategy is to file an IPR petition while simultaneously asserting non-infringement in district court and moving to stay the district court proceedings pending the PTAB outcome. If the PTAB invalidates the asserted claims, the district court case becomes moot. If the IPR is partially successful, the surviving claims are narrowed, often enough to strengthen a non-infringement position that was previously close. The PTAB proceedings also produce detailed prosecution history, including the patentee’s claim construction positions and prior art arguments, that feeds directly into the district court strategy. Filing timing matters critically. The one-year bar under 35 U.S.C. § 315(b) prohibits filing an IPR petition more than one year after service of a complaint for infringement. In Hatch-Waxman litigation, where the 30-month stay creates a defined timeline, petition filing should be planned from the moment the Paragraph IV certification is served, not after the lawsuit is filed.
Which Patents Are Most Vulnerable to PTAB Invalidity Challenges
The highest-return PTAB targets are follow-on patents, specifically those covering: (1) specific polymorphic forms of a known API, (2) formulation compositions where the excipient selection has prior art support, (3) methods of treatment claiming dose regimens or patient populations that were disclosed or suggested in the original clinical literature, and (4) metabolite patents where the metabolite was known or predictable from the parent compound. All four categories have higher-than-average invalidation rates both at the PTAB and in district court, and the prior art searches supporting IPR petitions for these patent types are comparatively efficient.
Key Patent Litigation Decisions Every Pharma IP Team Must Know
FTC v. Actavis: How Reverse Payment Settlements Changed Generic Entry Timelines
The Supreme Court’s 2013 decision in FTC v. Actavis held that “reverse payment” settlements, where a branded company pays a generic to delay market entry, can violate antitrust law and must be analyzed under the rule of reason. The decision did not ban such settlements but exposed them to meaningful antitrust scrutiny. The practical effect has been to change how branded and generic companies structure Paragraph IV settlements, shifting away from explicit cash payments toward non-cash value transfers such as licenses to other products, co-promotion agreements, or authorized generics arrangements. For companies planning a Paragraph IV strategy, the post-Actavis settlement landscape requires that any negotiated resolution be structured so that the value transferred to the generic challenger can be justified by independent business rationale beyond simply delaying competition. The antitrust risk of the settlement now runs directly to both parties’ counsel and boards.
Halo Electronics v. Pulse Electronics: What Enhanced Damages Risk Means for Non-Infringement Opinions
The Supreme Court’s 2016 ruling in Halo Elecs., Inc. v. Pulse Elecs., Inc. liberalized the standard for enhanced damages, making treble damages under 35 U.S.C. § 284 more accessible by focusing on the “subjective willfulness” of the infringer’s conduct. A defendant who knew about a patent and launched anyway, even with a good-faith belief in non-infringement, is potentially exposed to enhanced damages if a jury finds their conduct “egregious.” A formal, written non-infringement opinion from competent independent patent counsel remains the primary mechanism for demonstrating good-faith belief and reducing enhanced damages exposure. Post-Halo, the opinion cannot be a pro forma document. It must be thorough, technically detailed, and obtained before the infringing activity begins. An opinion secured during litigation, or one that ignores obvious claim arguments, provides little protection.
Revenue at Risk: How Patent Expiry Timelines Shape Commercial Strategy
For investors and commercial strategists, the most financially relevant question in pharmaceutical IP is not whether a patent is valid or infringed. It is when protection ends, and what happens to revenue in the quarters immediately following loss of exclusivity. Empirical data shows that branded small-molecule drugs typically lose 80 to 90 percent of their revenue within the first two years after first generic entry.
What Happens Financially After Loss of Exclusivity
The revenue cliff is steeper for small-molecule drugs than for biologics. A first generic entrant, with or without 180-day exclusivity from a first-filer certification, immediately captures significant market share. Price erosion begins instantly, and it is not recoverable. By the time multiple generic entrants are in the market, branded volume typically drops to a small fraction of pre-LOE levels, often held only by patients with strong brand preferences or formulary arrangements that protect branded access temporarily. For biologics, the post-LOE dynamic is different. Biosimilar penetration has historically been slower than generic small-molecule penetration, partly due to physician and patient switching reluctance, partly due to the lack of automatic substitution designations for most biosimilars until interchangeability status is obtained, and partly because biosimilar pricing discounts have been smaller than generic small-molecule discounts. Humira’s loss of exclusivity in the U.S. market in 2023 is the defining real-world case study: despite multiple approved biosimilars, AbbVie retained substantial market share through formulary contracts and a segmented pricing strategy that no generic entrant could have executed.
