Generic drug development is a high-stakes capital allocation decision disguised as a regulatory exercise. The Abbreviated New Drug Application (ANDA) pathway, codified under the Hatch-Waxman Act of 1984, reduced the barrier to generic entry by eliminating the need to replicate originator clinical trials. But ‘abbreviated’ is a misleading descriptor. A full ANDA dossier routinely exceeds 100,000 pages, the average review cycle still runs 18 to 36 months for first-cycle approvals, and the FDA’s own data show that roughly 60% of ANDAs receive a Complete Response Letter (CRL) before ultimate approval. For IP teams scoring a target molecule, and for portfolio managers deciding where to allocate development spend, understanding which variables actually predict a clean ANDA trajectory is the difference between capturing first-generic exclusivity and watching a competitor collect the 180-day prize.
This guide works through those variables systematically, from patent landscape topology to manufacturing site risk scores, with reference to real litigation, real drugs, and real regulatory events. The objective is an actionable analytical framework, not a regulatory overview.
Part I: Patent Landscape as the Primary Approval Predictor
Before a single bioequivalence experiment is run, the patent landscape of the reference listed drug (RLD) determines the economic viability, timing, and legal exposure of an ANDA program. Getting this analysis wrong costs years and tens of millions of dollars. Getting it right is the foundation of first-generic strategy.
Reading the Orange Book: What the Listings Actually Tell You
The FDA’s Orange Book (formally, the Approved Drug Products with Therapeutic Equivalence Evaluations) lists patents that the NDA holder has certified cover either the drug substance, the drug product, or a method of use. Each listed patent requires an ANDA applicant to submit one of four certifications: Paragraph I (no patent exists), Paragraph II (the patent is expired), Paragraph III (the applicant will not launch before patent expiry), or Paragraph IV (the patent is invalid, unenforceable, or will not be infringed by the generic product).
Paragraph IV certifications are where the economics concentrate. The first applicant to file a substantially complete ANDA with a Paragraph IV certification earns 180 days of marketing exclusivity from either the date of first commercial marketing or a court decision holding the challenged patent invalid or not infringed, whichever comes first. For a blockbuster with $1 billion-plus in U.S. net revenue, that 180-day window has historically been worth $200 million to $600 million in generic revenue. Mylan’s Paragraph IV filings against AstraZeneca’s Nexium (esomeprazole) patents in the early 2000s, Teva’s filing against Pfizer’s Lipitor (atorvastatin) compound patent, and Par Pharmaceutical’s filing against Novartis’s Gleevec (imatinib) formulation patents all followed this same calculus.
The critical analytical step is distinguishing between patents that are actually listable under 21 CFR 314.53 and those that have been improperly listed. Orange Book listings have been contested as improper on multiple grounds: method-of-use patents that do not claim a currently approved indication, process patents incorrectly listed as drug product patents, and patents claiming metabolites rather than the drug substance itself. Improper listings can be challenged via the FDA’s delisting petition process, a route that became significantly more accessible after the 2021 Supreme Court decision in Caraco Pharmaceutical Laboratories v. Novo Nordisk, which clarified that generic applicants can use counterclaims to force correction of overly broad use codes.
IP Valuation of the RLD Patent Estate: A Structural Assessment
Every molecule entering a generic development program deserves a structured IP valuation of the originator’s patent estate. This valuation has four components.
The first is patent term remaining. A compound patent expiring in 14 months is a categorically different target than one expiring in seven years, even if the product revenue profile looks similar. Patent term extension (PTE) under 35 USC 156 can add up to five years to a compound patent if the NDA holder applied promptly after approval, which most do. The Hatch-Waxman pediatric exclusivity provision adds six months to all Orange Book-listed patents when the innovator conducts FDA-requested pediatric studies under BPCA, blocking generic entry for that additional period regardless of Paragraph IV outcomes.
The second is patent quality, meaning the likelihood that any given patent would survive IPR or district court challenge. Patent quality correlates with several observable proxies: prosecution history length (longer prosecution with significant claim amendments signals prior art crowding), claim breadth relative to the actual commercial product, the presence of continuation patents that broaden or narrow the original claims, and whether the patents were subject to inter partes reexamination before the AIA replaced that process with IPR. Teva’s 2012 invalidation of Pfizer’s Lipitor reformulation patent (U.S. Patent 5,273,995) at the district court level came after years of prosecution history analysis that identified a narrow window for attack. The compound patent for atorvastatin itself had already expired; the reformulation patent was the final battleground.
The third component is the method-of-use patent landscape, which has become the primary evergreening tool for brand companies post-compound-patent. Drugs like AbbVie’s Humira (adalimumab), Novo Nordisk’s Ozempic (semaglutide), and Eli Lilly’s Mounjaro (tirzepatide) carry dense method-of-use patent estates that extend protection on specific indications well beyond the compound patent. An ANDA applicant can carve out patented indications via a ‘skinny label’ under Section viii of the Hatch-Waxman statute, but this strategy carries litigation risk after the Federal Circuit’s 2020 GlaxoSmithKline v. Teva decision, which held that promoting a skinny-label generic in a manner that encourages infringing use can sustain an induced infringement claim. That ruling effectively narrowed the skinny-label safe harbor and has since increased the complexity of method-of-use patent analysis for every ANDA filer.
The fourth component is the litigation track record of the NDA holder. Some originators, notably AbbVie, Allergan (prior to AstraZeneca acquisition), and Jazz Pharmaceuticals, have demonstrated consistent willingness to file Hatch-Waxman suits immediately upon receipt of a Paragraph IV notice letter, triggering the 30-month stay that delays ANDA approval even when the underlying patents are weak. That 30-month stay is a cash-flow weapon regardless of the merits, and it must be factored into any NPV model for a generic program.
