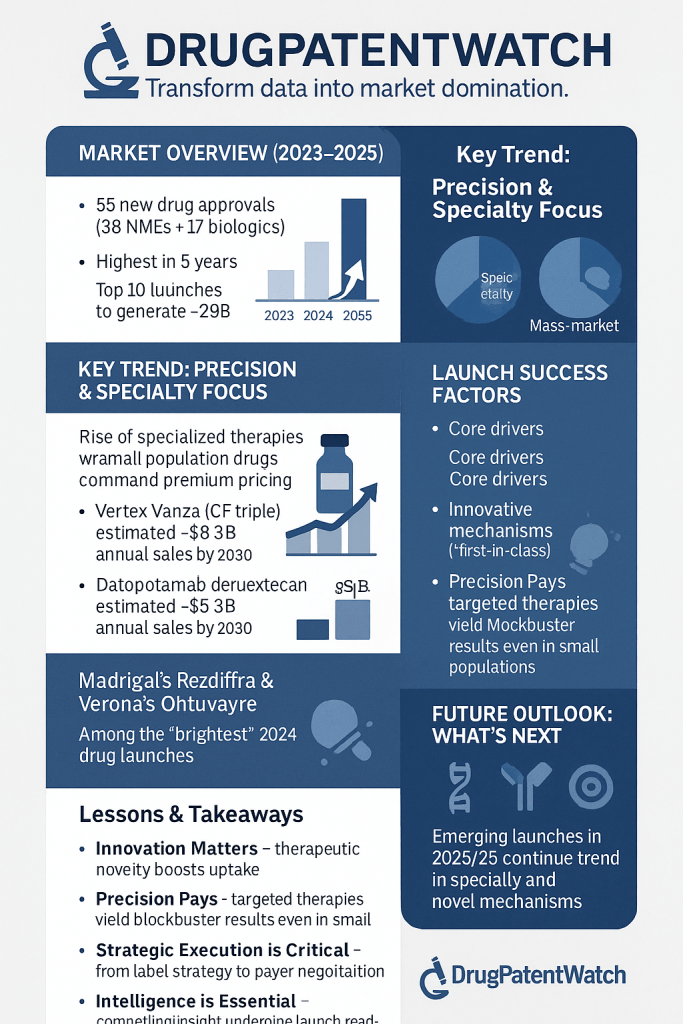
The FDA approved 55 new drugs in 2023, including 38 new molecular entities (NMEs) and 17 biologics license applications (BLAs), the most in a five-year span. That output continued into 2024, a year that produced some of the most commercially spectacular launches in the industry’s recent history. Merck’s Winrevair (sotatercept) opened at a $14,000-per-vial list price and generated $419 million in its first nine months on the U.S. market. Madrigal Pharmaceuticals launched Rezdiffra (resmetirom) as the first and only approved therapy for metabolic dysfunction-associated steatohepatitis (MASH) and reached 11,800 patients on therapy by year-end. Bristol Myers Squibb’s Cobenfy (xanomeline and trospium chloride) entered the market as the first genuinely new pharmacological mechanism for schizophrenia in more than 50 years. Vertex followed its Trikafta franchise with Alyftrek, the branded name for the vanzacaftor/tezacaftor/deutivacaftor combination, approved on January 2, 2025.
These four launches share a set of structural characteristics that go well beyond good clinical data or a skilled sales force. Each one entered the market with a carefully constructed intellectual property estate, a pricing strategy calibrated against payer access thresholds, and a label broad enough to sustain commercial expansion over a multi-year exclusivity window. The top 10 most anticipated launches of 2025 are collectively projected to generate $29 billion in cumulative revenue by 2030. Understanding what drove that potential requires moving past surface-level commercial analysis into the specific IP mechanics, clinical positioning decisions, and market access architecture that separate exceptional launches from adequate ones.
This article covers each major 2024-2025 launch in technical depth, with sub-sections on patent estate valuation, evergreening tactics, Paragraph IV exposure, and the commercial infrastructure decisions that will determine how durable each product’s revenue floor proves to be.
Part One: The 2024-2025 FDA Approval Cycle
Record Volume, Shifting Class Mix, and What It Means for Commercial Competition
The 55 approvals in 2023 were not evenly distributed. The NME-to-BLA ratio of roughly 2:1 reflects a deliberate industry shift toward first-in-class small molecules that can carry both compound and method-of-use patent coverage, rather than platform biologics that depend more heavily on 12-year BPCIA data exclusivity. That ratio matters commercially because NMEs are eligible for five-year New Chemical Entity (NCE) exclusivity under the Hatch-Waxman Act, triggering a clear ANDA Paragraph IV clock that generic manufacturers can anticipate and prepare for. Biologics carry their own exclusivity scaffold under the Biologics Price Competition and Innovation Act (BPCIA), but the competitive dynamics around biosimilar entry are substantially different: development costs are higher, interchangeability designations remain contested, and the 12-year data exclusivity period gives innovators a longer runway to build patient registries and payer contracts that create structural switching barriers.
What made 2024 different was not simply the volume of approvals but the concentration of first-in-class mechanisms. In Winrevair, Rezdiffra, Cobenfy, and Alyftrek, the market received four drugs that each occupied an essentially uncontested pharmacological space on the day of approval. That scarcity of immediate direct competition gave each launch team the pricing power and label flexibility that drugs entering crowded therapeutic categories rarely enjoy.
First-in-Class Mechanism as an IP and Commercial Asset
The term ‘first-in-class’ has commercial value primarily because it can be operationalized into a durable IP position. When a drug introduces a genuinely new mechanism of action, the initial compound patents typically cover not only the specific molecule but the broader chemical genus from which it was selected. Method-of-use patents filed early in clinical development can then lock up the specific indication before any competitor has sufficient data to attempt a Paragraph IV challenge against them. The combination of compound patents, formulation patents, dosing regimen patents, and method-of-use patents constitutes what patent professionals call a ‘patent thicket,’ though the term is misleading when applied to first-in-class compounds: when the mechanism itself is novel, the layers of protection are not defensive evergreening but the natural result of genuine innovation.
The Winrevair case illustrates this clearly. Sotatercept operates as an activin signaling inhibitor targeting the ALK1 pathway, a mechanism with no prior approved agent in PAH. The compound patents that Acceleron Pharma built before Merck’s acquisition covered not just sotatercept but the class of ligand traps designed to rebalance pro- and anti-proliferative signaling in pulmonary vascular smooth muscle cells. Those upstream patents, now owned by Merck, provide the kind of protection that cannot be designed around without abandoning the mechanism entirely.
Key Takeaways: The 2024-2025 Approval Cycle
The 2023-2024 approval surge reflects genuine scientific productivity concentrated in areas where multiple independent research programs converged on the same biological targets over a 10-to-15-year period. MASH, PAH, cystic fibrosis, and schizophrenia all share the characteristic that prior approved therapies addressed symptoms rather than disease mechanisms, leaving the biological underpinnings open for the kind of first-in-class innovation that commands premium pricing and broad FDA labels. The commercial implications extend forward: as these first-in-class drugs establish clinical practice patterns, the second and third entrants in each class will face not just Paragraph IV litigation but entrenched formulary positions and physician familiarity that are harder to dislodge than a patent.
