Paragraph IV Is Pharma’s Most Profitable R&D Strategy
The average cost to develop a new drug reached $2.23 billion in 2024, with an expected ROI of just 5.9%. […]
Paragraph IV Is Pharma’s Most Profitable R&D Strategy Read Post »
The average cost to develop a new drug reached $2.23 billion in 2024, with an expected ROI of just 5.9%. […]
Paragraph IV Is Pharma’s Most Profitable R&D Strategy Read Post »
Quick Summary: Obviousness-type double patenting (ODP) is a judicially created doctrine that prevents patent holders from stretching exclusivity by obtaining
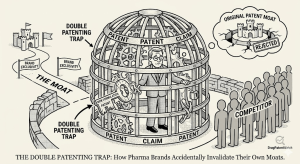
The Double Patenting Trap: How Pharma Brands Accidentally Invalidate Their Own Moats Read Post »
The answer to whether you should use PAT-INFORMED to find drug patent loss-of-exclusivity dates depends entirely on who you are,
PAT-INFORMED for Drug LOE Dates: What Procurement Agencies and Analysts Get Wrong Read Post »
Between September 2023 and May 2025, the Federal Trade Commission challenged more than 600 patent listings in the FDA’s Orange
Orange Book Delisting Wave: What the FTC Crackdown Means for Your Pipeline Read Post »
When a patient types “Is there a cheaper alternative to Eliquis?” into ChatGPT, they expect an accurate, up-to-date answer. What
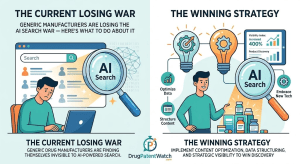
Do LLMs Recommend Branded Drugs More Often Than Generics? Read Post »
The Drug Price Competition and Patent Term Restoration Act of 1984, universally called Hatch-Waxman, created the legal architecture that now
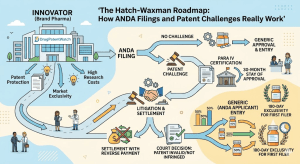
The Hatch-Waxman Roadmap: How ANDA Filings and Patent Challenges Really Work Read Post »
The primary compound patent on a drug is the one the industry talks about. It sets the nominal expiry date
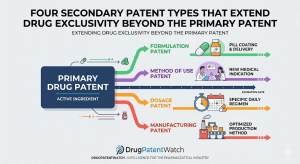
Four Secondary Patent Types That Extend Drug Exclusivity Beyond the Primary Patent Read Post »
There’s a particular moment in modern medicine that produces more confusion than almost anything else in healthcare. It happens somewhere
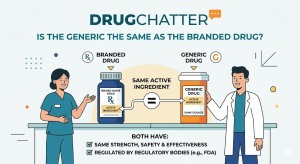
The day a competitor files a Paragraph IV certification against your lead molecule, you have 45 days to file suit

Sign in or create a free account to read this DrugPatentWatch article