Key Takeaways: Patent Landscape
The Orange Book listing is a starting point, not a complete picture. Proper IP valuation requires independent claim-by-claim analysis against the proposed generic product and process, prosecution history review, PTE and pediatric exclusivity calculations, and an assessment of the innovator’s litigation posture. A Paragraph IV filing without this groundwork is not a strategy; it is a guess.
Investment Strategy Note: Portfolio managers pricing generic pipeline assets should apply a probability-weighted NPV adjustment for each Orange Book-listed patent that has not yet been adjudicated. AbbVie’s Humira patent thicket, which included over 130 U.S. patents at peak, is the canonical example of how a single biologic’s IP estate can be structured to generate a 10-plus-year delay between biosimilar approval and actual competitive market entry. The same structural analysis applies to complex small-molecule generics: count the patents, assess their quality, model the litigation timeline, and price accordingly.
Part II: The ANDA Dossier — Technical Predictors of First-Cycle Approval
Reference Listed Drug Analysis: The Foundation of Bioequivalence Strategy
The RLD is the regulatory and scientific anchor of every ANDA. Before committing to a development program, the technical team must conduct a full characterization of the RLD that goes well beyond reading the package insert.
For oral solid dosage forms, characterization includes API solid-state form identification (polymorph, salt form, particle size distribution), excipient identification via analytical reverse engineering (HPLC-MS, FTIR, X-ray powder diffraction), dissolution profile generation across the FDA-recommended pH range (1.2, 4.5, and 6.8), and a stress stability study to identify the RLD’s degradation pathways and primary impurities. This last point is analytically critical: the FDA’s impurity guideline (ICH Q3B) requires that any degradation product in the generic above ICH qualification thresholds be identified and, if novel, toxicologically qualified. An impurity profile in the generic that differs substantially from the RLD’s profile is a near-automatic CRL.
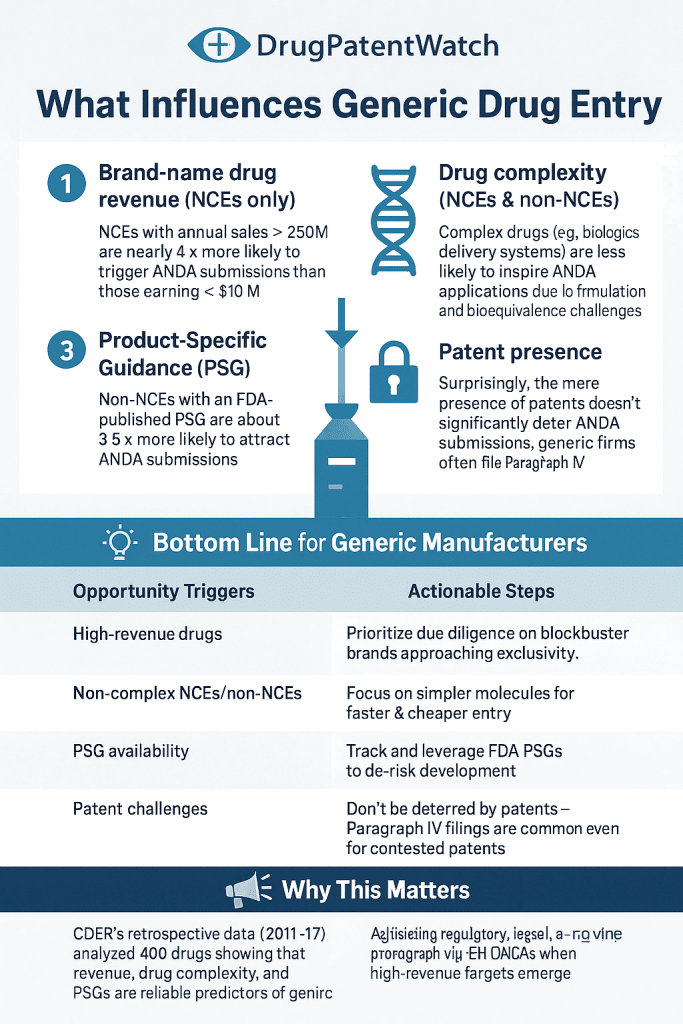
For complex dosage forms — modified-release tablets, transdermal patches, nasal sprays, inhalation products — the technical complexity compounds significantly. The FDA’s Product-Specific Guidances (PSGs), published for over 1,500 drug products, specify the recommended in vitro and in vivo study approaches for demonstrating bioequivalence. PSGs for products like tiotropium bromide inhalation powder (Spiriva), fluticasone/salmeterol inhalation powder (Advair Diskus), and budesonide/formoterol inhalation aerosol (Symbicort) require combinations of in vitro aerodynamic particle size distribution studies, in vitro drug deposition testing, pharmacokinetic studies, and, in some cases, pharmacodynamic endpoint studies. These multi-modal requirements make complex inhaler generics the most technically challenging small-molecule ANDA category, with typical development timelines of five to eight years before submission.
Bioequivalence Study Design: Where ANDAs Succeed or Fail
The FDA’s bioequivalence requirement — that the 90% confidence interval for the geometric mean ratio of the test to reference for Cmax and AUC must fall within 80-125% — functions as a binary gate. Either the study passes or it does not. There is no partial credit, and a failed bioequivalence study requires protocol redesign, reformulation, or both before a new study can be conducted.
