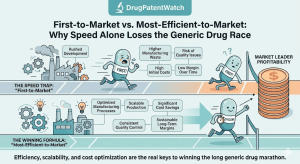
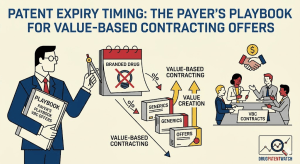
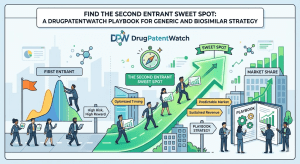
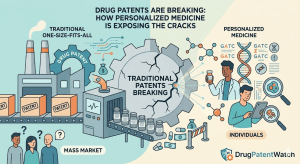
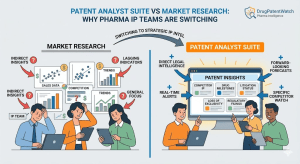
Data Beats Capital: Why the Real Challenger in Pharma IP Is the One Who Knows More
Every few years, a mid-size generic manufacturer files a Paragraph IV certification against a blockbuster drug, gets sued by a […]

Data Beats Capital: Why the Real Challenger in Pharma IP Is the One Who Knows More Read Post »