Part Two: Merck’s Winrevair (Sotatercept)
Clinical Profile, Label Architecture, and the PAH Market Structure
The FDA approved Winrevair on March 26, 2024, for adults with pulmonary arterial hypertension (WHO Group 1), making it the first activin signaling inhibitor ever approved in any indication. The Phase 3 STELLAR trial met both its primary endpoint (six-minute walk distance) and multiple secondary endpoints, including time to clinical worsening, WHO functional class improvement, and N-terminal pro-B-type natriuretic peptide reduction. The label Merck received covered add-on therapy to background PAH treatment, giving prescribers the flexibility to combine Winrevair with virtually any existing prostacyclin pathway agent, endothelin receptor antagonist, or PDE5 inhibitor without label restriction.
One prescribing cardiologist quoted in the commercial launch analysis described the label as containing ‘everything they wanted with a cherry on top,’ which translates into an unusually permissive prescribing framework that drove third-quarter 2024 sales to approximately $149 million, nearly double the figure from Winrevair’s initial quarter on market. By the end of 2024, total sales reached $419 million and prescriber and patient counts had grown roughly 40-fold relative to the first week of launch.
The ZENITH trial data, supporting Merck’s October 2025 label expansion, added an explicit claim for reducing the risk of clinical worsening events including hospitalization, lung transplantation, and death. The expanded label converts Winrevair from a functional-improvement drug into one with hard outcomes data, which is the critical threshold that many payer formulary committees require before moving a drug to preferred status. That regulatory milestone will extend the commercial trajectory well beyond the initial launch curve.
The PAH market that Winrevair entered was already substantial. Johnson & Johnson controlled approximately half of the then-$7.3 billion global PAH market through Uptravi (selexipag) and Opsumit (macitentan), with J&J’s Opsynvi combination tablet (macitentan plus tadalafil) approved in 2023. The market is projected to reach $12.2 billion by 2032. Winrevair’s addition of a mechanistically orthogonal drug to existing combination regimens, rather than competing head-to-head with existing agents, positions it as an additive revenue opportunity for the category rather than a market-share displacement story.
IP Valuation: Winrevair’s Patent Estate and Exclusivity Runway
Merck acquired sotatercept as the cornerstone of its $11.5 billion acquisition of Acceleron Pharma in November 2021. That transaction, at the time criticized by some analysts as expensive for a single Phase 3 asset, looks structurally different when examined through the IP lens. What Merck actually purchased was not just the clinical data package but the full patent estate covering the ALK1 activin signaling inhibitor class, including compound patents, formulation patents, and method-of-use patents that collectively protect Winrevair from biosimilar competition until approximately 2037.
Under the BPCIA, Winrevair carries 12 years of reference product exclusivity in the United States, running from the March 2024 approval date to approximately March 2036. In the European Union, the equivalent data exclusivity period is 10 years. The granted compound and treatment patents extend protection to 2037 in the U.S., providing roughly one additional year of IP protection beyond the BPCIA exclusivity floor. In practice, biosimilar development for a fusion protein of this structural complexity requires two to three years of preclinical characterization, analytical comparability work, and Phase 1 PK bridging studies before any biosimilar can file a 351(k) application, meaning the effective competitive entry window sits well into the mid-2030s at the earliest.
The IP valuation question for institutional investors is how to model the revenue contribution of Winrevair against the Keytruda patent cliff. Keytruda (pembrolizumab) generated approximately $29.5 billion in 2024 and faces loss of exclusivity in 2028. Merck has explicitly positioned Winrevair as a ‘main driver of growth’ in the post-Keytruda period, with William Blair projecting $4.9 billion in annual Winrevair sales by 2029, and industry analysts projecting peak annual sales between $4.9 billion and $11.4 billion depending on indication expansion assumptions. The variance in those forecasts reflects genuine uncertainty about how aggressively Merck pursues the Alzheimer’s psychosis indication, pulmonary hypertension related to heart failure with preserved ejection fraction (PH-HFpEF), and other exploratory programs that could materially expand the patient population.
The $14,000 per-vial list price, dosed at 0.3 mg/kg subcutaneously every three weeks and titrated to a target of 0.7 mg/kg, generates annual gross revenue per patient on the order of $200,000 to $280,000 depending on dose and patient weight. Net pricing, after rebates and patient assistance, runs materially lower, but the gross-to-net spread in PAH drugs has historically been narrower than in oncology because the payer base is concentrated in private commercial insurance and Medicare Part B, where the rebate dynamics are structurally different from Part D.
The Acceleron Acquisition as Structured IP Transaction
The Acceleron deal warrants examination as a case study in IP-driven M&A. Acceleron’s enterprise value at the time of Merck’s acquisition was built almost entirely on two assets: sotatercept and luspatercept (Reblozyl, already partnered with BMS). The sotatercept IP estate covered not only the clinical-stage compound but the full activin receptor type II fusion protein class, including manufacturing know-how, scalable Chinese hamster ovary (CHO) cell line production processes, and formulation trade secrets. These elements are not patentable in the same way that a compound structure is, but they function as practical exclusivity barriers because biosimilar manufacturers must independently develop equivalent production processes without access to Acceleron’s cell banking and purification protocols.
Merck’s willingness to pay 1.2 times Acceleron’s trailing 52-week average market price reflected, in part, the premium for acquiring these manufacturing trade secrets alongside the formal patent estate. A competitor developing a biosimilar to sotatercept would need to develop its own CHO expression system, characterize its own glycosylation profile, and demonstrate analytical comparability to Merck’s reference standard, all without access to Acceleron’s internal process knowledge. That practical barrier adds several years to the effective exclusivity period beyond what the formal BPCIA data exclusivity clock suggests.
Investment Strategy: Winrevair
For institutional investors, Winrevair’s risk-reward profile hinges on three variables: indication expansion speed, the net pricing trajectory as formulary competition evolves, and the pace at which Merck builds the prescriber base beyond the initial PAH specialist community. The ZENITH outcomes data resolves the first and most important uncertainty, providing the hard endpoint data that major health technology assessment bodies require for reimbursement decisions in Germany (under AMNOG), France, and the United Kingdom. Investors tracking the Merck story should monitor the pace of Winrevair net price relative to list price over the next two to three years: any significant deterioration in net pricing, particularly in the commercial insurance segment, would be an early signal that payer resistance is building and that the $4.9 billion-to-$11.4 billion sales peak range should be anchored at the lower end.
Part Three: Madrigal Pharmaceuticals and Rezdiffra (Resmetirom)
First-in-Disease Positioning in MASH
Madrigal received FDA approval for Rezdiffra (resmetirom) on March 14, 2024, for adults with noncirrhotic MASH with moderate to advanced liver fibrosis (F2 to F4 on the Metavir scale), making it the first drug ever approved for this indication. MASH, formerly known as nonalcoholic steatohepatitis (NASH), affects an estimated 16 to 20 million Americans with moderate to advanced fibrosis, and is projected to become the leading cause of liver transplantation in the United States within this decade. Before Rezdiffra, the standard of care was weight loss and metabolic risk factor management, with no pharmacological option carrying FDA approval.