The 80-125% window is wide enough for most immediate-release oral products but becomes genuinely difficult for drugs with high intrasubject variability. When intrasubject coefficient of variation (CV) for Cmax exceeds roughly 30%, a conventional two-period crossover design requires a prohibitively large sample size to achieve 80% power within the standard acceptance limits. The FDA’s Highly Variable Drug (HVD) guidance, finalized in 2020, allows scaled average bioequivalence (SABE) for these products, widening the acceptance limits proportionally to the RLD’s within-subject variability. This is not a free pass: the reference-scaled approach still requires a minimum sample size of at least 36 subjects and must be pre-specified in the protocol. For drugs with a narrow therapeutic index (NTI) — warfarin, cyclosporine, digoxin, levothyroxine — the FDA applies tighter standards, requiring that the 90% CI fall within 90-111.11% for primary PK parameters, making bioequivalence considerably harder to achieve without precise formulation matching.
The practical implication for ANDA planners is that bioequivalence study design is itself an IP-aware exercise. For an NTI drug whose RLD uses a proprietary controlled-release mechanism protected by formulation patents, a generic applicant may need to develop a technically distinct modified-release system that achieves the same PK profile without infringing the originator’s manufacturing process patents. That intersection of bioequivalence science and patent non-infringement analysis is where the most expensive errors occur.
Dissolution as a Bioequivalence Surrogate: BCS Classification and the Waiver Pathway
The Biopharmaceutics Classification System (BCS), developed by Gordon Amidon and colleagues in 1995 and adopted by FDA in the 2000 waiver guidance (updated in 2017 and again in draft form in 2021), classifies drugs along two axes: solubility at the highest clinical dose in 250 mL across pH 1 to 6.8, and intestinal permeability. BCS Class I compounds (high solubility, high permeability) are eligible for an in vivo bioequivalence study waiver if rapid dissolution (85% dissolved within 30 minutes in each of the three compendial pH buffers) can be demonstrated for both test and reference products. Class III compounds (high solubility, low permeability) became eligible for waivers under the updated 2017 guidance if rapid dissolution is demonstrated and excipients do not affect permeability.
The BCS waiver pathway, formally called a biowaiver, has substantial economic value. A pivotal in vivo bioequivalence study in healthy volunteers costs between $500,000 and $2 million depending on sample size, analytical complexity, and CRO fees. Eliminating that study reduces both cost and timeline by six to twelve months. For a Class I or Class III drug with a clear dissolution equivalence profile, pursuing the biowaiver is almost always the right economic call. The risk is that the FDA refuses the waiver request, typically because the agency determines that the product has a narrow therapeutic index, exhibits non-linear pharmacokinetics, or that excipient differences between the test and reference products are significant enough to affect permeability. A refused biowaiver late in the development cycle, after formulation lock but before submission, can trigger a 12- to 18-month delay.
For BCS Class II (low solubility, high permeability) and Class IV (low solubility, low permeability) compounds, dissolution profile similarity across pH conditions is not a bioequivalence surrogate but rather a formulation development tool. Demonstrating that test and reference dissolution profiles are similar across pH 1.2, 4.5, and 6.8 using the f2 similarity factor (f2 greater than 50) is a useful quality indicator, but it does not replace in vivo bioequivalence for these compounds. The in vivo study remains mandatory.
Analytical Method Validation: The Data Integrity Foundation
Every dataset in an ANDA submission rests on the analytical methods used to generate it. If those methods are not validated to ICH Q2(R1) standards, the FDA’s Office of Pharmaceutical Quality (OPQ) will issue a CRL before it even evaluates the bioequivalence conclusion. The validation parameters that most commonly generate deficiencies are selectivity (the ability to measure the analyte in the presence of degradation products and excipients without interference), lower limit of quantitation in bioanalytical methods (critical for drugs with low Cmax or those measured in plasma at trace concentrations), and stability of the analyte in the biological matrix across the full sample handling and storage cycle.
Bioanalytical method validation (BMV), governed by the FDA’s 2018 guidance, has specific additional requirements beyond pharmaceutical method validation. The FDA requires at least three concentration levels of quality control (QC) samples, at least 15 individual QC measurements distributed across runs, and that at least 67% of total QC samples and at least 50% of QC samples at each concentration level must be within 15% of nominal (20% at the LLOQ). Cross-validation is required when multiple analytical sites generate data within the same study, a common situation when sponsors use a CRO for sample analysis while maintaining in-house reference standard characterization.
Robustness testing, the deliberate introduction of small, systematic variations in analytical method parameters (mobile phase pH ±0.2, column temperature ±5 degrees, flow rate ±10%) to assess whether the method output changes unacceptably, is the parameter most frequently under-validated in ANDA submissions. Manufacturing processes inherently drift within specification, and a non-robust analytical method will produce results that appear to show product non-conformance when the product is actually within specification. OPQ has flagged non-robust methods as a contributing factor in manufacturing-related CRLs, because it cannot distinguish between genuine product quality deviations and analytical noise.
Current Good Manufacturing Practice: Manufacturing Site Risk as a Predictor
ANDA approval is not just a paper exercise. The FDA’s Office of Pharmaceutical Quality (OPQ) integrates manufacturing site inspection data into the approval decision via the Site Selection Model, which risk-scores facilities based on time since last FDA inspection, previous inspection classification (Official Action Indicated, Voluntary Action Indicated, or No Action Indicated), import alert status, and the compliance history of other applications associated with the same site.
