The Double Patenting Trap: How Pharma Brands Accidentally Invalidate Their Own Moats
Quick Summary: Obviousness-type double patenting (ODP) is a judicially created doctrine that prevents patent holders from stretching exclusivity by obtaining […]
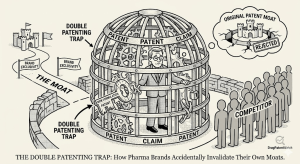
The Double Patenting Trap: How Pharma Brands Accidentally Invalidate Their Own Moats Read Post »