Resmetirom works as a liver-directed thyroid hormone receptor-beta (THR-beta) agonist. By selectively activating THR-beta receptors in hepatocytes rather than systemic THR-alpha receptors (which drive cardiac and bone effects), resmetirom reduces hepatic lipogenesis and stimulates mitochondrial fatty acid oxidation without the cardiovascular side effects that had stalled earlier systemic thyroid hormone analogs. The Phase 3 MAESTRO-NASH trial showed that resmetirom met both co-primary endpoints: NASH resolution with no worsening of fibrosis, and at least a one-stage improvement in fibrosis with no worsening of NASH activity score. Those co-primary endpoints, simultaneously, at 52 weeks, established the clinical bar for regulatory approval and now set the de facto standard against which any competitor must demonstrate equivalence or superiority.
Rezdiffra launched commercially in the second quarter of 2024. Net sales reached $103.3 million in the fourth quarter of 2024 alone and $180.1 million for the full year, with more than 11,800 patients on therapy by December 31, 2024. By Q3 2025, quarterly net sales had grown to $287.3 million, with more than 29,500 patients on therapy, a trajectory that implies a run-rate above $1 billion annualized within the first six quarters of commercial availability. The American Association for the Study of Liver Diseases (AASLD) updated its practice guidelines to recommend Rezdiffra as a first-line therapy for MASH, an endorsement that carries formulary access implications across hospital systems and pharmacy benefit managers.
IP Valuation: Rezdiffra’s Orange Book Architecture and the 2044-2045 Patent Clock
Rezdiffra’s IP estate reflects a textbook example of the method-of-use patent layering that companies use to extend the effective exclusivity period beyond the initial compound patent expiration. Resmetirom as a chemical entity carries existing patent coverage, but the commercially critical IP is the suite of method-of-use and dosing regimen patents that Madrigal has systematically added to the FDA’s Orange Book since the March 2024 approval.
In July 2025, the USPTO issued a Notice of Allowance for a patent covering Rezdiffra’s commercial weight-threshold dosing regimen as described in the FDA-approved label, with protection running through September 30, 2044. That patent (U.S. Patent No. 12,377,104, titled ‘Methods for treating a fatty liver disease’) was listed in the Orange Book in August 2025. A subsequent patent was added to the Orange Book providing protection into February 2045. The weight-threshold dosing claim is strategically important: the FDA-approved label specifies dosing based on patient weight categories (80 mg for patients under 100 kg, 100 mg for those 100 kg or above), meaning that any generic manufacturer seeking approval for the full label indication must either design around the dosing claim or challenge it with a Paragraph IV certification.
The Paragraph IV clock for Rezdiffra starts on March 14, 2028 (four years after approval, the NCE-1 date under Hatch-Waxman). Generic manufacturers can file ANDAs with Paragraph IV certifications at that point, but any contested Paragraph IV triggers a 30-month stay of generic approval, pushing the earliest possible generic entry to roughly September 2030. Given that the compound’s generic launch date is estimated at September 17, 2033 based on the full patent and exclusivity stack, Madrigal has approximately nine years of practical exclusivity from the April 2024 commercial launch date.
The competitive threat is not primarily from small-molecule generics over the near term but from branded competitors entering the MASH space. GLP-1 receptor agonists (semaglutide in particular) have Phase 3 programs in MASH, and obeticholic acid (Intercept/Iqvia Therapeutics) previously failed to gain approval but remains under development. Madrigal’s 2025 licensing agreement with CSPC Pharma for an oral GLP-1 receptor agonist (MGL-2086) is a direct strategic response to the combination therapy opportunity: Rezdiffra’s THR-beta mechanism and a GLP-1 mechanism are pharmacologically additive in addressing the steatotic and fibrotic components of MASH, and a fixed-dose combination would reset the Orange Book listing and potentially provide a new exclusivity runway into the early 2050s.
IP Valuation: The Orange Book Listing as a Strategic Moat
Madrigal’s approach to Orange Book prosecution illustrates the method-of-use patent strategy at its most operationally precise. The key distinction between a compound patent and a method-of-use patent in the context of pharmaceutical exclusivity is that method-of-use patents cover specific therapeutic uses, dosing schemes, and patient selection criteria as described in the approved label. When those claims are tightly aligned with the label language, a generic manufacturer cannot simply rely on a ‘skinny label’ (section viii carve-out) to avoid them without also carving out the primary indication, which in Rezdiffra’s case is the only indication.
With the weight-threshold dosing patent covering the FDA-approved label through 2044 and compound patents providing coverage into the early 2030s, the layered structure means that any generic manufacturer faces at least three separate patent challenges simultaneously: the compound patent, the method-of-use patent for MASH treatment, and the dosing regimen patent. The cost and uncertainty of litigating three independent Orange Book-listed patents with different expiration dates through a Paragraph IV challenge exceeds the expected profit margin of a first-filer generic in a specialty drug with a currently finite patient population. That economic calculus, rather than any single patent’s strength, constitutes the real moat.
Key Takeaways: Rezdiffra
Madrigal’s transformation from a single-asset clinical-stage company to a commercial-stage entity generating over $1 billion in annual revenue run-rate within six quarters of launch is attributable to three structural factors operating simultaneously. First, the first-in-disease regulatory position eliminated the formulary access negotiation that drugs entering established therapeutic categories face; payers had no comparator and no basis for rebate demands from an alternative. Second, the AASLD guideline endorsement of first-line status provided clinical cover for gastroenterologists and hepatologists to prescribe without waiting for comparative data against a non-existent standard of care. Third, the Orange Book patent strategy, with method-of-use coverage running to 2044-2045, provided investors with a credible long-duration revenue forecast that justified the capital required to build a commercial infrastructure from scratch.
Investment Strategy: Rezdiffra
The MAESTRO-NASH OUTCOMES trial, which could read out with clinical outcomes data (liver-related events, transplantation, death) this decade, is the single most important catalyst for Rezdiffra’s long-term commercial trajectory. A positive readout would establish Rezdiffra as the only MASH therapy with hard outcomes data in F2-F4 patients, triggering formulary renegotiations across major PBMs and potentially supporting a price increase or reduced rebate requirement. The European commercial launch, beginning with Germany in 2025, adds a revenue stream that was not yet reflected in the Q3 2025 run rate and expands the addressable patient population substantially. Investors should track the net sales to gross sales ratio over the next four to six quarters as payer mix evolves: if net-to-gross deteriorates faster than patient volume grows, it signals that payer-mandated step therapy requirements are limiting the addressable population more than the company’s guidance implies.
Part Four: Vertex’s Alyftrek (Vanzacaftor/Tezacaftor/Deutivacaftor)
The Serial Innovation Model in Cystic Fibrosis
Vertex has spent more than a decade executing a deliberate lifecycle management strategy in cystic fibrosis that uses each successive CFTR modulator approval to build a new IP layer while sustaining or expanding the commercial patient population. The sequence runs from Kalydeco (ivacaftor, 2012) to Symdeko (tezacaftor/ivacaftor, 2018) to Trikafta (elexacaftor/tezacaftor/ivacaftor, 2019) to Alyftrek (vanzacaftor/tezacaftor/deutivacaftor, approved January 2, 2025). Each transition added clinical benefit, reset the Orange Book patent clock, and extended Vertex’s effective monopoly over the CFTR modulator category.
