Two Markets, Two Fates: Why Branded Generics Dominate Emerging Economies and Get Crushed in the US
Branded generics — off-patent drugs sold under a proprietary name rather than an INN — are a $280 billion global […]
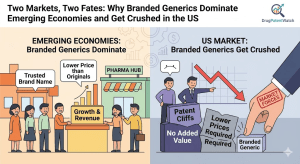
Branded generics — off-patent drugs sold under a proprietary name rather than an INN — are a $280 billion global […]
The compound is twenty years old. The molecule patent expires in eighteen months. Forty ANDA filers are lined up. The
The Oral-to-Injectable Flip: How to Use Patent Gaps to Reinvent a Therapy Class Read Post »
When a brand-name drug loses patent protection, it typically sheds 80% of its revenue within 12 months. That is not

Reformulation: Pharma’s Best Defense Against Generic Commoditization Read Post »
How the smartest pharmaceutical IP teams build exclusivity that outlasts the molecule by a decade — and what happens when
Stop Copying Molecules: Patent Drug Delivery Systems Instead Read Post »
There is a class of pharmaceutical asset that rarely shows up in earnings calls but drives billions in brand revenue
Somewhere in a brand-name drug company’s lifecycle management plan, there is usually a line item that reads something like: ‘Evaluate

The FDC Patent Goldmine: How Two Expired Patents Can Equal One New One Read Post »
The FDA Orange Book publishes patent expiry dates. Analysts read them, model them into revenue forecasts, and brief executives on

The FDA Orange Book Is Dead Weight: The New Pharmaceutical Patent Intelligence Playbook Read Post »
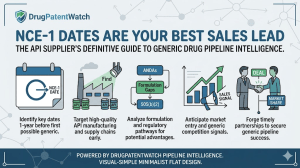
How to turn a single FDA regulatory date into a structured, repeatable, competitively timed sales funnel for active pharmaceutical ingredient
When a pharmaceutical brand launches an authorized generic, the press release typically says something about “patient access” or “expanding options.”
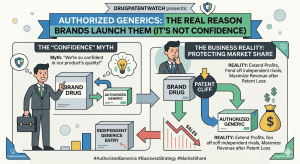
Authorized Generics: The Real Reason Brands Launch Them (It’s Not Confidence) Read Post »
Sign in or create a free account to read this DrugPatentWatch article