A manufacturing site classified as OAI at any point in the 24 months preceding an ANDA approval decision is a near-automatic block. The FDA will not approve a new application for an OAI facility until the facility has submitted an adequate corrective action and preventive action (CAPA) plan and, in most cases, has been re-inspected with a more favorable outcome. For generic manufacturers with manufacturing operations concentrated in India and China — which supply roughly 80% of APIs and a significant share of finished dosage form manufacturing for U.S. generics — this creates meaningful approval risk that is entirely decoupled from the scientific quality of the ANDA itself.
Sun Pharmaceutical’s Halol facility received an OAI classification in 2014 following FDA inspections that identified data integrity violations and inadequate investigation of out-of-specification results. The Halol situation delayed approval of multiple ANDAs for more than two years and contributed to Sun’s share price declining roughly 35% between the inspection findings and the subsequent resolution. Ranbaxy’s manufacturing problems at Dewas, Paonta Sahib, and Toansa between 2008 and 2014 resulted in import alerts covering those facilities, rendered hundreds of ANDAs non-approvable without site transfer, and ultimately contributed to the company’s 2013 guilty plea and $500 million settlement with the U.S. Department of Justice for criminal violations of cGMP requirements.
The lesson from those episodes is that manufacturing site selection is as much a regulatory decision as a commercial one. ANDA applicants should independently audit proposed manufacturing sites against FDA warning letter databases, the FDA’s Site Inspection Reports where available via FOIA, and the agency’s Drug Quality Reporting System data before filing. For API suppliers, a separate ANDA-specific evaluation of the Drug Master File (DMF) holder’s recent compliance history is warranted. An API-level manufacturing deficiency can delay approval of finished dosage form ANDAs that reference that DMF, even when the finished-dose facility itself has a clean compliance record.
The FDA’s GDUFA II commitments, operational since FY2018, established performance goals of 90% of standard ANDAs receiving a first-action within 12 months of submission for Fiscal Year 2022 and beyond. GDUFA III, which covers FY2023-2027, maintained those goals while adding enhanced commitments on complex generic review timelines. Despite these performance targets, the average total approval time from original submission to approval for ANDAs receiving at least one CRL still runs approximately 36 months, based on FDA Office of Generic Drugs (OGD) annual report data through 2024. Applications that enter the queue without manufacturing site issues, with clean bioequivalence data packages, and with resolved patent certifications (Paragraph I or II) approve substantially faster, often within the 12-month GDUFA target for standard ANDAs.
Key Takeaways: Technical Predictors
The primary technical factors predicting first-cycle ANDA approval are: a clean bioequivalence study with confidence intervals comfortably within the 80-125% window (not just barely inside), validated analytical methods that pass OPQ review without data integrity flags, a formulation with a dissolution profile that matches the RLD across all compendial pH conditions, and a manufacturing site with NAI or VAI classification on its most recent FDA inspection. Any single one of these failing is sufficient to generate a CRL. All four failing simultaneously — which happens regularly when generic manufacturers rush submissions to capture an early ANDA queue position — results in multi-year approval delays that effectively forfeit the 180-day exclusivity the filing was designed to capture.
Part III: The Hatch-Waxman Litigation Gauntlet
The 30-Month Stay: Economics Before Law
When an NDA holder receives a Paragraph IV notice letter from an ANDA applicant, it has 45 days to file a patent infringement suit in federal district court. If it does, the FDA is automatically stayed from approving the ANDA for 30 months, or until the court enters a final judgment of invalidity or non-infringement, whichever comes first. The 30-month stay has no requirement that the suit have merit. It is automatic upon filing, which means it functions primarily as a delay instrument, not a legal determination.
Brand companies have exploited this mechanism systematically. AbbVie filed patent suits against every biosimilar applicant for Humira between 2016 and 2023, effectively using a combination of IPR petitions (in some cases), district court stays, and settlement agreements to manage the timing of biosimilar entry. While Humira biosimilar litigation technically falls under the BPCIA’s ‘patent dance’ rather than Hatch-Waxman, the economic logic is identical: litigation creates delay, delay preserves exclusivity, and exclusivity converts to revenue.
For small-molecule generics, the 30-month stay creates a predictable pattern. An ANDA filer with a strong invalidity or non-infringement case will often seek declaratory judgment (DJ) jurisdiction in a plaintiff-friendly district (historically the District of Delaware or the District of New Jersey), push for an aggressive claim construction and summary judgment schedule, and attempt to resolve the case before the 30 months expire. An ANDA filer with a weaker case, or one seeking primarily to use the litigation to establish settlement leverage, may adopt a more passive litigation posture.
Settlement agreements in Hatch-Waxman cases, particularly those involving authorized generics or payment transfers from brand to generic, have faced scrutiny under FTC Act Section 5 since the Supreme Court’s 2013 FTC v. Actavis decision. That ruling held that reverse-payment settlements (where the brand pays the generic to delay market entry) are not automatically immune from antitrust challenge and must be assessed under a rule-of-reason standard. In practice, post-Actavis settlements have migrated away from explicit cash payments toward equivalent-value exchanges: co-promotion agreements, API supply arrangements, authorized generic licenses, and shared exclusivity carve-outs. The FTC continues to monitor these structures, and ANDA programs premised on settlement as the exit strategy rather than litigation success must account for antitrust exposure in the deal structuring.