Alyftrek, the commercial name for the vanza triple that Vertex had been developing under its previous working name, received Priority Review based on data from the SKYLINE 102, SKYLINE 103, and RIDGELINE 105 pivotal trials. The primary endpoint was non-inferiority on absolute change from baseline in ppFEV1 (percent predicted forced expiratory volume in one second) relative to Trikafta. Alyftrek met that endpoint, and on the key secondary endpoint of sweat chloride reduction, it demonstrated superiority: in SKYLINE 102, sweat chloride decreased by 7.5 mmol/L with Alyftrek versus an increase of 0.9 mmol/L for Trikafta. In SKYLINE 103, Alyftrek reduced sweat chloride by 5.1 mmol/L compared to 2.3 mmol/L for Trikafta. Sweat chloride reduction to below the diagnostic threshold for CF (60 mmol/L) is widely regarded as the best surrogate for disease modification, and Alyftrek’s superiority on that measure provides the clinical narrative for label differentiation from Trikafta.
The drug received Fast Track, Priority Review, and Orphan Drug designations from the FDA. Vertex used a priority review voucher to accelerate the review timeline from the standard 10 months to 6 months, enabling the January 2, 2025 PDUFA date instead of a mid-2025 target. The priority review voucher represented a capital allocation decision: Vertex was willing to spend a voucher worth several hundred million dollars in the secondary market to move the Alyftrek launch forward by four months, reflecting the company’s internal estimate of the revenue at stake during that window.
IP Valuation: Alyftrek’s Role in Vertex’s CF Evergreening Strategy
The ‘vanza triple’ represents Vertex’s most aggressive execution of a technology-driven evergreening strategy in recent pharmaceutical history. Evergreening, broadly defined, refers to the practice of obtaining new patents on modified versions of an approved drug to extend market exclusivity beyond the life of the original compound patent. The term carries a pejorative connotation when applied to trivial reformulations or minor salt changes, but Vertex’s CF program does not fit that characterization: each successive modulator generation introduces genuinely superior CFTR correction or potentiation, as evidenced by the sweat chloride data.
The structural IP argument for Alyftrek as a lifecycle management asset rather than a defensive evergreening maneuver rests on three points. Vanzacaftor is a new chemical entity, not a modification of elexacaftor; it has a distinct molecular structure and a different binding mode within the CFTR protein. Deutivacaftor is a deuterated analog of ivacaftor, where strategic hydrogen-to-deuterium substitution at the metabolically labile positions of the ivacaftor scaffold reduces CYP3A4-mediated first-pass metabolism and extends the half-life sufficiently to support once-daily dosing (Trikafta requires twice-daily dosing). The once-daily dosing claim is patentable as a formulation and method-of-use innovation and is clinically meaningful for patient compliance in a chronic disease requiring lifelong treatment. Tezacaftor is retained from the Trikafta combination, providing Vertex with manufacturing continuity and supply chain economies.
From an IP clock perspective, Alyftrek’s new compound patents for vanzacaftor and the deutivacaftor deuterium substitution extend Orange Book coverage well beyond Trikafta’s existing expiration dates. The Trikafta compound and formulation patents expire on dates ranging from 2024 through the early 2030s depending on the specific patent, and Vertex has faced sustained Paragraph IV challenges from generic manufacturers targeting those filings. Alyftrek’s new NDA, with vanzacaftor as a new chemical entity carrying NCE exclusivity, provides five years of data exclusivity from the January 2025 approval date, meaning generic manufacturers cannot file ANDAs challenging Alyftrek’s compound patents until January 2030 at the earliest. The full patent stack is likely to extend effective exclusivity into the late 2030s.
Alyftrek is projected to generate $8.3 billion in annual sales by 2030, supported by Vertex’s established global reimbursement relationships for Trikafta and the straightforward patient-switching narrative: physicians managing the roughly 90% of CF patients eligible for CFTR modulator therapy have strong clinical and compliance rationale (superior sweat chloride, once-daily dosing) to transition current Trikafta patients to Alyftrek as formulary negotiations complete.
The CFTR Technology Roadmap: Beyond Small Molecules
Vertex’s pipeline beyond Alyftrek maps two parallel development tracks. The first targets the remaining approximately 10% of CF patients who either have mutations not responsive to CFTR modulator therapy or who lack functional CFTR protein expression entirely, numbering more than 5,000 patients. For these patients, Vertex is developing a nebulized mRNA therapy that would provide temporary CFTR protein expression through inhaled mRNA delivery, bypassing the mutation-specific limitation of CFTR correctors and potentiators. This program is in early Phase 1 development.
The second track pursues next-generation small molecules that could further improve the efficacy ceiling established by Alyftrek, potentially enabling sweat chloride normalization across a broader patient population, including children younger than 6 years (the RIDGELINE study includes cohorts in the 2-to-5-year age group). Each new approval in younger patients resets Orphan Drug Exclusivity calculations and opens pediatric exclusivity extensions of six months, adding incremental IP protection.
Key Takeaways: Alyftrek
Vertex’s CF franchise is the most complete executed example of IP-integrated lifecycle management in modern pharmaceutical history. The company has successfully transitioned the CF patient population through three modulator generations in 13 years, each time capturing a new exclusivity period while improving clinical outcomes. The once-daily Alyftrek formulation provides a concrete clinical differentiation from Trikafta that facilitates patient switching and payer acceptance. The pipeline investments in mRNA and next-generation modulators for young pediatric patients ensure that Vertex maintains patent coverage in CF well into the 2040s even as the current Alyftrek NDA exclusivity matures.
Investment Strategy: Alyftrek
The Alyftrek transition thesis is straightforward for institutional investors: Vertex carries a dominant global CF market position, Alyftrek’s once-daily convenience profile creates switching momentum, and the compound’s NDA exclusivity provides a clear timeline through at least 2030. The key risk is not generic competition in the near term but the pace and completeness of global reimbursement coverage transitions from Trikafta to Alyftrek. Germany’s AMNOG process, France’s Haute Autorite de Sante, and the UK’s NICE each require independent submissions demonstrating added therapeutic benefit relative to the existing comparator (Trikafta), and any rejection of the ‘added benefit’ claim would force Vertex into cost-control negotiations that could compress European net pricing. Investors should treat European HTA submission outcomes in 2025-2026 as the primary valuation catalyst for the Alyftrek transition.
Part Five: Bristol Myers Squibb’s Cobenfy (Xanomeline and Trospium Chloride)
A New Pharmacological Mechanism After 50 Years
Cobenfy received FDA approval on September 27, 2024, for the treatment of schizophrenia in adults. The drug combines xanomeline, a dual M1- and M4-preferring muscarinic acetylcholine receptor agonist, with trospium chloride, a peripherally restricted muscarinic antagonist that does not appreciably cross the blood-brain barrier. The rationale for the combination is that xanomeline activates M1 and M4 receptors centrally (the therapeutic target) while also activating peripheral muscarinic receptors (producing nausea, gastrointestinal disturbance, and cardiovascular effects). Trospium chloride, because it does not cross the blood-brain barrier, blocks xanomeline’s peripheral muscarinic activity without antagonizing its central therapeutic effects.