The Role of Pediatric Exclusivity, Patent Term Extensions, and Regulatory Exclusivities in LOE Timing
U.S. patent term extensions (PTEs) under 35 U.S.C. § 156 can add up to five years to a patent’s term to compensate for FDA regulatory review time. The extension is calculated based on half the clinical investigation period plus the full regulatory review period, subject to caps. For drugs with long clinical development timelines and standard regulatory review periods, PTEs routinely extend primary compound protection into the mid-2030s for compounds approved in the late 2010s. Pediatric exclusivity, an additional six months attached to any existing exclusivity or patent protection when a sponsor completes pediatric studies required by the FDA, is a standard lifecycle management tool. The cost of conducting the required pediatric studies is almost always dwarfed by the revenue protected by six additional months of exclusivity on a top-selling drug.
New Chemical Entity (NCE) exclusivity: 5 years from approval for new molecular entities; generic ANDA cannot be filed until year 4 for Paragraph IV certifications
New Clinical Investigation (NCI) exclusivity: 3 years for new indications, formulations, or routes requiring new clinical studies
Orphan Drug exclusivity: 7 years for rare disease indications; blocks approval of same drug for same indication
Biologics exclusivity: 12 years reference product exclusivity under the BPCIA; 4-year filing exclusivity
Pediatric exclusivity: 6-month extension attached to any existing patent or exclusivity period
Patent Term Extension under 35 U.S.C. § 156: up to 5 additional years for regulatory review time
Why Manufacturing Complexity Creates Patent Moat Alternatives for Biologics
For large-molecule drugs, the patent thicket is only one layer of protection. Manufacturing complexity itself functions as a barrier to entry that is independent of, and often more durable than, any single patent claim. The production of a monoclonal antibody involves cell line selection and development, bioreactor conditions, purification cascade design, and post-translational modifications that collectively define the molecule’s biological behavior. Replicating that process from a public patent disclosure is, in practice, extremely difficult. This manufacturing moat is why biosimilar development programs routinely cost $100 million to $300 million and take eight to twelve years from initiation to approval. The Biologics Price Competition and Innovation Act (BPCIA) created a regulatory framework that reflects this reality, providing 12 years of reference product exclusivity precisely because Congress recognized that biologics require a different competitive timeline than small molecules.
Why GLP-1 Manufacturing Complexity Matters for Assessing Generic Entry Risk
The GLP-1 receptor agonist class illustrates how manufacturing complexity interacts with IP protection. Semaglutide, Ozempic and Wegovy’s active ingredient, is a modified GLP-1 peptide analog with a C18 fatty acid chain attached via a linker to position 26. The synthesis involves solid-phase peptide synthesis at scale, multiple purification steps, and a site-specific acylation that is technically demanding to execute with high yield and consistent purity. Even after primary patent protection expires, the manufacturing complexity of replicating the exact impurity profile and the clinical bridging data required for an ANDA or biosimilar pathway create a significant barrier. Investors assessing generic entry risk for GLP-1 drugs should weight manufacturing complexity alongside patent expiry timelines. A compound whose primary patent expires in 2031 but whose synthesis requires specialized process chemistry equipment and multi-year process development has a materially different competitive timeline than a small molecule with equivalent patent life but a straightforward synthetic route.
How Paragraph IV Litigation Determines Generic Entry Timelines and Drug Valuations
A Paragraph IV certification is a generic entrant’s declaration that a listed Orange Book patent is invalid, unenforceable, or will not be infringed by the generic product. Filing a Paragraph IV ANDA triggers a statutory 30-month stay of FDA approval if the brand company sues within 45 days. The 30-month stay is, effectively, a congressionally mandated extension of market exclusivity that runs concurrent with patent litigation.
How First-Filer 180-Day Exclusivity Changes Generic Entry Economics
The first ANDA applicant to submit a Paragraph IV certification earns 180 days of generic exclusivity upon triggering certain conditions, most commonly either launching “at risk” or winning the litigation. During that 180-day window, no other generic can enter the market. For a drug generating $3 billion in annual U.S. revenue, 180-day exclusivity on a product priced at 20 to 30 percent below brand is worth hundreds of millions of dollars, making first-filer status one of the most valuable positions in the pharmaceutical industry. The strategic race to be first filer, combined with the litigation dynamics of the 30-month stay, explains why Paragraph IV litigation timelines and litigation outcomes are among the most financially relevant data points for pharmaceutical equity analysis. A favorable settlement granting the first-filer a specific entry date, even years before patent expiry, can be worth as much as an outright litigation win.