Inter Partes Review as a Pre-ANDA Tool
The America Invents Act of 2011 established inter partes review (IPR) as an administrative challenge to patent validity before the Patent Trial and Appeal Board (PTAB). For generic manufacturers, IPR has become a significant pre-ANDA or parallel-to-ANDA tool. A successful IPR petition that results in institution, followed by a Final Written Decision invalidating the challenged patent claims, eliminates those claims from the Orange Book landscape without triggering a 30-month stay.
The strategic use of IPR by generic and biosimilar manufacturers has been extensive. Mylan filed IPR petitions against AbbVie’s Humira composition-of-matter and formulation patents starting in 2016. While PTAB ultimately declined to institute review on most of the petitions (finding insufficient grounds to challenge AbbVie’s claims), the process generated public claim construction analysis that informed subsequent district court litigation strategy. Hikma and Fresenius Kabi have used IPR petitions against method-of-use patents for injectable drugs as a cost-effective alternative to full Paragraph IV litigation.
The key limitation of IPR as an ANDA strategy is timing. An IPR must be filed within one year of the generic applicant being served with a complaint alleging infringement of the patent at issue (the one-year bar under 35 USC 315(b)). An ANDA filer that receives a Paragraph IV notice triggering a brand lawsuit and then waits more than 12 months to file an IPR on the asserted patent forfeits the IPR route entirely for that patent. Coordinating the IPR timeline with the litigation schedule and ANDA amendment filings is a core task for the legal-regulatory-IP team.
Post-Actavis Settlement Structures and Market Entry Timing
The practical outcome of most Hatch-Waxman litigation is a negotiated settlement, not a trial. Post-Actavis, these settlements increasingly specify entry dates rather than payments, often structured as ‘at risk’ entry rights (the generic can launch before final patent expiry at its own infringement risk) or definitive licensed-entry agreements where the brand grants the generic a license to sell as of a specified date.
Authorized generics, where the brand manufactures the same product under an NDA and sells it through a generic subsidiary or licensing partner without Orange Book patent exclusivity, are a frequent settlement feature and a direct competitive threat to the first-filer’s 180-day exclusivity. When AstraZeneca settled with generic manufacturers over Nexium patents, it maintained the right to launch an authorized generic of esomeprazole through its subsidiary, which directly competed with the settling first-filers during their 180-day window and substantially eroded the exclusivity’s economic value.
Modeling a generic program’s NPV requires explicit assumptions about: the probability of litigation, the expected duration and outcome of any suit, the likelihood of an authorized generic competing during the 180-day window, and the probability that settlement produces a licensed-entry date that is earlier than what Paragraph III would have yielded. All of those assumptions require inputs from both patent counsel and business development, which is why high-value ANDA programs at companies like Teva, Mylan (now Viatris), Aurobindo, and Sun Pharma are managed by cross-functional teams with permanent legal-commercial integration, not sequential handoffs.
Key Takeaways: Hatch-Waxman Litigation
The 30-month stay is economically motivated delay, and generic applicants should plan around it from filing day. Paragraph IV certification strategies must account for post-Actavis antitrust scrutiny of any settlement, the IPR timeline constraints, and the authorized generic risk during the 180-day exclusivity window. For high-value targets, the litigation analysis is as determinative of program economics as the bioequivalence timeline.
Investment Strategy Note: When evaluating a generic company’s pipeline, discount any Paragraph IV filing against an NDA holder with a track record of rapid-firing suits and authorized generic deployment. The effective value of a 180-day exclusivity period, even on a $1 billion revenue drug, drops dramatically if the brand simultaneously captures 40-60% of generic market share via an authorized generic. The Nexium settlement precedent should be the benchmark for discount assumptions, not the theoretical first-generic economics.
Part IV: Complex Generics — The High-Barrier, High-Reward Frontier
What Makes a Generic ‘Complex’
The FDA defines complex generics across five complexity dimensions: complex active ingredients (peptides, polymeric mixtures, complex mixtures, proteins), complex formulations (liposomes, colloids, modified-release formulations), complex routes of delivery (inhalation, nasal, ophthalmic, dermal, transdermal), complex drug-device combination products, and drugs with complex drug substance/drug product interrelationships. Products that qualify as complex generics include inhalation products like Advair Diskus (fluticasone/salmeterol), injectable liposomal formulations like Doxil (doxorubicin HCl liposome), transdermal systems like Duragesic (fentanyl), and complex polymeric mixtures like glatiramer acetate (Copaxone).
Complex generics take five to ten years to develop, cost $15 million to $50 million in development expenditure before submission, and carry substantially higher approval risk per submission cycle than standard small-molecule immediate-release generics. But the reward structure is commensurate: the market for complex generics that finally do win approval is typically concentrated among two to four competitors rather than twelve to twenty, producing sustained price premiums of 30-60% above standard generic economics for three to five years post-launch. Mylan’s approval of the first generic Advair Diskus in 2019, after years of failed attempts by multiple applicants, illustrates both the difficulty of the pathway and the market concentration that follows for those who navigate it successfully.
Inhaler Generics: The Device-Drug Combination Problem
Metered-dose and dry-powder inhalers present the most technically demanding bioequivalence demonstration challenges in the complex generic category, because the drug delivery depends not just on the formulation but on the device’s aerodynamic performance. The FDA requires that inhaler generic applicants demonstrate in vitro equivalence of aerodynamic particle size distribution (APSD), device resistance, spray content uniformity, drug delivery rate, and actuator/chamber deposition, in addition to in vivo pharmacokinetic or pharmacodynamic bioequivalence.