Every antipsychotic approved for schizophrenia in the preceding five decades had operated through dopamine D2 receptor antagonism or partial agonism. Cobenfy’s mechanism is entirely orthogonal: it does not block D2 receptors at all. This distinction is clinically meaningful in two respects. First, it predicts a side effect profile without the extrapyramidal symptoms, tardive dyskinesia, and prolactin elevation that complicate long-term D2 antagonism, which affects tolerability and long-term adherence. Second, it opens the question of whether Cobenfy might address symptom domains, particularly cognitive and negative symptoms, that respond poorly to D2 blockade, which is the subject of ongoing Phase 3 trials in adjunctive therapy settings.
The EMERGENT clinical program (EMERGENT-2, EMERGENT-3, and long-term open-label extensions) established efficacy on Positive and Negative Syndrome Scale (PANSS) total scores and Clinical Global Impression of Severity (CGI-S) measures in placebo-controlled trials. Phase 3 programs in Alzheimer’s disease-associated psychosis (ADEPT-1 and ADEPT-2) are active, with the potential to expand the approved indication into a substantially larger patient population. A positive ADEPT readout would represent the most material near-term revenue catalyst for BMS’s Cobenfy program.
The $14 Billion Karuna Acquisition as a Structured IP Bet
Bristol Myers Squibb closed its acquisition of Karuna Therapeutics for $14 billion in March 2024, six months before Cobenfy’s approval. The acquisition premium reflected BMS’s assessment of the IP and commercial value of the KarXT program, which analysts projected could reach $6 billion or more in annual sales across schizophrenia and Alzheimer’s psychosis indications.
The IP situation Karuna brought to BMS was complex. Xanomeline had been discovered by Eli Lilly in the 1980s, licensed to Karuna’s predecessor entity by Lilly in 2012, and the Cobenfy combination concept was developed at PureTech Health before Karuna became independent. The novel IP in Cobenfy is not the individual components, both of which are prior art, but the combination itself: the specific claim that co-administering a peripheral muscarinic antagonist (trospium) enables therapeutic muscarinic agonism (xanomeline) in the CNS without the peripheral tolerability problems that had halted xanomeline’s development in the 1990s. That combination concept is patentable as a method-of-use and composition-of-matter claim, and Karuna’s patent strategy focused on building that combinatorial IP estate.
PureTech retained royalty rights to Cobenfy under its founding entity agreements, specifically approximately 2% royalties on net annual sales above $2 billion. The FDA approval triggered $29 million in milestone payments to PureTech. That royalty structure provides a publicly traded reference point for tracking Cobenfy’s net sales trajectory: once BMS reports sales above the $2 billion threshold, PureTech’s royalty payments will provide an independent revenue verification data point for investors.
NCE Exclusivity Risk and the Paragraph IV Clock
Cobenfy’s FDA exclusivity classification was a contested question before approval. Karuna indicated before launch that it anticipated NCE exclusivity for Cobenfy, arguing that because xanomeline had never previously received FDA approval as a drug product, the xanomeline component qualified as a new active moiety under 21 CFR 314.108(b)(2). If that analysis holds, Cobenfy receives five-year NCE exclusivity, preventing any ANDA filing with a Paragraph IV certification before September 2029.
The alternative classification is three-year exclusivity under 21 CFR 314.108(b)(4), applicable to new clinical investigations essential to approval, which would permit ANDA filings as early as September 2027. The difference between five-year NCE exclusivity and three-year exclusivity is not merely two years of protected revenue; it is also the difference between a Paragraph IV challenge filed with limited clinical data in 2027 versus one filed after Cobenfy has had four years to establish prescribing patterns, payer contracts, and a patient population that creates practical switching barriers.
The composition-of-matter patent covering the combination of xanomeline and trospium and the method-of-use patents covering schizophrenia treatment are the next layer of defense after the regulatory exclusivity period. Because trospium chloride has been a generic drug since 2004 (originally approved as Sanctura), and because xanomeline as an isolated compound is prior art, the combination patents are the only IP asset that can delay generic combination entry. Those patents are subject to Paragraph IV challenges by any generic manufacturer that files an ANDA for the combination product, and the strength of those claims, particularly the degree to which the combination represents a non-obvious solution to xanomeline’s tolerability problem, will be tested in litigation once the Paragraph IV clock opens.
BMS’s IP team faces the challenge of building an Orange Book listing for Cobenfy that creates enough method-of-use coverage to require potential Paragraph IV challengers to address multiple claims simultaneously. The Alzheimer’s psychosis indication, if obtained, would generate a new method-of-use patent listing and potentially a new period of three-year exclusivity for the new indication, layering additional IP on top of the existing schizophrenia estate.
Key Takeaways: Cobenfy
Cobenfy’s value proposition rests on two separable claims: that its mechanism produces antipsychotic efficacy without the extrapyramidal and metabolic liabilities of D2 antagonism, and that its M1/M4 agonism may address cognitive and negative symptom domains that current antipsychotics leave untreated. The first claim is supported by Phase 3 data. The second is being tested in the ongoing EMERGENT adjunctive program and the Alzheimer’s psychosis ADEPT trials. The IP complexity, particularly the contested NCE exclusivity question and the reliance on combination patents covering prior-art components, makes Cobenfy’s exclusivity runway shorter and more legally vulnerable than Winrevair’s or Rezdiffra’s. BMS purchased a mechanism franchise, not just a single indication.
Investment Strategy: Cobenfy
The Alzheimer’s disease psychosis indication is the key commercial catalyst: the AD psychosis population is substantially larger than the adult schizophrenia population (BMS has described it as a multifold expansion of the addressable market), and a positive ADEPT readout would trigger a supplemental NDA that could receive three years of exclusivity for the new clinical evidence. Investors should closely track the NCE exclusivity classification, which BMS has not definitively confirmed publicly, and the timeline of any Paragraph IV certifications against Cobenfy’s Orange Book-listed patents, which would provide the first concrete signal of generic manufacturers’ IP assessment. The BMS royalty payment to PureTech serves as an independent revenue verification mechanism once sales cross $2 billion annually.
Part Six: The Five Structural Pillars of a Successful Drug Launch
Market Access Architecture Starts in Phase 2
The single most consistent differentiator between exceptional drug launches and adequate ones is not the sales force or the marketing budget. It is the quality of market access planning that begins during Phase 2 clinical development and drives pricing, clinical endpoint selection, health economic modeling, and payer communication strategy long before an NDA is filed.
The misalignment between market access strategy and overall launch planning is a documented failure mode. A substantial proportion of pharmaceutical companies build market access teams years before launch but fail to connect those teams’ work to clinical trial endpoint selection, label strategy, or health economic modeling in a way that produces coherent value evidence packages. The result is that the payer-facing value story is assembled retroactively from clinical data that was not designed with payer requirements in mind, which creates gaps in economic evidence that payers exploit in formulary negotiations.
For the 2024-2025 class of successful launches, the pattern ran differently. Madrigal’s MAESTRO-NASH trial was designed with AASLD guideline requirements in mind from the outset, including the simultaneous co-primary endpoint structure (NASH resolution AND fibrosis improvement) that mirrors the clinical evidence standard guideline committees use. Merck’s STELLAR trial included the six-minute walk distance as primary endpoint and clinical worsening events as key secondary endpoints, providing both functional and outcomes evidence in a single pivotal program. Vertex’s SKYLINE program used head-to-head comparator data against Trikafta specifically to generate the ‘added benefit’ evidence that European HTA bodies require for Alyftrek to receive reimbursement on preferred terms.