How Branded Companies Use Citizen Petitions and FDA Orange Book Listing to Extend Exclusivity
Patent thickets, the accumulation of multiple overlapping patents covering different aspects of a drug product in the FDA Orange Book, are the primary branded company tool for extending practical exclusivity beyond the primary compound patent’s expiration. The strategic logic is that a generic entrant must certify against every listed patent, and even if the primary API patent is undefendable, litigation on secondary formulation or method-of-use patents can extend the 30-month stay and delay final FDA approval. Citizen petitions filed with the FDA during the generic approval process, challenging the adequacy of bioequivalence studies or raising manufacturing concerns, are a separate but related tactic. The FDA is required to respond before approving the ANDA, and while FDA has taken steps to reduce the strategic misuse of citizen petitions, they remain an available delay mechanism that sophisticated brand companies use alongside patent litigation.
Selecting and Deploying Technical Expert Witnesses in Chemical Patent Cases
Technical expert witnesses are central to chemical patent litigation at every stage. The judge at a Markman hearing is almost never a chemist. Without a credible expert who can translate structure-activity relationships, stereochemistry, reaction kinetics, and analytical chemistry into accessible, persuasive language, even a legally correct non-infringement argument can fail because the court does not fully understand the science supporting it.
What Distinguishes a Useful Expert from a Credentialed Witness
Credentials are the floor. The ceiling is teaching ability and credibility under cross-examination. A professor of medicinal chemistry with an impressive publication record who cannot explain a HPLC chromatogram to a non-specialist judge is less useful than a slightly less credentialed expert who can make the court understand exactly why the missing functional group matters. The best experts in pharmaceutical patent cases are those who have worked in industry as well as academia, understand both the bench science and the commercial context, and can be both technical when required and accessible when the audience needs clarity. Involving the expert early, before discovery closes, allows them to help shape the technical theory of the case, identify weaknesses in the opposing expert’s anticipated positions, and contribute to claim construction briefing. An expert whose first substantive contribution is their expert report is under-utilized.
Common Investor Questions About Chemical Patent Non-Infringement Risk
What is the difference between a non-infringement opinion and a freedom-to-operate analysis?
A freedom-to-operate analysis is the broader commercial process: identifying all potentially blocking patents across the relevant landscape, assessing their scope, and determining whether and how your product can proceed to market without infringing them. A non-infringement opinion is a formal, written legal opinion from patent counsel, focused on specific patents, that concludes your product does not infringe those patents’ claims. An FTO analysis may produce one or more formal non-infringement opinions for the highest-risk patents, but it also produces a ranked landscape of risk across many patents. The formal opinion is what protects against enhanced damages in willful infringement proceedings; the broader FTO analysis informs product development and business strategy.
How does a non-infringement position affect the financial modeling of generic entry?
Significantly. A credible, well-documented non-infringement position changes the litigation risk profile that a brand company sees when evaluating whether to sue a generic entrant. If the generic can demonstrate a clean missing element in the primary compound patent, the brand’s expected litigation outcome shifts from likely win to uncertain, and the settlement calculus changes accordingly. Earlier licensed entry dates, reduced royalty rates, and authorized generic agreements are all more achievable when the generic’s non-infringement position is credible. For financial modeling, the generic entry date assumption is the variable that most affects net present value of the generic program, and a strong non-infringement position shortens that timeline materially.
After Amgen v. Sanofi, which biologic patents are most at risk?
Broad, functionally-defined genus claims for monoclonal antibodies, bispecifics, and other biologics whose specifications identify only a handful of exemplified structures while claiming thousands or millions of functional variants. The risk is highest where the claim defines the genus purely by what the antibody does (binds residue X, blocks interaction Y) rather than what it is structurally. Patents with limited sequence data, few working examples, and heavy reliance on a “roadmap” or screening method rather than concrete synthetic or expression guidance are priority targets for enablement challenges. Small-molecule patents are also affected where broad structural genus claims lack representative species and identifiable common structural features linking genus members.
Can a well-reasoned non-infringement opinion fully protect against willful infringement findings post-Halo?