For fluticasone/salmeterol inhalation powder (Advair Diskus), the FDA’s 2019 revised PSG requires a Q1/Q2 sameness requirement — the generic must contain the same qualitative and quantitative excipient blend as the RLD — combined with a device equivalence demonstration and a PK study in healthy volunteers. Multiple applicants failed this combination over the preceding decade. Perrigo, Cipla, and Sandoz all received CRLs for Advair generic applications at various points between 2013 and 2019, citing failures in device characterization or formulation matching. Mylan’s approval came after the company developed a proprietary manufacturing process for lactose carrier blending that reproduced the Diskus’s APSD profile within FDA tolerances.
The IP valuation dimension of inhaler generics deserves specific attention. GlaxoSmithKline’s Advair franchise carried a patent estate that included compound patents (expired), formulation patents covering the specific powder blend and particle engineering process, device patents covering the Diskus mechanism, and method-of-use patents for specific asthma and COPD indications. The compound patents’ expiration did not open the product to generic competition because the formulation and device patents extended practical exclusivity. GSK’s strategy of patenting the manufacturing process and device mechanics rather than just the API is now standard practice for inhaler originators, and it is the primary reason why inhaler generic development timelines are measured in years, not months.
Liposomal Injectables: Characterization Without a Molecular Definition
Liposomal drug products, where an API is encapsulated within phospholipid bilayer vesicles to modify its distribution and reduce toxicity, are complex because the ‘drug product’ is not a single molecular entity with a defined structure. The bioequivalence of a generic liposomal product depends not only on the API identity and concentration but on the vesicle size distribution, lipid composition, encapsulation efficiency, zeta potential, and the dynamic balance between encapsulated and non-encapsulated API fractions. All of those parameters affect the product’s pharmacokinetic behavior in vivo.
Doxil (doxorubicin HCl liposome injection, Janssen) — the original liposomal chemotherapy approved in 1995 — had no approved generic until Sun Pharmaceutical received approval for Lipodox in 2013, following an extended manufacturing shortage of Doxil that prompted FDA to expedite review under the shortage mitigation pathway. Even after Lipodox’s approval, questions about exact equivalence persisted in oncology practice, because the FDA’s PK-based bioequivalence standard for liposomal doxorubicin does not require equivalence of tissue-level doxorubicin concentrations, which are the pharmacologically relevant endpoint in oncology. This clinical nuance is not just academic: oncologists at some institutions maintained separate formularies for Doxil and generic equivalents based on this uncertainty, and that prescribing behavior affected generic substitution rates and market penetration.
The FDA’s 2018 guidance on drug products containing nanomaterials acknowledged the characterization challenge explicitly, requiring physicochemical characterization data alongside PK bioequivalence for liposomal products. Subsequent PSGs for liposomal amphotericin B (AmBisome), vincristine sulfate liposome (Marqibo), and cytarabine/daunorubicin liposome (Vyxeos) each specify a unique combination of in vitro characterization tests and in vivo data requirements, making each liposomal generic effectively a novel technical challenge rather than a standard application of an established bioequivalence paradigm.
Glatiramer Acetate: Complex Mixture Bioequivalence and the Copaxone Saga
Glatiramer acetate (Copaxone, Teva) is a mixture of synthetic polypeptides composed of four amino acids in a specific molar ratio, with molecular weights ranging from 5,000 to 9,000 daltons. Because it is not a single molecular entity, standard pharmaceutical equivalence criteria — identical API, identical molecular weight, identical pharmacokinetic profile — cannot be applied directly. The FDA classified glatiramer acetate as a complex mixture and required a combination of physicochemical characterization (amino acid composition, molecular weight distribution, antigenicity), pharmacokinetic analysis (which is difficult because the drug does not have a simple systemic PK profile), and immunological data to support bioequivalence.
Teva’s patent protection strategy for Copaxone is one of the most analyzed evergreening case studies in pharmaceutical IP. After the compound patents on glatiramer acetate itself expired, Teva filed patents covering specific dosing regimens: the three-times-weekly 40 mg/mL dose introduced in 2014, protecting it from generic competition even as the daily 20 mg/mL formulation faced generic entry from Mylan and Sandoz. The three-times-weekly dose regimen patents were challenged in IPR proceedings, and PTAB invalidated the key claims in 2017 on obviousness grounds, finding that the dose regimen would have been obvious to a skilled formulator given existing clinical data. The Federal Circuit affirmed in 2018, and multiple generic versions of the 40 mg/mL product entered the market. Teva lost approximately $1 billion in annual Copaxone revenue between 2017 and 2020 as a direct result of generic entry.
For generic applicants, the Copaxone litigation history offers a precise roadmap: when an originator’s primary protection shifts to dosing regimen patents rather than compound or formulation patents, the IPR obviousness attack has a high historical success rate. Dosing optimization studies rarely generate patent claims that can withstand challenge on the basis that the optimal dose would not have been obvious to a person of ordinary skill given the existing literature.
Key Takeaways: Complex Generics
Complex generic programs are capital-intensive multi-year projects, not fast-follower opportunities. The technical barriers are real: inhaler APSD matching, liposomal characterization, complex mixture equivalence, and drug-device combination testing each require specialized expertise and often require proprietary manufacturing process development. But the reward structure — concentrated markets, sustained price premiums, and high barriers to subsequent competitive entry — justifies the investment for manufacturers with the technical capabilities and balance sheets to absorb multi-cycle CRL risks. The patent analysis for complex generics must extend beyond compound patents to encompass device patents, manufacturing process patents, and dosing regimen patents, each of which has been used effectively to extend practical exclusivity well beyond the primary patent term.