The technical tool set for early market access integration includes HEOR modeling using decision-analytic simulation frameworks (commonly Markov models or partitioned survival models), targeted payer advisory boards in major markets during Phase 2b to test value messaging, and systematic engagement with national and regional formulary bodies to understand their specific evidentiary requirements before the Phase 3 design is locked.
Pricing Strategy: The Winrevair $14,000-Per-Vial Decision
Pharmaceutical pricing decisions in rare and specialty disease are shaped by the intersection of willingness-to-pay thresholds across payer segments, international reference pricing (IRP) implications, net-to-gross spread optimization, and the long-term forecasted revenue curve under different access scenarios. Winrevair’s $14,000-per-vial list price was described in commercial analyses as ‘higher than expected,’ yet the drug achieved strong payer access on the commercial side from launch. That outcome was not accidental.
PAH is a rare disease with a well-established precedent for high-cost therapies: prostacyclin analogs (epoprostenol, treprostinil) and endothelin receptor antagonists have list prices in the six-figure annual cost range, and payers managing PAH patient populations have developed coverage and utilization management frameworks calibrated to that price range. A new mechanism entering at a price consistent with existing PAH agents, rather than attempting to redefine the category price ceiling, faces less formulary access friction than a drug in a lower-cost therapeutic area attempting to establish a new price point without comparator precedent.
The Quantzig case study framework for pharmaceutical launch pricing illustrates the systematic methodology: comprehensive market research to establish stakeholder willingness-to-pay by segment, competitive analysis to identify the price floor below which underprice risk exceeds the access benefit, predictive modeling of volume response curves under different net pricing scenarios, and risk assessment of international reference pricing blowback if U.S. list prices anchor international submissions below sustainable thresholds. The companies that achieve optimal launch pricing outcomes treat the exercise as a quantitative optimization problem rather than a strategic negotiation, accepting that the output may be a price point that feels counterintuitive relative to internal cost-plus benchmarks but that maximizes net present value of the exclusivity window.
Competitive Intelligence and Scenario Planning
Structured competitive intelligence, extending beyond standard secondary research into conference coverage, regulatory submission monitoring, and clinical trial endpoint analysis, is a separable capability from standard market research. The Deallus case study for PharmaA’s rare disease launch (referenced in the source material) describes a continuous competitive monitoring program that covered conference abstracts, orphan drug approval scenarios, and competitor drug deep dives, combined with cross-functional workshops that distributed competitive knowledge throughout the organization rather than siloing it in the business development function.
The commercial value of pre-launch competitive intelligence lies in its ability to de-risk label strategy decisions and pricing scenarios against a realistic competitive horizon. A company launching into a ‘sole approved therapy’ position, as Rezdiffra did, prices and positions differently from one that projects a second entrant within 18 to 24 months of its own approval. Getting that competitive horizon wrong in either direction has direct P&L consequences: underestimating competitive entry speed leads to price points that become unsustainable when a comparator arrives, while overestimating it leads to defensive access concessions (step therapy carve-outs, preferred placement concessions) that unnecessarily sacrifice revenue during the exclusivity window.
For the 2024-2025 launch class, competitive intelligence affected several specific decisions. Madrigal’s parallel clinical investment in the MAESTRO-NASH OUTCOMES trial, while expensive, reflected the recognition that GLP-1 receptor agonists were entering Phase 3 MASH programs and that outcomes data would be the long-term competitive moat. Merck’s decision to pursue the ZENITH outcomes trial rapidly after STELLAR approval reflected similar competitive awareness: J&J’s Opsynvi combination tablet, approved in 2023, had no comparable outcomes data, and securing the hard endpoint label language before any competitor could do so establishes Winrevair’s formulary priority on clinical grounds.
Label Breadth as a Commercial Asset
The FDA label, specifically the indication language, dosing flexibility, and contraindication profile, is a commercial asset that requires active investment during clinical development to maximize. Label breadth determines which patient segments are eligible for the drug under standard of care, which payer tiers the drug occupies, and how much clinical flexibility prescribers have in selecting patients without off-label exposure.
The Winrevair label is the clearest 2024 illustration of this principle. Approval across the full WHO functional class spectrum (II through IV), without restriction to background therapy combinations, without biomarker-based patient selection requirements, and with home administration capability (subcutaneous injection that patients can self-administer) gave prescribers and payers a product with broad eligibility criteria and low administrative burden. The physician description of the label as having ‘everything they wanted with a cherry on top’ translated directly into rapid formulary access and high prescription rates in the initial quarters.
Narrow labels, by contrast, require commercial teams to work around restrictions or engage in off-label promotion (which creates regulatory and legal risk), and they give payers explicit formulary management levers: step therapy requirements, prior authorization criteria keyed to label-specified patient characteristics, and quantity limits tied to approved dosing ranges. The investment in broad label clinical evidence, which typically means running additional arms in pivotal trials or supplementary studies to capture data across patient subgroups, pays compound returns through the commercial access period.
Digital and Omnichannel Commercial Execution
The COVID-19 pandemic fundamentally altered the infrastructure of pharmaceutical commercial execution. The move from field-force-dominant to hybrid engagement models, combining in-person clinical detailing with digital medical education, real-world evidence programs, patient journey tools, and remote access for non-targeted prescribers, was accelerated by necessity and has not reversed. The 2024-2025 launch class executed against this hybrid model as standard operating procedure rather than as an adaptation.
Omnichannel execution in pharmaceutical launches encompasses several technically distinct components. Medical education programs for key opinion leaders (KOLs) at academic medical centers operate on a different timeline and channel mix from reach-and-frequency detailing of community prescribers. Patient identification and identification support tools, often built on claims data analytics and electronic health record (EHR) flags, help community physicians identify undiagnosed or undertreated patients who match the approved label criteria. Speaker bureau programs and peer-to-peer education, long the backbone of specialty drug adoption, have been supplemented with virtual advisory boards and digital congress coverage that allows broader prescriber exposure to clinical data without travel.
The quantitative benchmark most often cited for omnichannel launch execution is a 23% higher market share at 12 months post-launch for companies with advanced analytics capabilities relative to those with less developed data infrastructure. That premium reflects not the technology itself but the speed and precision of targeting decisions: knowing which physicians are seeing the highest volume of eligible patients, which are currently undertreating due to unawareness of the drug, and which require medical rather than commercial engagement narrows the resource allocation problem and compresses the adoption S-curve.
Part Seven: IP Strategy as Launch Infrastructure
The Layered Patent Estate: Compound, Formulation, Method-of-Use, and Dosing Claims
A pharmaceutical patent estate at the time of commercial launch typically consists of four distinct patent layers, each with a different expiration date, a different vulnerability to Paragraph IV challenge, and a different role in delaying generic or biosimilar entry. Understanding the interaction between these layers is fundamental to IP valuation work.
The compound patent covers the specific molecular structure of the active pharmaceutical ingredient (API). For NMEs, it is filed during preclinical or Phase 1 development, typically carrying a 20-year term from the priority date. By the time of approval (which typically occurs 8 to 12 years after the earliest priority date), the effective remaining compound patent term is 8 to 12 years, which the Patent Term Extension (PTE) mechanism under 35 U.S.C. 156 can extend by up to five years to compensate for the time spent in regulatory review.