Not fully, but substantially. Post-Halo, enhanced damages are available for “egregious” infringement: conduct that was objectively reckless in the sense that the defendant knew about the patent and proceeded without a plausible defense. A thorough, pre-launch non-infringement opinion from independent counsel is the best available evidence that the defendant had a good-faith basis for believing their product was non-infringing. Courts have consistently held that such opinions weigh heavily against enhanced damages findings. The opinion must be genuinely independent, technically rigorous, and obtained before the launch decision, not prepared after litigation begins as a post-hoc rationalization.
What happens to a non-infringement defense if the court construes a key claim term more broadly than expected?
Adverse claim construction is a real risk, and managing it is part of litigation strategy. Before the Markman hearing, you should have a fallback position: if the court adopts the patentee’s broader construction, does the doctrine of equivalents analysis change? Are your PHE, ensnarement, and vitiation arguments still available and effective under the broader construction? Is there an invalidity argument that becomes stronger if the claim is construed broadly, because a broader claim is more likely to be anticipated or obvious? A non-infringement defense built on a single claim construction theory with no fallback is fragile. Building out multiple positions, each with its own evidentiary support, is how you survive an adverse Markman ruling and keep the case alive.
How do patent thickets around biologic drugs like Humira affect biosimilar entry timelines?
Humira’s patent thicket is the textbook case. AbbVie built a portfolio exceeding 250 patents around adalimumab, covering not just the molecule but its manufacturing process, formulation, methods of use, and dosing regimens. Biosimilar developers had to either challenge or design around each layer of that thicket. AbbVie settled with most biosimilar developers for staggered entry dates beginning in 2023 in the U.S., years after European biosimilar entry, on terms that included royalties and licensing conditions. The financial result for AbbVie was billions of additional protected revenue. The lesson for biosimilar developers is that the cost of Paragraph IV-equivalent challenges under the BPCIA’s patent dance must be modeled against the revenue value of earlier entry. For Humira-class biologics, the settlement math favored accepting a delayed but licensed entry rather than bearing the full litigation cost of fighting every patent in the thicket.
Key Takeaways
Non-infringement is an offensive commercial tool. Its primary audience is opposing counsel and the business team behind them, not a judge. A well-documented missing element ends cases before trial and shifts settlement leverage.
The Markman hearing decides more cases than trial. Claim construction determines the patent’s legal scope. Winning the interpretation of a single claim term can make infringement legally impossible regardless of what happens later.
Prosecution history is your best evidentiary source. File wrappers contain admissions, definitions, and narrowing amendments that lock patentees into narrow claim interpretations. Analyze every patent in the family, not just the asserted patent.
PHE, ensnarement, and vitiation form a triad. Build all three defenses against the doctrine of equivalents from the start. Each uses different evidence: internal prosecution history, external prior art, and claim language, respectively.
The enantiomer patentability argument becomes a trap in litigation. Every statement a patentee made about unexpected superiority to secure a single-enantiomer patent becomes evidence that the other enantiomer is not an insubstantial equivalent.
Post-Amgen, broad genus claims are high-value targets. Functional genus claims that cover more than the patentee actually made and tested are vulnerable to enablement attacks, and the patentee’s attempt to defend enablement by arguing predictability simultaneously weakens their non-obviousness position.
FTO analysis must run continuously from lead identification through launch. The cost of a design-around at the chemistry stage is a fraction of the cost at Phase 2 or later. Every development milestone should trigger an FTO update.
IPR and district court strategies should be coordinated from the first ANDA notification. The one-year filing bar under § 315(b) requires planning IPR petitions immediately after service of complaint, not reactively.
Investment Strategy: How Patent Non-Infringement Intelligence Affects Pharmaceutical Asset Valuation
For portfolio managers and hedge funds holding pharmaceutical equities or assessing drug asset acquisitions, non-infringement analysis is a direct input to net present value modeling. The two most NPV-sensitive variables in any pharmaceutical asset model are the peak revenue assumption and the LOE date. The LOE date, in turn, is determined partly by patent expiry and partly by the outcome of Paragraph IV litigation. A branded company with a primary compound patent expiring in 2031 but a secondary formulation patent expiring in 2035, for which the non-infringement case for generic products is weak, has a materially different competitive moat than an identical revenue compound where generics have a clean non-infringement position against the secondary patents. The difference in modeled NPV between a 2031 LOE and a 2035 LOE on a $5 billion revenue drug is in the billions of dollars.
What Paragraph IV Filing Activity Signals About Competitive Timelines









")
