Investment Strategy Note: Complex generic pipeline assets should be valued using real-option methodology rather than simple NPV, because the probability of approval per submission cycle is meaningfully lower than standard generics and the value of ultimately achieving approval is substantially higher. A complex generic program that has already received one CRL and resolved the cited deficiencies has a substantially higher approval probability than a first-submission estimate would suggest, and that probability improvement should be reflected in the valuation.
Part V: Regulatory Intelligence as Competitive Advantage
FDA Priority Review Programs and the ANDA Queue
The FDA’s Office of Generic Drugs (OGD) manages ANDA prioritization through a tiered system. Applications for drug products on the FDA’s Drug Shortage list receive expedited review under the shortage prioritization policy. Applications for drug products with fewer than three approved generics (the ‘competitive generic therapy’ or CGT designation, established under the FDA Reauthorization Act of 2017) receive a 180-day marketing exclusivity upon approval of the first applicant, analogous to the Paragraph IV first-to-file exclusivity but applicable regardless of patent status. Applications for drug products identified in the FDA’s Complex Drug Substances list or subject to a priority ANDA designation also receive accelerated review.
CGT designation is an underutilized tool in generic pipeline strategy. For molecules where fewer than three generic applications are approved but where no active Paragraph IV litigation is preventing entry (perhaps because the remaining Orange Book patents have already been adjudicated, or because the NDA holder chose not to file suit), the CGT pathway provides both expedited review and a temporary market exclusivity that justifies development investment even for lower-revenue products. Applicants that systematically scan for CGT-eligible targets, particularly products coming off shortage lists or products with abandoned ANDAs from prior filers, can build a pipeline of high-approval-probability, low-competition targets that generate reliable cash flow even without first-generic exclusivity on blockbusters.
Complete Response Letters: Pattern Recognition for Approval Prediction
FDA CRL data, published in aggregate in OGD’s annual reports, reveals a consistent pattern of deficiency types. Manufacturing and facility deficiencies account for roughly 40% of first-cycle CRLs. Bioequivalence deficiencies (failed studies, inadequate study design, insufficient data) account for approximately 25%. Chemistry, manufacturing, and controls (CMC) deficiencies — primarily labeling errors, stability data gaps, and container closure system documentation — account for roughly 20%. Patent and exclusivity issues, primarily incorrect or incomplete certification statements, account for approximately 10%, with the remaining 5% distributed across miscellaneous administrative deficiencies.
The practical value of this breakdown is that it concentrates pre-submission quality assurance effort appropriately. A manufacturing site audit and compliance check conducted three to six months before submission filing is far more economically valuable than a final formatting review of the administrative sections. An independent bioequivalence data package review by an outside expert biostatistician — not the CRO that conducted the study — is worth the cost because it simulates the FDA reviewer’s scrutiny more closely than an internal review does.
The FDA’s published analysis of ANDA major deficiencies (released as a data summary through OGD) identifies that the most common CMC deficiencies involve container closure system testing (particularly for injectable products where leachables and extractables data from the primary packaging must be fully characterized), drug substance specifications (inadequate control of polymorphic form for BCS Class II drugs), and dissolution method development (inadequate discriminatory power of the dissolution method, which is a problem because a non-discriminatory method cannot reliably detect formulation differences between batches).
Post-Approval Considerations: Maintaining the Approval
ANDA approval is not the end of the regulatory relationship with the FDA; it is the beginning of a sustained post-market compliance obligation. Any post-approval change to the approved formulation, manufacturing site, testing methodology, container closure system, or labeling must be reported to the FDA under the prior approval supplement (PAS), changes being effected in 30 days (CBE-30), or annual report categories, depending on the significance of the change. A manufacturing site change from a validated site to a new facility requires a PAS, which triggers a new manufacturing site inspection and can take 12 to 18 months to approve.
The FDA’s Generic Drug User Fee Amendments (GDUFA) III framework introduced enhanced commitments for post-approval supplement review timelines that align with the original ANDA review goals, but PAS reviews involving facility changes remain subject to the same inspection-based delays that affect original ANDA approvals. For generic manufacturers managing large portfolios — Teva’s global generics business, for example, includes over 3,500 marketed products — the post-approval compliance burden is itself a significant operational infrastructure investment.
Key Takeaways: Regulatory Intelligence
Systematic FDA database monitoring — including OGD’s publicly available ANDA action data, warning letter databases, 483 inspection observation records, and the Orange Book’s daily updated patent and exclusivity listings — is the operational core of a competitive generic intelligence function. Companies that update their regulatory intelligence on a rolling basis, rather than at annual strategy reviews, identify CGT opportunities, competitive application filings, and manufacturing site risk flags months ahead of competitors who rely on periodic reports. The difference between filing an ANDA six months before a competitor and filing six months after, on a high-value Paragraph IV target, is routinely the difference between 180 days of exclusivity and zero.
Investment Strategy Note: When assessing a generic manufacturer’s regulatory capability, the most predictive metric is not the size of the ANDA portfolio but the first-cycle approval rate. A company with 200 ANDAs approved at a 65% first-cycle rate is a better regulatory execution machine than one with 400 approvals at a 35% first-cycle rate. The regulatory efficiency measure — total review time from submission to approval divided by the GDUFA performance goal — is an underused benchmark in generic company diligence that directly predicts future development productivity and capital efficiency.