The formulation patent covers the specific dosage form, excipient combination, delivery system, or dosing frequency used in the approved drug product. These patents are most valuable when the formulation provides a clinical benefit (extended release, reduced food effect, improved bioavailability) that would require a generic manufacturer to use the same formulation to demonstrate bioequivalence. Deutivacaftor’s once-daily dosing advantage over ivacaftor in Alyftrek exemplifies this: the deuterium substitution is both a compound and a formulation innovation, making it simultaneously an NME and a formulation improvement.
The method-of-use patent covers the specific therapeutic indication, patient population, and dosing regimen as described in the FDA-approved label. These are the most commercially durable patents because they can be filed late in development (based on pivotal trial data), listed in the Orange Book immediately at approval, and layered across multiple indications as label expansions occur. Rezdiffra’s weight-threshold dosing regimen patent through 2044 is a method-of-use filing that would have been impossible to draft before the Phase 3 dose-finding data was available.
The dosing regimen patent is a subcategory of method-of-use, specifically covering the dose, frequency, titration schedule, and patient selection criteria as approved in the label. When the dosing regimen is clinically essential to the drug’s efficacy or safety profile, which is demonstrable from Phase 3 data, these patents are especially resistant to design-around attempts because a generic manufacturer filing an ANDA must demonstrate bioequivalence to the approved dosage form, which inherently means using the approved dosing regimen.
Biologic Exclusivity Under the BPCIA: 12-Year Data Protection as a Revenue Moat
The BPCIA’s 12-year reference product exclusivity period runs from the date of first FDA approval and prevents any biosimilar applicant from relying on the innovator’s data to support a 351(k) application during that window. This is categorically different from the Hatch-Waxman NCE exclusivity: it cannot be circumvented by a biosimilar developer who independently generates equivalent safety and efficacy data, because the exclusivity protection attaches to the reference product’s regulatory status, not to the underlying clinical data.
For Winrevair, the BPCIA exclusivity runs to March 2036. For any biologic approved in the 2023-2024 cohort, the 12-year clock provides a revenue floor that is immune to early Paragraph IV-type challenges. The practical consequence is that a biologic innovator can plan commercial investments, manufacturing capacity expansions, and indication extension clinical programs with a revenue floor guaranteed through the exclusivity period. That planning certainty is a qualitative asset that does not appear explicitly in patent valuation models but that directly affects the capital efficiency of a biologic franchise.
The ‘patent dance’ provisions of the BPCIA, which govern information exchange between the reference product sponsor and a biosimilar applicant after the 351(k) application is filed, create additional procedural timelines that typically add 18 to 36 months to the effective market entry date for biosimilars even after the 12-year exclusivity expires. Combined with the analytical complexity of demonstrating biosimilarity to a fusion protein like sotatercept, the practical biosimilar entry window for Winrevair sits closer to 2038 to 2040 than the 2036 exclusivity expiration date suggests.
Evergreening Tactics: A Technology Roadmap for IP Lifecycle Extension
‘Evergreening’ encompasses a range of IP prosecution and product development strategies that extend market exclusivity beyond the compound patent’s original expiration date. The tactics span a spectrum from genuinely innovative product improvements (new indications supported by Phase 3 data, pediatric formulations, fixed-dose combinations with synergistic clinical benefit) to less defensible strategies (trivial salt changes, polymorph patents on forms that provide no clinical benefit, minor reformulations that do not change bioavailability).
The regulatory framework distinguishes between these strategies through the Orange Book listing criteria: a patent must relate to the approved drug product and must be a patent that could reasonably be asserted against a competitor making, using, or selling the drug. The FDA does not adjudicate patent validity, but it does review whether a submitted patent meets the listing criteria, and incorrectly listed patents carry legal and regulatory exposure under the False Claims Act and FDA regulations.
For the 2024-2025 launch class, the following evergreening strategies are either active or available:
Rezdiffra carries a combination therapy potential with oral GLP-1 (MGL-2086) that, if developed into a fixed-dose combination product, would generate a new NDA, a new NCE exclusivity period (if the GLP-1 component qualifies), and a new Orange Book listing. The CSPC Pharma licensing agreement covering an oral GLP-1 derivative is the first step in that program.
Alyftrek’s deutivacaftor deuterium substitution illustrates deuterium chemistry as an evergreening tool: deuterium-labeled versions of approved drugs can achieve patent protection as new chemical entities if the deuterium substitution produces measurable clinical differences (extended half-life, reduced metabolite toxicity, improved tolerability). The FDA has recognized deuterium-substituted drugs as eligible for NME designation, as demonstrated by CTP-543 (deutetrabenazine, Austedo) for tardive dyskinesia.
Cobenfy’s active Phase 3 program in Alzheimer’s disease psychosis is an indication-expansion evergreening strategy: a positive ADEPT readout would support a supplemental NDA and three years of exclusivity for the new clinical investigation, extending the effective commercial exclusivity period.
Winrevair’s pediatric development program, covering PAH in pediatric patients, would, if successful, generate six months of pediatric exclusivity under the Best Pharmaceuticals for Children Act (BPCA), appended to all existing patent terms and the BPCIA exclusivity period. For a drug with Winrevair’s revenue trajectory, six months of exclusivity extension is worth several hundred million dollars in protected revenue.
The Paragraph IV Threat: Anticipating Generic Challenges from Day One
A Paragraph IV certification is a generic manufacturer’s legal declaration that the Orange Book-listed patent is either invalid or will not be infringed by the proposed generic product. Filing a Paragraph IV triggers automatic 30-month stay of the ANDA approval decision, during which the brand and generic parties litigate patent validity and infringement. First ANDA filers with Paragraph IV certifications receive 180 days of generic marketing exclusivity upon approval, creating a competitive first-mover advantage that has led to an entire industry of ‘Paragraph IV specialty’ generic manufacturers who systematically target brand drugs for early patent challenges.
The strategic response to Paragraph IV threats begins at the IP prosecution stage: by filing multiple independent Orange Book-listed patents with different expiration dates and different claim structures (compound, formulation, method-of-use, dosing regimen), the innovator ensures that any Paragraph IV filer must certify against all listed patents simultaneously and litigate each independently. The cost and complexity of parallel patent litigation against four or five independent Orange Book-listed patents, each requiring separate expert testimony, claim construction, and validity analysis, materially increases the litigation cost for generic challengers and reduces the expected value of first-filer exclusivity.
For the 2024-2025 drugs, the Paragraph IV risk varies substantially by product. Rezdiffra’s NCE-1 date is March 2028 (four years after approval), and the compound’s estimated generic launch date of September 2033 reflects the full IP stack including the 2044-2045 method-of-use patents. The 30-month stay following any 2028 Paragraph IV filing would run to approximately September 2030, at which point the compound’s remaining compound patent term and the method-of-use patents would be litigated. Cobenfy faces an earlier Paragraph IV window (September 2027 or 2029 depending on NCE exclusivity resolution) and relies more heavily on the combination composition-of-matter claims, which will be rigorously tested once the challenge window opens.
