SHIONOGI INC. v. LUPIN LIMITED
SHIONOGI v. LUPIN (2:26-cv-06218): Litigation Summary, Patent Estate Impact, and Generic Launch Risk Shionogi Inc. v. Lupin Limited, case number […]

SHIONOGI INC. v. LUPIN LIMITED Read Post »
SHIONOGI v. LUPIN (2:26-cv-06218): Litigation Summary, Patent Estate Impact, and Generic Launch Risk Shionogi Inc. v. Lupin Limited, case number […]
SHIONOGI INC. v. LUPIN LIMITED Read Post »
A patent expiry for a blockbuster drug is not a legal event. It’s a commercial trigger. The twelve months before

Patent Analyst Suite vs Market Research: Why Pharma IP Teams Are Switching Read Post »
The biosimilar wave was supposed to be simple. Blockbuster biologics lose patent protection, cheaper copies enter the market, prices collapse,

The assumption is logical: if a tool costs nothing, you save money using it. That arithmetic breaks down badly when
Free Patent Databases Are Costing Pharma Teams $10K an Hour in Manual Labor Read Post »
Every year, generic pharmaceutical companies write off hundreds of millions of dollars on programs that were commercially viable on paper
Patent Data to Justify a $50M Generic Drug R&D Spend: The Exact Playbook Read Post »
The pharmaceutical industry loses about 80% of a brand’s revenue within twelve months of generic entry. For a blockbuster generating
The OTC Switch Window: Why You Need to Track Patents 10 Years Out Read Post »
When AbbVie watched its first Humira biosimilar land on the US market in January 2023, the company had already spent

Bio-Betters Are Eating Biosimilars: The Strategic Shift Reshaping Biologic Drug Patents Read Post »
Branded generics — off-patent drugs sold under a proprietary name rather than an INN — are a $280 billion global
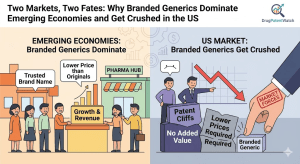
The compound is twenty years old. The molecule patent expires in eighteen months. Forty ANDA filers are lined up. The
The Oral-to-Injectable Flip: How to Use Patent Gaps to Reinvent a Therapy Class Read Post »
Get fresh news and insights, drug patent expirations & more…