Part VI: Biosimilars and the Parallel Approval Architecture
The 351(k) Pathway and What ‘Interchangeability’ Actually Means
Biologics license applications for biosimilars follow a parallel but distinct regulatory architecture under Section 351(k) of the Public Health Service Act, established by the Biologics Price Competition and Innovation Act (BPCIA) of 2010. The BPCIA’s analytical framework for demonstrating biosimilarity requires that the proposed biosimilar be ‘highly similar’ to the reference biologic ‘notwithstanding minor differences in clinically inactive components,’ and that there be ‘no clinically meaningful differences’ in safety, purity, and potency. These standards are more demanding and more ambiguous than the small-molecule ANDA bioequivalence standard, because biologics are structurally complex enough that exact molecular identity between manufacturer batches is not achievable even by the originator.
Biosimilar interchangeability is a higher regulatory standard than biosimilarity. An interchangeable biosimilar must meet the biosimilarity standard and must also be shown to produce the same clinical result as the reference product in any given patient, and — for products administered more than once — that alternating or switching between the interchangeable and the reference product presents no greater risk than continued use of the reference product. The practical significance of interchangeability is that pharmacists in most U.S. states can substitute an interchangeable biosimilar for the reference product without prescriber intervention, analogous to small-molecule generic substitution.
Boehringer Ingelheim’s Cyltezo (adalimumab-adbm) became the first interchangeable biosimilar to AbbVie’s Humira upon FDA designation in 2021, following completion of a switching study that enrolled approximately 240 patients across three treatment sequence alternation cycles. That data package cost an estimated $50 to $80 million to generate. The commercial payoff from interchangeability designation, in the form of formulary substitution at pharmacy level, depends on state law implementation: all 50 states have now enacted laws permitting interchangeable biosimilar substitution, but the notification and patient opt-out requirements vary. In 2023, when seven adalimumab biosimilars launched on the U.S. market, most achieved formulary positioning through negotiated rebate structures with PBMs rather than purely through interchangeability substitution.
The BPCIA ‘Patent Dance’ and Its Strategic Implications
The BPCIA’s patent disclosure and negotiation process, colloquially called the ‘patent dance,’ requires the biosimilar applicant to provide the reference product sponsor (RPS) with its application and manufacturing process information within 20 days of FDA accepting the application for review. The RPS then has 60 days to identify patents it believes would be infringed by the biosimilar, and the parties negotiate a subset to litigate in an initial infringement action. This process was intended to front-load patent resolution before commercial launch, but the Supreme Court’s 2017 Sandoz v. Amgen decision held that participation in the patent dance is optional, not mandatory. Biosimilar applicants that elect not to participate can still be sued by the RPS, but the suit timeline shifts to a post-approval posture.
AbbVie’s litigation strategy against adalimumab biosimilar applicants included assertions of over 100 U.S. patents on Humira, creating what patent analysts called a ‘patent thicket’ designed primarily to generate negotiation complexity rather than to litigate each patent on its merits. All major biosimilar applicants — Samsung Bioepis, Sandoz, Mylan/Viatris, Coherus, Fresenius Kabi, and Boehringer Ingelheim — ultimately settled with AbbVie under confidential license agreements that permitted U.S. market entry in January 2023, approximately four years after European biosimilar adalimumab launch. AbbVie collected approximately $21 billion in U.S. Humira revenue in 2022, the year before biosimilar entry, making the patent thicket strategy one of the most economically successful IP defense executions in pharmaceutical history.
Key Takeaways: Biosimilars
Biosimilar interchangeability requires a switching study and carries a development cost 5-10x higher than a standard biosimilarity package. The commercial payoff from that investment depends heavily on formulary access and PBM negotiations, not just FDA designation. The BPCIA patent dance is optional but waiving participation shifts litigation risk to a post-approval timeline, which suits biosimilar applicants pursuing aggressive commercial launch strategies. AbbVie’s Humira thicket is the benchmark for biologic IP defense; any biosimilar or complex generic program targeting a high-revenue biologic must model patent resolution timelines conservatively and structure deal economics around late-entry scenarios.
Conclusion: A Predictive Scoring Framework for ANDA Programs
Predicting ANDA approval likelihood is a multi-variable analytical problem, not a binary patent-expiry timing exercise. The variables that most reliably predict successful ANDA outcomes are: patent estate quality and remaining term (with PTE and pediatric exclusivity adjustments), NDA holder litigation posture and authorized generic history, bioequivalence feasibility for the specific formulation and patient population, manufacturing site compliance history for all proposed sites and API suppliers, the completeness and accuracy of the labeling package and patent certification, and the FDA’s current review queue characteristics for the relevant product type.
For any specific ANDA target, these variables can be scored, weighted, and combined into a program probability estimate that is more accurate and more actionable than simple patent-expiry calendaring. IP teams and portfolio managers who build this kind of structured predictive framework — updated continuously with current Orange Book, OGD, and FDA compliance data — consistently outperform those who rely on static patent expiry databases and periodic market research reports. The 180-day first-generic exclusivity is still among the most reliable value-creation mechanisms in pharmaceutical development. Capturing it requires getting the patent analysis right, the bioequivalence strategy right, the manufacturing site selection right, and the submission quality right, simultaneously. That is a cross-functional challenge, and the companies that solve it systematically are the ones that consistently dominate the generic pipeline.




(2) Pathway")
(2) Pathway: Unlocking a Hybrid Strategy for Drug Innovation")
(2) Approval Strategy: The Complete Step-by-Step Guide to a Successful NDA Submission")





(2) and 505(b)(1) Regulatory Pathways")







(2) Beats the Branded Generic Every Time — Unless It Doesn't")
 Rejections, CRL Patterns, and CMC Pitfalls")