Key Takeaways: IP Strategy
The drugs that generate the highest NPV during their exclusivity windows are those whose IP teams treat the Orange Book as a living commercial document rather than a regulatory filing obligation. Filing method-of-use patents tied precisely to label language, pursuing indication expansions that generate new exclusivity layers, and building combination therapy pipelines that reset the exclusivity clock are all executable strategies for the 2024-2025 approval cohort. The Paragraph IV litigation risk is manageable but not eliminable: the goal is to raise the cost and complexity of challenges to the level where the expected value to a generic challenger falls below the litigation investment, and to build commercial switching barriers through payer formulary contracts and physician relationships that render the first generic entry economically limited even when it eventually arrives.
Part Eight: The $29 Billion Pipeline: 2025’s Most Anticipated Launches
Datopotamab Deruxtecan (Dato-DXd): AstraZeneca and Daiichi Sankyo
Datopotamab deruxtecan (Dato-DXd, branded as Datroway) is a TROP2-directed antibody-drug conjugate (ADC) developed jointly by AstraZeneca and Daiichi Sankyo. The FDA approved it in January 2025 for previously treated unresectable or metastatic HR-positive, HER2-negative breast cancer. Analysts project annual sales reaching $5.9 billion by 2030, reflecting the large addressable population in HR-positive breast cancer and the clinical differentiation provided by the ADC format.
Dato-DXd carries compound patents on both the TROP2-targeting antibody and the DXd payload component, plus formulation patents on the linker-payload conjugation chemistry. The DXd payload (an exatecan derivative) is protected by Daiichi Sankyo’s broader ADC platform patents, which cover the tetrapeptide cleavable linker and the DXd cytotoxic payload across multiple ADC products including trastuzumab deruxtecan (Enhertu). The breadth of the DXd platform patent estate means that any biosimilar manufacturer seeking to develop a Dato-DXd follow-on must navigate both the TROP2 antibody IP and the linker-payload platform IP, creating a two-layer patent challenge.
The Enhertu precedent (trastuzumab deruxtecan, approved across multiple cancer types since 2019) provides a commercial roadmap: AstraZeneca and Daiichi Sankyo have demonstrated they can execute multi-indication lifecycle management in ADCs, with each new indication approval generating a new NDA supplemental, new clinical evidence exclusivity, and additional physician experience data that creates prescribing momentum. The same model applied to Dato-DXd in HR-positive breast cancer positions the drug for a label expansion program covering earlier lines of therapy, non-small cell lung cancer (where Phase 3 data are available), and other TROP2-expressing tumor types.
Ohtuvayre (Ensifentrine): Verona Pharma
Ensifentrine (Ohtuvayre), approved by the FDA in June 2024, is the first inhaled non-steroidal bronchodilator/anti-inflammatory approved in COPD in more than two decades. It works as a dual inhibitor of phosphodiesterase-3 (PDE3) and phosphodiesterase-4 (PDE4), producing both bronchodilation (through PDE3 inhibition in airway smooth muscle) and anti-inflammatory effects (through PDE4 inhibition in immune cells) with a single molecule. That dual mechanism, delivered via nebulization, differentiates Ohtuvayre from both LABAs and inhaled corticosteroids, occupying a distinct pharmacological space.
Verona Pharma, a company with no prior commercial infrastructure, executed its own launch in parallel with Madrigal’s Rezdiffra as a case study in first-in-mechanism positioning by a small-to-midsize company. The IP estate covering the PDE3/4 dual inhibitor mechanism is fundamental to Ohtuvayre’s commercial longevity: because no other approved drug inhibits both enzymes simultaneously with a single molecule, competitors cannot enter this mechanistic space without either licensing Verona’s patents or running their own Phase 3 programs against a now-established comparator.
The 2025 Anticipated Launch Cohort: Revenue Projections and IP Context
The 10 most anticipated launches of 2025 collectively project $29 billion in revenue by 2030. That figure is achievable but carries significant variance depending on indication-specific HTA outcomes in major European markets, the pace of U.S. payer coverage transitions, and competitive entry timelines. The drugs most likely to anchor the high end of that forecast range are those, like Alyftrek and Dato-DXd, with pre-established payer relationships from predecessor drugs in the same franchise, large addressable patient populations, and multi-indication expansion pipelines that can sustain revenue growth after initial label penetration plateaus.
The drugs with higher revenue variance are first-in-disease launches (like a potential ALK2 inhibitor for fibrodysplasia ossificans progressiva) where the patient population is genuinely rare, payer coverage frameworks do not exist at launch, and the clinical differentiation narrative must be built from scratch with no comparator. These launches carry higher per-patient pricing to compensate for market size limitations, but their total addressable market constrains the absolute revenue ceiling regardless of pricing.
Key Takeaways: The 2025 Launch Pipeline
The $29 billion aggregate forecast reflects a shift in the commercial center of gravity in pharma away from blockbuster mass-market drugs (where pricing pressure from PBMs, IRA negotiation, and international reference pricing compresses net revenues) toward rare and specialty biologics and first-in-class small molecules where the IP estate is durable, the payer negotiating position is stronger, and the per-patient economics justify the infrastructure investment. For investors, the most reliable predictor of whether a specific launch will meet its forecast is not the Phase 3 p-value but the breadth of the label, the quality of the Orange Book filing strategy, and the depth of HTA preparation in European markets where outcomes data requirements increasingly govern pricing outcomes.
Conclusion: What the 2024-2025 Class Teaches About IP-Integrated Launch Strategy
The commercial performance of Winrevair, Rezdiffra, Cobenfy, and Alyftrek in their first 12 to 18 months on the market reflects strategies that were locked in years before approval. Each drug entered the market with a first-in-class mechanism, a label built to maximize prescribing flexibility, a pricing strategy calibrated against the specific payer dynamics of its therapeutic category, and an IP estate designed to convert the regulatory exclusivity period into a multi-decade revenue-generating asset.
The common IP architecture across the four drugs includes a compound patent providing baseline protection, method-of-use patents layered onto the Orange Book at or shortly after approval, dosing regimen patents tied precisely to FDA-approved label language, and active pipeline programs (combination therapies, pediatric indications, indication expansions) that will reset portions of the exclusivity clock before the initial compound patents mature.
The commercial architecture includes market access preparation that began in Phase 2, pricing decisions modeled against payer access threshold scenarios rather than internal cost-plus benchmarks, label strategy that invested in clinical evidence breadth rather than minimum regulatory sufficiency, and competitive intelligence programs that anticipated competitor timelines with enough precision to inform both clinical program design and pricing strategy.
For pharma IP teams, the operational takeaway is that Orange Book maintenance is not a post-approval administrative function. It is an active competitive strategy that requires coordinating patent prosecution timelines with clinical development milestones, indication expansion plans, and combination therapy programs in a way that maximizes the IP-protected revenue window. For institutional investors, the clearest signal of a launch likely to exceed projections is the quality of the IP estate, specifically whether the company has method-of-use patents tied to label language that would survive a Paragraph IV challenge, combined with the breadth of the FDA label and the quality of European HTA preparation that will determine net pricing outside the United States. The 2024-2025 class set a high benchmark on all four dimensions. Future launches will be evaluated against it.
Sources: DrugPatentWatch, FDA Orange Book, company SEC filings (Merck, Madrigal, Vertex, BMS, Verona Pharma), Markman Advisors IP analysis, DelveInsight, BioSpace, PharmExec, STAT News, and public HTA submissions.